Synthesis and evaluation of partly fluorinated polyelectrolytes as components in 19F MRI-detectable nanoparticles†
Leena
Nurmi
a,
Hui
Peng
b,
Jukka
Seppälä
a,
David M.
Haddleton
c,
Idriss
Blakey
b and
Andrew K.
Whittaker
*b
aDepartment of Biotechnology and Chemical Technology, Aalto University, PL 16100, 00076, Aalto, Finland. E-mail: leena.nurmi@tkk.fi
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, 4072, Australia. E-mail: a.whittaker@uq.edu.au; Fax: +61-7-33463973; Tel: +61-7-3346388
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 14th May 2010
Abstract
A series of partly fluorinated polyelectrolytes were synthesized by transition metal mediated living radical polymerization and evaluated for their applicability as corona-forming components in 19F MRI-detectable nanoparticles in aqueous solutions. The polymers were statistical and block copolymers of trifluoroethyl methacrylate (TFEMA) and 2-(dimethylamino)ethyl methacrylate (DMAEMA). The polymers were either directly dissolved in water (statistical copolymers), or assembled into aqueous nanoparticles with PTFEMA cores and P(TFEMA-co-DMAEMA) coronas (block copolymers). The polymer composition, polymer charge density, solution ionic strength and solution pH were varied. The 19F spin–lattice (T1) and spin–spin (T2) relaxation times and 19F image intensities of solutions of the polymers were measured and related to polymer structure and aqueous conformation. The 19F NMR T2 relaxation times were found to be highly indicative of the 19F imaging performance. Maintaining sufficient mobility of the 19F nuclei was important for obtaining images of high intensity. 19F mobility could be increased by preventing their aggregation in water by exploiting electrostatic repulsion between monomer units.
Introduction
Recently, there has been growing interest in the development of fluorinated compounds capable of being tracked in vivo using 19F magnetic resonance imaging (19F MRI). With the advent of commercially available 19F MRI coils, 19F imaging is achievable in the clinical setting. When the 19F image is superimposed on the familiar 1H density image, the location of a fluorinated compound, e.g. therapeutic particle or cell, can be determined in a non-invasive manner. 19F is an attractive tracking nucleus for MRI due to its high sensitivity and the absence of a confounding 19F background signal within the body.The requirements for a successful 19F MRI tracking agent include that it should have a high fluorine content, the fluorine nuclei should have appropriate NMR properties as discussed below, and preferably there should be a single peak in the 19F NMR spectrum. Such compounds have the potential to provide high image intensity without the need for selective excitation sequences. A variety of fluorinated compounds have been studied, ranging from small fluorocarbon molecules1 to oligomeric emulsions2 and quite recently, to polymeric nanoparticles and particulates.3–7 The targeted applications have varied on a broad scale, including a multitude of in vivo experiments. For example, Higuchi et al.8 have reported the imaging of fluorinated amyloidophilic compounds in living mice as potential markers for the early stages of Alzheimer's disease, and Janjic et al.2 have visualized the migration of T-cells loaded with oligomeric perfluoropolyether (PFPE) emulsions.
