The effect of dendronisation of arylamine centred chromophores on field effect transistor performance
Ellen J.
Wren
,
Karyn
Mutkins
,
Muhsen
Aljada
,
Paul L.
Burn
*,
Paul
Meredith
* and
George
Vamvounis
Centre for Organic Photonics and Electronics, The University of Queensland, Brisbane, Queensland, Australia 4072. E-mail: Meredith@physics.uq.edu.au; p.burn2@uq.edu.au
First published on 1st June 2010
Abstract
We report the design and synthesis of new dendronised hole transport materials and their first use in organic field effect transistors (OFETs) as solution processed amorphous channels. The dendrimers were comprised of a triphenylamine centre, core chromophores containing one or two thiophene units per arm, and first generation biphenyl dendrons with 2-ethylhexyloxy surface groups attached. The dendronised materials were found to be more easily processed than their non-dendronised equivalents. Top contact OFETS were fabricated and found to have hole mobilities in the saturated regime of 1.7 × 10−6 cm2 V−1 s−1 and 1.1 × 10−5 cm2 V−1 s−1 for dendrimers with one and two thiophene units in each arm of the chromophore, respectively. The devices had threshold voltages of around 10 V and ON/OFF ratios in the range 102 to 103. The OFET results demonstrate that for amorphous films it is important that the chromophore is as large as possible to allow for maximal intermolecular interactions.
Introduction
Conjugated dendrimers have had a significant impact on organic optoelectronic devices such as light-emitting diodes1–5 and more recently photovoltaic cells.6–10 A key strength of dendritic materials is that they are macromolecular compounds but because of their branching can be considered as being comprised of discrete molecular chromophores. The former property means that dendrimers can be solution processed in a similar manner to conjugated polymers while the latter allows the controlled design of individual chromophores akin to small molecule semiconductors. Conjugated dendrimers are composed of three main components: the core to which one or more dendrons (the branched units) are attached, and surface groups. In conjugated dendrimers the surface groups provide solubility and hence play an important role in the processing of the materials. The dendrimers are defined by their generation, which is the number of levels of branching in the dendrons. The dendrons themselves can be either insulating, playing a purely structural role,11 or electroactive.12,13 In many of the studies reported to date the core has been the optoelectronic chromophore but it is important to note that it also provides the basic dimensionality of the dendrimer. For example, bi(fluorene) cores lead to more planar dendrimers at low generation14 while a fac-tris(2-phenylpyridyl)iridium(III) core forms the basis of a three dimensional dendrimer.4 These structural components give rise to a number of important materials properties. For example, dendrimers with the same surface groups can be processed under essentially the same conditions. In addition, incorporation of poorly soluble small molecules into a dendritic architecture can enable them to be solution processed. These characteristics have been illustrated extensively for organic light-emitting diodes (OLEDs)15 and to a lesser extent photovoltaic cells but have not been explored in any detail for organic field effect transistors (OFETs).The main efforts on field effect transistors have focused on small molecules with devices fabricated by evaporation, and solution processable conjugated polymers. However, more recently there have been a number of reports of OFETs that have solution processable small molecules with very good device performance.16 In contrast we are aware of only a few reports of dendrimers being used in an active layer of an OFET.17–19 For example, in ref. 17 the active channel layer was formed from a second-generation PAMAM dendronised C60 deposited using the Langmuir–Blodgett technique and the OFETs had mobilities of the order 2.7 × 10−3 cm2 V−1 s−1. There have also been a number of reports of OFETs containing star shaped materials with three or more arms emanating from the central unit [see for example ref. 20], with mobilities generally in the range 10−5–10−2 cm2 V−1 s−1 for solution processed devices. In our development of light-emitting materials we have utilised time-of-flight methods to measure charge mobility and found that dendrimer films formed from simple solution processing can have good hole mobilities. For example, a first generation dendrimer with a bi(9,9-di-n-hexylfluorene) core, biphenyl dendrons and 2-ethyhexyloxy surface groups had a hole mobility of around 2 × 10−4 cm2 V−1 s−1.21 Also, a highly dendronised iridium(III) complex cored dendrimer with carbazole based dendrons and fluorenyl surface groups had a hole mobility of ∼1 × 10−3 cm2 V−1 s−1 at room temperature.13 In both cases the films were not annealed and hence these results suggest that dendrimers could be interesting materials for the active channel layers in OFETs.