Fluorinated polymeric nanoparticles have recently received attention as potential 19F MRI tracking compounds, as they have large structural design potential when compared to traditional systems such as emulsions and solutions of smaller molecules. The diversity of polymeric structures achievable with modern synthetic methods allows the incorporation of a multitude of fluorine nuclei within a nanoparticle, potentially leading to high image intensity. However, the hydrophobic nature of fluorinated groups makes them susceptible towards aggregation in water, and the resulting reduction in molecular mobility can lead to loss of signal intensity. Specifically, the spin–spin relaxation time (T2) of polymeric nuclei is heavily influenced by local motions, with nuclei in constrained environments exhibiting faster T2 relaxation, and therefore broader signals having poorer signal-to-noise ratio (SNR), than identical nuclei in less-constrained environments.9
A number of studies have been published on 19F MRI of polymeric nanoparticles. Du et al.3,4 formed particles by grafting poly(acrylic acid-co-trifluoroethyl methacrylate) copolymer coronas from a small hydrophobic core. They conducted phantom 19F MRI and relaxation time measurements of the aqueous particle solutions, and observed that the T2 of the fluorine nuclei was reasonably long (T2 = 50–56 ms) and the particles were detectable by 19F MRI. Nyström et al.6 formed particles from poly(acrylic acid)-b-poly(styrene-co-pentafluorostyrene) block copolymers. In this study, the fluorine nuclei were incorporated within the hydrophobic core-forming block, and it was noticed that no 19F signal could be detected from the aqueous particle solution. This indicated that the rigid core significantly restricted the mobility of the core nuclei. Peng et al.5 formed nanoparticles from block copolymers of poly(acrylic acid) with varying fluorinated hydrophobic blocks. Again, the 19F nuclei were incorporated within the cores of the particles. However, in this case it was noticed that by lowering the glass transition temperature (Tg) of the core by choosing low Tg components, detectable 19F signal could be obtained from the aqueous nanoparticle solutions. The longest T2 and highest phantom image intensity were obtained with particles of poly(acrylic acid)-b-poly(trifluoroethyl acrylate-co-butyl acrylate) block copolymers (T2 = 1.75 ms).
The previous studies of nanoparticle systems with 19F nuclei within the coronas of the particles have shown promising results.3,4 To further improve the imaging performance of such particles, the 19F content in the corona could be increased in order to obtain higher image intensity. However, increasing the 19F density also increases the hydrophobicity of the corona, eventually resulting in aggregation and loss of signal intensity. Our hypothesis is that, in the case of water-soluble polymers (i.e. nanoparticles stabilized by fluorinated hydrophilic coronas), the highest 19F MRI intensity will be obtained with polymeric materials that combine high fluorine content and efficient aqueous solvation of the fluorinated segments.
One way to limit the aggregation and increase the mobility of the fluorinated segments in water is copolymerization with charged monomer units. The aqueous conformation of polyelectrolytes containing hydrophobic groups is controlled on one hand by the long-range repulsive electrostatic interactions between charged groups and on the other hand by the short-range attractive hydrophobic interactions between hydrophobic groups. Depending on the relative strengths of these forces, hydrophobic polyelectrolyte chains can adopt various conformations in solution, ranging from fully extended chains to collapsed globules.10–12 Similarly, particles with relatively hydrophobic polyelectrolyte coronas can have extended or collapsed conformations (Scheme 1).
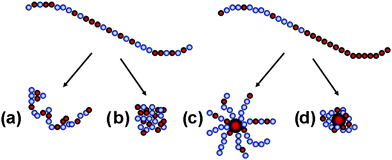 | ||
| Scheme 1 Possible conformations in aqueous solutions of hydrophobic polyelectrolytes (left) and block polyelectrolytes assembled to nanoparticles (right). (a) Extended coil, (b) compact globule, (c) extended nanoparticle and (d) compact nanoparticle. | ||
The balance between the repulsive and attractive interactions in hydrophobic polyelectrolytes in solution is determined by various parameters, including the relative contents of charged and hydrophobic groups, the polymer concentration, solution ionic strength and solution pH. The effects of these parameters on the solution conformation of hydrophobic polyelectrolytes have been widely studied theoretically13,14 and experimentally.15–18
The conformation of the polymer chains has a large impact on the NMR properties of the polymeric nuclei. Measurements of T2 relaxation times along with other NMR techniques have previously been used as a method of probing the conformational restriction of polymeric segments in solutions.19–23
In this current study we have synthesized partially fluorinated polyelectrolytes and evaluated their applicability as 19F MRI agents. The main objective of the study was to observe how changes in polymer composition and solution conditions (including pH and ionic strength) affect the aqueous conformation and subsequently 19F MRI signal intensity of water-soluble polyelectrolyte chains. The polyelectrolytes examined in this work were both statistical and block copolymers of trifluoroethyl methacrylate (TFEMA) and 2-(dimethylamino)ethyl methacrylate (DMAEMA). The statistical polyelectrolytes were directly dissolved in water, whereas the block polyelectrolytes were assembled into aqueous nanoparticles with kinetically frozen PTFEMA cores and P(TFEMA-co-DMAEMA) coronas. The 19F spin–lattice (T1) and spin–spin (T2) relaxation times and 19F image intensities of solutions of the polymers were measured.