In this article we describe the preparation of a family of materials (Fig. 1) that contain triarylamines at their centre and their use as the active layer in OFETs. Triarylamine based compounds have been used extensively as hole-transport materials in applications such as photocopiers, OLEDs and more recently solar cells. We discovered that such materials could also be used as the light-emitting layer in an OLED.22 These previous results of triarylamine based compounds have led to their investigation for the use as the active layers in OFETs.23 We report the synthesis of a new family of OFET materials that are comprised of a triphenylamine centre linked to either first generation dendrons or a simple phenyl ring via one or two thiophene rings. We reveal the effect of the number of thiophene units between the triphenylamine at the centre and the distal unit (the alkoxyphenyl or dendron), and the role the dendron plays on charge transport. The triphenylamine centre was chosen as it is almost planar and in previous work we have shown that the ‘active chromophore’ is extended through the nitrogen atom thus giving a large electroactive moiety with the greatest possibility of intermolecular chromophore interactions.24,25 A large electroactive chromophore should improve intermolecular interactions in the solid-state, and hence charge transport. The thiophene units were incorporated as a structural motif as sulfur–sulfur interactions have been shown to be important for good charge mobility in organic semi- and superconductors.
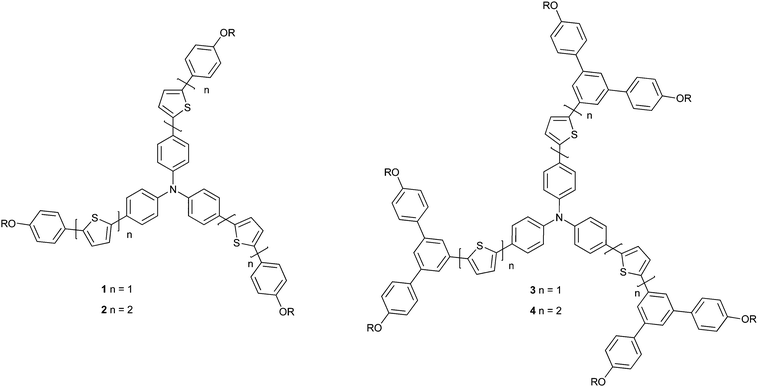 | ||
| Fig. 1 Structures of the materials used in the active channel of the OFETs. | ||
Results and discussion
Synthesis and physical properties
The synthetic pathway to the materials is shown in Scheme 1. In each case a convergent route was followed with the three extended thiophene containing units added in the last step. For the formation of the star compounds 1 and 2 (Fig. 1) the first step was the formation of the boronate ester 6 (Scheme 1) from 4-(2-ethylhexyloxy)phenylbromide 5, which was achieved by formation of the Grignard followed by reaction with trimethylborate, hydrolysis of the dimethylborate with aqueous acid, and finally esterification of the formed boronic acid with ethylene glycol. Under these conditions 6 was isolated in a yield of 65%. We prefer to use the boronate esters rather than the boronic acids as it avoids the formation of dimers and trimers and the material is easier to handle. The advantage