Experimental
Materials
Copper(I) bromide (Aldrich, 98%) was purified according to the method of Keller and Wycoff.24 2-(Dimethylamino)ethyl methacrylate (DMAEMA) (Aldrich, 98%) and 2,2,2-trifluoroethyl methacrylate (TFEMA) (Fluorochem) were passed through basic alumina to remove the inhibitor. N-(n-Pentyl)-2-pyridylmethanimine25 and benzyl 2-bromoisobutyrate26 were synthesized using methods described in the literature. All the other reagents were of analytical grade and used as received.Synthesis of the copolymers
Characterization
19F spin–spin (T2) relaxation times were measured using the CPMG pulse sequence, which had from 2 to 256 180° pulses in the echo train. A total of 12 data points were collected. The echo times (τ) were separately optimized for each measurement and the values used were between 25 µs and 5 ms. The majority of the data collected could be described by a single exponential function. In some cases, where separate short and long decay rates were clearly present, the curve was better described by the sum of two exponential relaxation decays. Spin–lattice (T1) relaxation times were measured using the standard inversion-recovery pulse sequence, with 16 values of inversion times used. A single T1 relaxation time was observed for all samples. Note that single measurements of T1 and T2 relaxation times were generally performed, although several samples were measured more than once. The repeatability of the measurements was confirmed as better than 15%.
19F images were collected using the 3D spin-echo pulse sequence. The field of view was 20 × 20 mm2, and the slice thickness was 2.5 mm in a 128 × 128 × 8 matrix. The echo time was 4.8 ms and the repetition time was three seconds. The results presented here were all acquired by co-adding two acquisitions resulting in a constant imaging time of one hour and 20 minutes.
Preparation of the aqueous polymer solutions
The statistical copolymers containing quaternized METAI units were dissolved directly in water (salt-free or 0.25 M NaCl). The polymer concentration was either 1 w/v% or 2.5 w/v%. The solutions were stirred for 24 h at ambient temperature. The statistical copolymers containing non-quaternized DMAEMA were not soluble in water without adjustment of the pH. The polymers were dispersed in water (salt-free or 0.25 M NaCl), and HCl was added to obtain the desired pH. The polymer concentration was 1 w/v%. The solutions were stirred for 24 h at ambient temperature.The block copolymers containing METAI units were dissolved directly in water (salt-free or 0.25 M NaCl). An elevated dissolution temperature of 80 °C was chosen, which is above the glass transition temperature of PTFEMA (Tg,PTFEMA = 74 °C, as determined by DSC). The formed nanoparticle solutions were stirred for 3 h at 80 °C and then for 24 h at room temperature. The polymer concentration was either 1 w/v% or 2.5 w/v%. The particle sizes of the nanoparticles were determined by DLS.
The block copolymers containing non-quaternized DMAEMA were not directly soluble in water. The solutions were prepared by first dissolving 20 mg of block copolymer in 2 ml of acetone. Water, 2 ml, was added drop-wise with a syringe pump over the course of 1.5 h, under constant stirring. The formed nanoparticle solutions in 1 : 1 acetone : water were stirred for 24 h, after which they were dialyzed against water for three days to remove the acetone. After dialysis, the concentration of the solutions was adjusted to 1 w/v% by allowing the excess water to evaporate slowly under a slight N2 flow, until only 2 ml of solution were left. The particle sizes of the formed nanoparticles were determined by DLS.
The size of the PTFEMA-b-P(TFEMA-DMAEMA) nanoparticles as a function of pH was studied with diluted polymer solutions. The polymer concentration was 0.005 w/v%, and a background NaCl concentration of 0.01 M was used in the solutions. The solutions were prepared by first dissolving 20 mg of block copolymer in 2 ml of acetone. Water, 2 ml, was added drop-wise with a syringe pump over the course of 1.5 h, under constant stirring, to induce the formation of nanoparticles. 16 ml of water were then added quickly to provide a solution of 0.1 w/v% polymeric nanoparticles in 90 : 10 water : acetone mixture. The nanoparticle solutions were stirred for 24 h, after which they were dialyzed against water for three days to remove the acetone. After dialysis, 4 ml of 0.1 M NaCl solution, filtered with 0.25 µm filter, were added. Finally, approximately 15 ml of water were added to obtain a batch solution with 20 mg of polymer in 40 ml of water with 0.01 M NaCl (polymer concentration 0.05 w/v%). From this batch solution, the 0.005 w/v% polymer solutions at different pH values were prepared by diluting with aqueous 0.01 M NaCl solution (filtered with 0.25 µm filters) and by adjusting the pH with drop-wise addition of dilute aqueous solutions of HCl or NaOH. Particle sizes were determined by DLS.
Results and discussion
Synthesis of the copolymers
In this study, amphiphilic partly fluorinated polyelectrolytes were synthesized in order to evaluate their potential as 19F MRI-detectable compounds. The structures of the monomer units are presented in Scheme 2 and the properties of the polymers listed in Table 1. The polymers were either statistical or block copolymers with varying proportions of 2,2,2-trifluoroethyl methacrylate (TFEMA) as the hydrophobic fluorine containing monomer unit and 2-(dimethylamino)ethyl methacrylate (DMAEMA) or its quaternized form [2-(methacryloyloxy)ethyl] trimethyl ammonium iodide (METAI) as the electrostatically charged, cationic monomer unit.![The monomers used in the copolymers. (a) 2,2,2-Trifluoroethyl methacrylate (TFEMA), (b) 2-(dimethylamino)ethyl methacrylate (DMAEMA), (c) quaternized DMAEMA, [2-(methacryloyloxy)ethyl] trimethyl ammonium iodide (METAI).](/image/article/2010/PY/c0py00035c/c0py00035c-s2.gif) | ||
| Scheme 2 The monomers used in the copolymers. (a) 2,2,2-Trifluoroethyl methacrylate (TFEMA), (b) 2-(dimethylamino)ethyl methacrylate (DMAEMA), (c) quaternized DMAEMA, [2-(methacryloyloxy)ethyl] trimethyl ammonium iodide (METAI). | ||
| Sample | Composition | TFEMA content (mol%) | F content/mmol g−1 | M n /g mol−1 (NMR) | M w/Mnb |
|---|---|---|---|---|---|
| a Calculated from 1H NMR spectra by comparing end-group peaks to appropriate side-chain peaks. b By SEC with DRI detection. c Measured before quaternization. | |||||
| Statistical copolymers | |||||
| C-12 | P(TFEMA17-co-DMAEMA122) | 12 | 2.3 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.39 |
| C-24 | P(TFEMA34-co-DMAEMA109) | 24 | 4.5 | 22900 |
1.39 |
| C-51 | P(TFEMA75-co-DMAEMA73) | 51 | 9.3 | 24100 |
1.24 |
| QC-51 | P(TFEMA75-co-METAI73) | 51 | 6.6 | 34400 |
1.24c |
| Block copolymers | |||||
| B-20 | PTFEMA39-b-P(DMAEMA197-co-TFEMA12) | 20 | 3.8 | 39500 |
1.55 |
| B-47 | PTFEMA91-b-P(DMAEMA122-co-TFEMA19) | 47 | 8.9 | 37700 |
1.43 |
| QB-47 | PTFEMA91-b-P(METAI122-co-TFEMA19) | 47 | 6.1 | 55000 |
1.43c |
TFEMA was chosen as the fluorine-containing monomer for all the polymers, as it has three equivalent fluorine nuclei, resulting in a single peak of potentially high signal-to-noise ratio (SNR) in the 19F NMR spectrum, and as it is less hydrophobic than more fluorinated monomer units. DMAEMA and its quaternized form METAI were chosen as the electrostatically charged monomers. DMAEMA is a basic monomer unit, and therefore the degree of cationic charge in the DMAEMA-containing polymers in aqueous solutions was dependent of pH, and increased with decreasing pH. The pKa value of DMAEMA monomer is 8.4.28 The average pKa of PDMAEMA, defined as the pH at which 50% of the DMAEMA units are protonated, is dependent on e.g. polymer molecular weight, polymer architecture and the ionic strength of the solution. For multibranched PDMAEMA star polymers in salt-free solutions, average pKa values as low as 5.9 have been measured.29 In contrast, METAI monomer unit, which has additional methyl group attached to the DMAEMA amine group by a post-polymerization reaction, is cationically charged independent of pH.