of using the glycolate ester compared to the pinacolate esters we have traditionally used is that they are much less expensive although they are a little less stable and a small amount of hydrolysis is observed during chromatography over silica. The boronate ester 6 was then reacted with 5-bromo-2,2′-bithiophene26 under Suzuki conditions to give 9 in a 50% yield. The thiophene containing materials 727 and 9 were brominated with N-bromosuccinimide to give 8 and 10 in yields of 98% and 97% respectively. The final step in the formation of 1 and 2 was the Suzuki coupling of 8 and 10 with tris[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]amine28 with 1 being formed in a 60% yield and 2 being isolated in a 58% yield.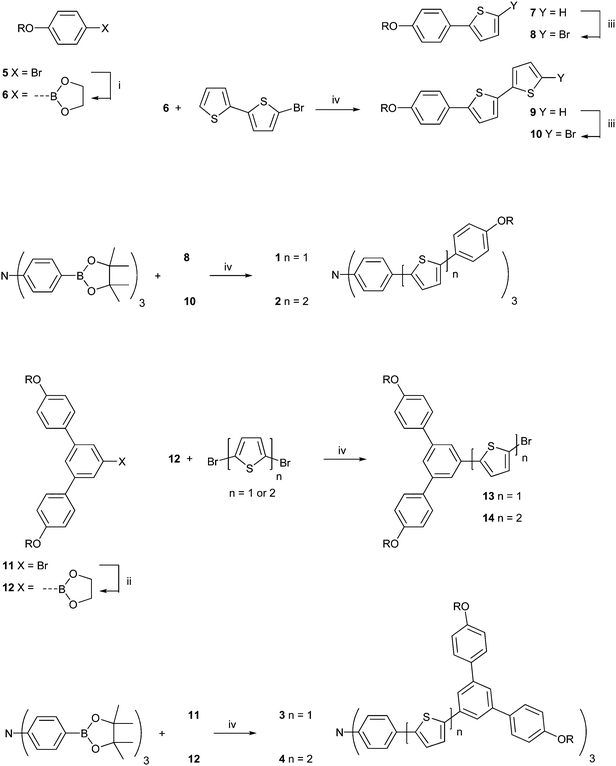 | ||
| Scheme 1 Reagents and conditions: (i) Mg turnings, THF, reflux, 1 h; then B(OMe)3, 0 °C to rt 12 h; then 3 M HCl(aq), rt, 2 h; then ethylene glycol, toluene, reflux Dean–Stark conditions, 12 h; (ii) n-butyllithium, THF, −78 °C, Ar(g); then B(OMe)3, −78 °C to rt 12 h; and then 3 M HCl(aq), rt, 2 h; then ethylene glycol, toluene, reflux Dean–Stark conditions, 8 h; (iii) N-bromosuccinimide, THF, 0 °C, 20 min; (iv) Pd(PPh3)4, 2 M Na2CO3(aq), EtOH, toluene, reflux, Ar(g), 24 h. | ||
The equivalent first generation dendrimers, 3 and 4, were formed by a similar process to that of 1 and 2. Compound 11 was reacted with n-butyllithium to form the anion, which was then treated with trimethylborate. Subsequent hydrolysis with dilute hydrochloric acid followed by reaction with ethylene glycol under Dean–Stark conditions gave the corresponding boronate ester focused dendron 12 in a 92% yield. To add a single thiophene ring 12 was reacted with 2,5-dibromothiophene under Suzuki conditions and this gave a mixture of the desired 13 and the debrominated material. The debrominated compound was then treated with N-bromosuccinimide to selectively brominate the α-position of the thiophene to give an overall yield of 13 of 45% over the two steps. Compound 12 was then reacted with 5,5′-dibromo-2,2′-bithiophene in a similar two step procedure to form 14 in an isolated yield of 49%. To complete the synthesis of the two dendrimers 13 and 14 were reacted with tris[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]amine under Suzuki conditions to give 3 and 4 in the good yields of 72% and 62% respectively. All four materials were thermally stable with decomposition temperatures above 400 °C (determined by thermal gravimetric analysis).