The copolymers were polymerized by transition metal mediated living radical polymerization in toluene with CuBr/N-(n-pentyl)-2-pyridylmethanimine as catalyst.30,31 The method provided copolymers with controlled molecular weights and reasonably low molecular weight distributions (Table 1). DMAEMA was used as a hydrophilic monomer in statistical copolymers C-12, C-24 and C-51 and in block copolymers B-20 and B-47. METAI was used in statistical copolymer QC-51, which was a completely quaternized version of C-51 and in block copolymer QB-47, which was a completely quaternized version of B-47.
The block copolymers were polymerized in a sequential manner; the TFEMA block was polymerized first and DMAEMA was added when 70–80% TFEMA conversion was reached. Due to the preparation method, the second block was a statistical copolymer, and 20–30% of TFEMA present in the polymer was in the hydrophilic block.
The reactivity ratios of the TFEMA/DMAEMA monomer pair were measured by Jaacks method27 and they were found to be rTFEMA = 0.76 and rDMAEMA = 0.81. This indicates that there was no tendency towards formation of blocks within the statistical copolymers or the statistical hydrophilic blocks of the block copolymers. For additional information on the polymerization of the PTFEMA/PDMAEMA copolymers, including polymerization kinetics, calculation of molecular weights from the 1H NMR spectra and measurement of reactivity ratios, see ESI†.
Properties of the aqueous solutions of the copolymers
The polymer concentration in the aqueous solutions was 1 w/v%. The relatively high concentration was chosen to allow the 19F NMR and 19F MRI measurements. The statistical copolymers were dissolved directly in water. The block copolymers were assembled into aqueous nanoparticles, either through solvent exchange from acetone, or by direct dissolution at elevated temperature. In order to study the effect of solution conditions on the 19F NMR performance of the polymers, the non-quaternized statistical copolymers were studied at varying pH and salt conditions.
Details of the polymer solutions are presented in Table 2, along with the results of the measurements performed with the solutions. In addition, estimations of the polymer conformations (collapsed or extended), based on the combined measurement results, are presented in Table 2 for each polymer solution. However, these conformation estimations represent only relative qualities between the samples, and do not indicate the absolute extent of the collapse or extension of the chains.
| Sample | Solution conditions | D H/nm (DLS) | 19F T1/ms | 19F T2/ms | SNR in 19F MRI | Expected conformation of the copolymer in solution | |
|---|---|---|---|---|---|---|---|
| pH | [NaCl]/M | ||||||
| a Not measured. b The result in parentheses is for more concentrated, 2.5 w/v% solution. c Dissolved by solvent exchange from acetone. d Relative standard deviation was <2%. e Dissolved at elevated temperature (80 °C). f A broad size distribution was obtained, the extreme values are reported. The DLS measurement was done in 0.1 M NaCl. g Measured with 2.5 w/v% solution. | |||||||
| Statistical non-quaternized copolymers, with varying solution pH and ionic strength | |||||||
| C-12 | 8 | 0 | 320 | 13 | No signal | Collapsed globule | |
| 8 | 0.25 | 310 | 12 | —a | Collapsed globule | ||
| 6.5 | 0 | 520 | 140 | 3.9 | Extended coil | ||
| 7.3 | 0.25 | 460 | 76 | —a | Extended (intermediate) coil | ||
| 2 | 0 | 550 | 140 | 3.9 | Extended coil | ||
| 2 | 0.25 | 520 | 160 | —a | Extended coil | ||
| C-24 | 6.5 | 0 | 430 | 81 | 6.0 | Extended (intermediate) coil | |
| 2 | 0 | 540 | 150 | 6.2 | Extended coil | ||
| 2 | 0.25 | 490 | 120 | —a | Extended coil | ||
| C-51 | 2 | 0 | 360 | 11 | 6.9/(14)b | Collapsed globule | |
| 2 | 0.25 | 320 | 8.4 | 5.5 | Collapsed globule | ||
| Other samples | |||||||
| QC-51 | 0.25 | 330 | 11 | Weak signal/(9.2)b | Collapsed globule | ||
| B-20c | 0 | 45d | 340 | 69% 0.12 31% 11 | No signal | Collapsed nanoparticle | |
| B-47c | 0 | 29d | 400 | 86% 0.22 14% 11 | No signal | Collapsed nanoparticle | |
| QB-47e | 0.25 | 20–1000f | 480g | 89% 0.73 11% 81g | No signal/(3.4)b | Extended nanoparticle | |
In addition to the solution experiments described in Table 2, a set of DLS experiments were conducted, where the hydrodynamic diameters of B-47 nanoparticles were studied as a function of pH. However, these experiments were conducted with more dilute, 0.005 w/v% nanoparticle solutions, and also the nanoparticle preparation conditions were slightly different from those applied in Table 2. Therefore, the results are expected to give only a qualitative estimation on how the B-47 particles described in Table 2 behave if the pH is altered. The hydrodynamic diameters of the B-47 particles obtained at varying pH conditions are shown in Table 3.
The results in Table 3 show that the DH of the particles approximately doubled when the pH was lowered from 9 to 3.
The corona blocks of the particles consisted of a statistical copolymer of TFEMA and DMAEMA containing 13 mol% of TFEMA units (Table 1). The blocks had low charge density at pH 9 whereas they had high charge density at pH 3.
Since the particles had highly hydrophobic cores which were below the glass transition temperature (Tg,TFEMA = 74 °C as measured by differential scanning calorimetry) they were expected to be kinetically trapped structures.32,33 Therefore the change in DH is not expected to derive from changes in the aggregation number, as the pH adjustment was done as the final step, after the particles were formed. Alternatively, the change in the DH must derive from the significant expansion of the corona chains with increasing charge density, as suggested in Scheme 1. Based on these results it can be expected that qualitatively similar expansion with increasing charge density could also take place in the particles of the other block copolymer samples, as well as in the statistical copolymer coils. Similar responsiveness to changes in pH has been visualized for PDMAEMA in an AFM study of PDMAEMA cylindrical brushes.15
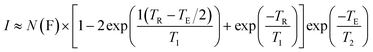 | (1) |
In eqn (1)I is the image intensity, N(F) a measure of the number of fluorine nuclei in the sensitive volume of the imager, TR and TE are the pulse sequence repetition and echo times, respectively.
Eqn (1) states that the most effective imaging agents will have short T1 relaxation times, long T2 relaxation times and high fluorine contents. Both T1 and T2 are affected by the mobility of the nuclei in question, in this case fluorine. The relaxation time results of the solutions are presented in Table 2 and the fluorine contents of the polymers are presented in Table 1.
The SNR of the phantom 19F images, describing the image intensity, is also presented in Table 2. The images were taken with a spin-echo imaging sequence, where the echo time was 4.8 ms and the repetition time was 3 s. The imaging time was restricted to 80 min. For some samples the polymer concentration in the solutions was increased from the 1 w/v% used in the relaxation measurements to 2.5 w/v% in order to get images of higher SNR. The SNR results of these more concentrated solutions are presented in parentheses in Table 2.
As a preliminary overview of the 19F NMR and 19F MRI results in Table 2 it can be seen that differences in both relaxation rates were observed with changes in the polymer structures and solution conditions. The values of T1 ranged from 310 ms to 550 ms. However, more significant differences in the values of T2 were observed between the samples, where variation greater than two orders of magnitude was observed: from values less than 1 ms to 160 ms. The relatively small variation between T1 values indicates that the spectral density of motion of the fluorine nuclei