Electronic properties and transistor performance
It is important for good OFET performance to have the minimum offset in energy between the source and drain electrodes and the energy levels of the organic semiconductor in the active channel. The materials of this work lend themselves to p-type OFETs and hence we used cyclic voltammetry to determine the oxidation potentials. The E1/2 for the first oxidation of 1 was found to be 0.2 V and that for 2, 3 and 4 was 0.3 V against the ferrocenium/ferrocene couple. The E1/2s correspond to ionization potentials of 5.0 eV and 5.1 eV respectively given that the ionization potential of ferrocene has been reported to be 4.8 eV.29 Ionisation potentials around 5 eV mean that gold is a suitable contact for the source and drain electrodes in p-type channel operation.A top contact-bottom gate OFET (see Experimental section) architecture was used to test the new semiconducting materials. Fig. 2–5 show the electrical characteristics of devices containing semiconducting channels of 1–4 respectively. In all cases the top panes correspond to plots of IDSversusVDS for a range of gate voltages. From these plots we can see consistent p-type transistor behavior, low leakage currents and clear saturation. From the IDSversusVDS plots the transfer curves [VGversusIDS and (IDS)1/2, bottom panels in Fig. 2–5] for specific source-drain voltages have been derived. Using the IDSversusVDS plots, the transfer curves and eqn (1) and (2) (in the Experimental section) we calculate the key FET performance parameters including carrier mobilities from the linear and saturated regime (µlin and µsat respectively), the threshold voltage for transistor behavior (VT) and the ON/OFF ratio, and these are summarized in Table 1. It is important to note that the values reported in Table 1 represent the averages from multiple devices fabricated with each material and that the plots in Fig. 2–5 all correspond to devices with the same W/L (channel width to length ratio) for direct comparison.
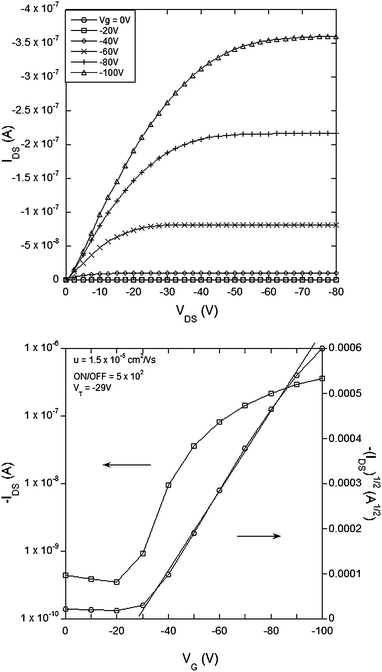 | ||
| Fig. 2 Electrical characteristics of a field effect transistor with compound 1 (small molecule thiophene) as the semiconducting channel with L = 60 µm, W = 46 mm, W/L = 767: (top) IDSvs.VDS and (bottom) transfer curve for VDS = −80 V. | ||
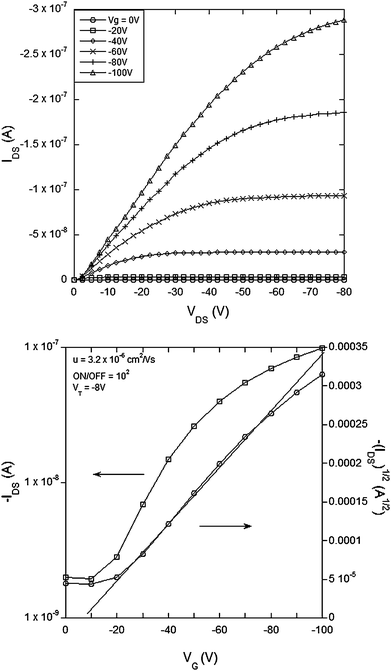 | ||
| Fig. 3 Electrical characteristics of a field effect transistor with compound 2 (small molecule bithiophene) as the semiconducting channel with L = 60 µm, W = 46 mm, W/L = 767: (top) IDSvs.VDS and (bottom) transfer curve for VDS = −80 V. | ||
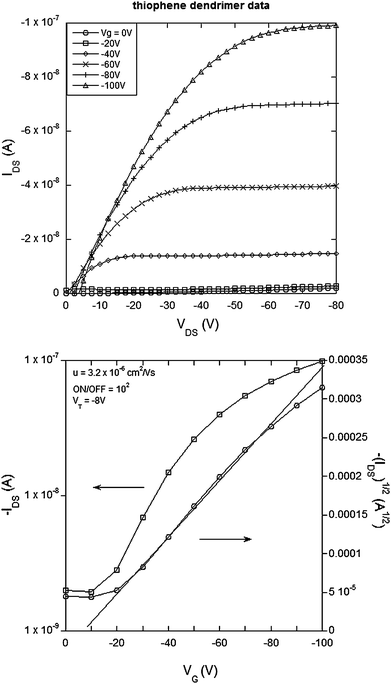 | ||
| Fig. 4 Electrical characteristics of a field effect transistor with compound 3 (thiophene dendrimer) as the semiconducting channel with L = 60 µm, W = 46 mm, W/L = 767: (top) IDSvs.VDS and (bottom) transfer curve for VDS = −80 V. | ||
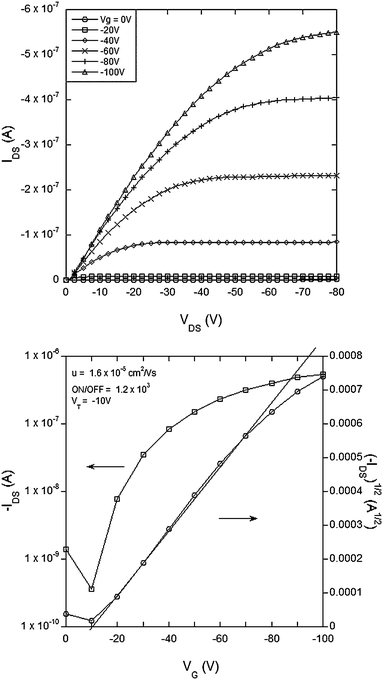 | ||
| Fig. 5 Electrical characteristics of a field effect transistor with compound 4 (bithiophene dendrimer) as the semiconducting channel with L = 60 µm, W = 46 mm, W/L = 767: (top) IDSvs.VDS and (bottom) transfer curve for VDS = −80 V. | ||
| Material | µ sat (standard deviation)/cm2 V−1 s−1 | µ sat,max/cm2 V−1 s−1 | µ lin (standard deviation)/cm2 V−1 s−1 | µ lin,max/cm2 V−1 s−1 | V T/V | ON/OFF |
|---|---|---|---|---|---|---|
| 1 | 1.0 × 10−5 (0.2 × 10−5) | 2 × 10−5 | 1.3 × 10−5 (0.4 × 10−5) | 2 × 10−5 | −8.6 | 1.6 × 103 |
| 2 | 3.7 × 10−5 (0.7 × 10−5) | 6.4 × 10−5 | 4.5 × 10−5 (0.76 × 10−5) | 7 × 10−5 | −7 | 5.6 × 103 |
| 3 | 1.75 × 10−6 (0.9 × 10−6) | 2.9 × 10−6 | 2.8 × 10−6 (1.1 × 10−6) | 4.5 × 10−6 | −8 | 5.3 × 102 |
| 4 | 1.3 × 10−5 (0.3 × 10−5) | 1.65 × 10−5 | 1.5 × 10−5 (0.2 × 10−5) | 1.9 × 10−5 | −10 | 1.9 × 103 |
| P3HT | 0.0147 (2.7 × 10−3) | 0.017 | 0.0142 (2 × 10−3) | 0.017 | −0.5 | 1 × 105 |
To calibrate our device architecture and fabrication procedures we used poly(3-n-hexylthiophene) (P3HT) as a model p-type channel material. The P3HT devices had mobilities of the order 10−2 cm2 V−1 s−1 and ON/OFF ratios of up to 1 × 105. The mobilities of materials 1 to 4 were smaller than the P3HT but respectable for amorphous spin-coated films, and were in the range 10−6–10−5 cm2 V−1 s−1. The threshold voltages were around 10 V and the ON/OFF ratios were between 102 and 103. The mobility results show two general trends: first, increasing the number of thiophene units in the core increases the charge mobility for both the dendronised and non-dendronised materials; and second, the attachment of the dendron only reduces the measured mobility by a small amount (Table 1). For all four materials the electroactive chromophore is comprised of all three arms due to the delocalization of the π-system across the central nitrogen atom.25 The improvement in charge mobility on adding the extra thiophene into 2 and 4 relative to 1 and 3 is therefore due to the improved overlap of the arms of adjacent dendrimers. This is particularly important for amorphous films. The slight decrease in mobility on addition of the first generation biphenyl-based dendrons is due to the fact that the insulating dendrons hold the chromophores on neighbouring dendrimers on average further apart and hence increases the hopping distance. However, the poorer charge transport is more than offset by the improvement in processing ability for the dendrimers which we qualitatively compared by measuring relative processability for 20 mg ml−1 solutions of each molecule in toluene mixed at room temperature. It was also more difficult to form films of the non-dendronised materials 1 and 2, and particularly 2 reliably. In contrast the dendronised compounds 3 and 4 allowed facile formation of good quality thin films.
Conclusion
In summary we have designed and synthesized a family of amine centred materials that differ in the number of thiophene units between the nitrogen and the groups at the surface. We found that when the electroactive chromophore was dendronised it was more easily processed. The materials were used as the semiconductor in OFETs demonstrating for the first time that conjugated dendrimers can be used as the active layers in such a device. The study also illustrates important design criteria for charge transport in amorphous materials, namely that it is important to design the chromophore to have maximal overlap between adjacent molecules in the channel.Experimental
General methods
Column chromatography was performed using the flash chromatography technique over silica. Solvent mixtures are stated as volume/volume proportions. Tetrahydrofuran was distilled from sodium and benzophenone under a nitrogen atmosphere before use. Where used light petroleum was 40–60 °C fraction.1H and 13C NMR spectra were recorded using a 400 MHz Oxford Instruments or a 500 MHz Bruker spectrometer in deuterated chloroform; ThH = thienyl H, spH = surface phenyl H, G1-bpH = dendron branching phenyl H. Coupling constants are quoted to the nearest 0.5 Hz. Microanalyses were carried out in Microanalysis Laboratory of the School of Chemistry and Molecular Biosciences, The University of Queensland or at the Microanalysis Laboratory of the School of Human Sciences, London Metropolitan University. UV-Visible absorption measurements were recorded with a Cary Varian 5000 UV-Vis-NIR spectrophotometer. Melting points were measured in a glass capillary on a BUCHI Melting Point B-545 and uncorrected. Thermalgravimetric analysis (TGA) was carried out on a Mettler-Toledo TGA/DSC 1 STARe System at the Australian National Fabrication Facility (Queensland Node). Decomposition temperatures (Td) are reported for a 10% decrease in mass. Mass spectra were recorded on either an Applied Biosystems Voyager matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) from 2-[(2-E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) in positive reflection mode or on a Finnigan MAT 900XL-TRAP (EI 70 eV) electron impact ionisation.Electrochemistry was performed using a BAS Epsilon electrochemistry station with a glassy carbon working, Ag/AgCl reference, and platinum counter electrodes. All measurements were made at room temperature on samples at a 1 mM concentration in dichloromethane (HPLC grade), with 0.1 M tetra-n-butylammonium tetrafluoroborate as the electrolyte. The solutions were deoxygenated with argon and the ferricenium/ferrocene couple was used as standard.30 The scan rate was 100 mV s−1 and in all cases several scans were carried out to confirm the chemical reversibility of the redox processes.
Bottom-gate, top contact OFETs using 1–4 as the channel semiconductors were fabricated and tested in a glove box (MBRAUN) under a nitrogen environment at 23 °C with oxygen and water levels between 0.1 and 0.3 ppm respectively. For direct comparison and validation of the fabrication process, a series of transistors were also made using standard poly-3-n-hexylthiophene (95% RR, Merck). The general fabrication process was as follows: a heavily doped n-silicon wafer (University Wafer, P(100) 1–10 ohm cm−1 SSP 500um Prime Grade) coated with chromium (5 nm) and gold (40 nm) was used as the gate electrode. The evaporation thicknesses were monitored with a calibrated quartz crystal monitor. Thermally grown SiO2 (350 nm), whose thickness was confirmed using a NanoSPEC 210 thickness measurement system, was used as the gate dielectric. The SiO2 layer was cleaned in a Class 1000 clean room by sonication in acetone (>99.8% by GC) then 2-propanol (>99.9% by GC) for 20 minutes at room temperature, dried with a stream of nitrogen, and then exposed to UV/ozone for 30 minutes (MBRAUN with OP 73). The SiO2 surface was modified with hexamethyldisilazane (HMDS) by covering the substrate surface with 300 µL of neat HMDS and leaving it to stand for 60 seconds before spinning at 800 rpm for 60 seconds. This modification serves to cap potential Si–OH trap sites on the silica surface. The substrate was then baked at 110 °C for 20 minutes on a hot plate to remove excess HMDS. The semiconductor layer was formed by spin-coating filtered (0.25 µm PTFE filter) dendrimer solutions (20 mg mL−1, 9.1 × 10−4 to 18.1 × 10−4 M) in toluene (>99.7%, anhydrous) at 1500 rpm for 60 seconds. The film thicknesses were measured using a Dektak 150 profilometer and were found to be approximately 55 nm. Finally, interdigitated gold source/drain electrode pairs were deposited by vacuum evaporation through an in-house prepared shadow mask, creating a series of OFETs with channel length L = 60 µm and width W = 46 mm. The materials used were annealed before testing at the following temperatures 1 = 40 °C, 2 = 83 °C, 3 = 46 °C, and 4 = 59 °C. These temperatures were found to produce the optimal OFET performances.
The OFETs were characterized using an Agilent B1500A Semiconductor Device Analyser and an SA-6 Semi-Auto Prober. The mobility in the saturation regime was extracted in the usual manner from eqn (1),31 where W is the channel width, L is the channel length, IDS is the drain current, Ci is the capacitance per unit area of the dielectric layer, and VG and VT are the gate voltage and threshold voltage. VT was determined from the relationship between the square root of IDS at the saturation regime and VG of the device by extrapolating the measured data to IDS = 0. The mobility in the linear regime was extracted from eqn (2), where the ON/OFF ratios were calculated from the transfer curves at a VD of −80 V and VG of 0 to −100 V.
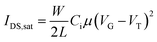 | (1) |
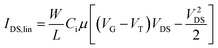 | (2) |
Synthesis
Acknowledgements
This work was funded by the Australian Research Council (PLB Federation Fellowship), the Queensland State Government (PM Smart State Senior Fellowship), and the University of Queensland (Strategic Initiative—Centre for Organic Photonics and Electronics). GV is an ARC Australian Research Fellow. We also acknowledge use of equipment of the Australian Nanofabrication Facility Queensland Node (ANFF-Q) funded by the Australian Federal Government National Collaborative Infrastructure Strategy (NCRIS).References
- J. Li and D. Liu, J. Mater. Chem., 2009, 19, 7584 RSC.
- S.-C. Lo, N. A. H. Male, J. P. J. Markham, S. W. Magennis, P. L. Burn, O. V. Salata and I. D. W. Samuel, Adv. Mater., 2002, 14, 975 CrossRef CAS.
- T. D. Anthopoulos, M. J. Frampton, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2004, 16, 557 CrossRef CAS.
- S.-C. Lo, T. D. Anthopoulos, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2005, 17, 1945 CrossRef CAS.
- S.-C. Lo, R. N. Bera, R. E. Harding, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2008, 18, 3080 CrossRef CAS.
- B. L. Rupert, W. J. Mitchell, A. J. Ferguson, M. E. Köse, W. L. Rance, G. Rumbles, D. S Ginley, S. E. Shaheen and N. Kopidakis, J. Mater. Chem., 2009, 19, 5311 RSC.
- W. J. Mitchell, A. J. Ferguson, M. E. Köse, B. L. Rupert, D. S. Ginley, G. Rumbles, S. E. Shaheen and N. Kopidakis, Chem. Mater., 2009, 21, 287 CrossRef CAS.
- B.-K. An, P. L. Burn and P. Meredith, Chem. Mater., 2009, 21, 3315 CrossRef CAS.
- B.-K. An, R. Mulherin, B. Langley, P. Burn and P. Meredith, Org. Electron., 2009, 10(7), 1356 CrossRef CAS.
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097 CrossRef CAS.
- J. M. Lupton, I. D. W. Samuel, R. Beavington, P. L. Burn and H. Bässler, Adv. Mater., 2001, 13, 258 CrossRef CAS.
- K. A. Knights, S. G. Stevenson, C. P. Shipley, S.-C. Lo, S. Olsen, R. E. Harding, S. Gambino, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2008, 18, 2121 RSC.
- S. Gambino, S. G. Stevenson, K. A. Knights, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2009, 19(2), 317 CrossRef.
- H. Cavaye, A. R. G. Smith, M. James, A. Nelson, P. L. Burn, I. R. Gentle, S.-C. Lo and P. Meredith, Langmuir, 2009, 25(21), 12800 CrossRef CAS.
- P. L. Burn, S.-C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070 CrossRef CAS.
- N. Kawasaki, T. Nagano, Y. Kubozono, Y. Sako, Y. Morimoto, Y. Takaguchi, A. Fujiwara, C.-C. Chu and T. Imae, Appl. Phys. Lett., 2007, 91, 243515 CrossRef.
- A. Mishra, C.-Q. Ma and P. Bauerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS.
- N. Negishi, Y. Ie, M. Taniguchi, T. Kawai, H. Tada, T. Kaneda and Y. Aso, Org. Lett., 2007, 9(5), 829 CrossRef CAS.
- K. H. Kim, Z. Chi, M. J. Cho, J.-I. Jin, M. Y. Cho, S. J. Kim, J.-S. Joo and D. H. Choi, Chem. Mater., 2007, 19, 4925 CrossRef CAS.
- J. P. J. Markham, T. D. Anthopoulos, I. D. W. Samuel, G. J. Richards, P. L. Burn, C. Im and H. Bässler, Appl. Phys. Lett., 2002, 81, 3266 CrossRef CAS.
- D. Ma, J. M. Lupton, R. Beavington, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2002, 12, 507 CrossRef CAS.
- M. Sonntag, K. Kreger, D. Hanft, P. Strohriegl, S. Setayesh and D. de Leeuw, Chem. Mater., 2005, 17, 3031 CrossRef CAS.
- L.-O. Pålsson, R. Beavington, M. J. Frampton, J. M. Lupton, S. W. Magennis, J. P. J. Markham, J. N. G. Pillow, P. L. Burn and I. D. W. Samuel, Macromolecules, 2002, 35, 7891 CrossRef.
- J. M. Lupton, I. D. W. Samuel, P. L. Burn and S. Mukamel, J. Phys. Chem. B, 2002, 106, 7647 CrossRef CAS.
- K.-J. Jung, S. B. Kang, J.-E. Won, S.-E. Park, K. H. Park, J. K. Park, S.-G. Lee and Y.-J. Yoon, Synlett, 2009, 490 CAS.
- S. Vaidyanathan, F. Dötz, H. E. Katz, U. Lawrentz, J. Granstrom and E. Reichmanis, Chem. Mater., 2007, 19, 4676 CrossRef CAS.
- J. Cremer and P. Bauerle, J. Mater. Chem., 2006, 16, 874 RSC.
- P. I. Djurovich, E. I. Mayo, S. R. Forrest and M. E. Thompson, Org. Electron., 2009, 10, 515 CrossRef CAS.
- G. Gritzner and J. Kuta, Electrochim. Acta, 1984, 29, 869 CrossRef.
- S. M. Sze, K. K. Ng, Physics of Semiconductor Devices, John Wiley and Sons, 3rd edn, 2007 Search PubMed.
- S.-C. Lo, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Macromolecules, 2003, 36, 9721 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |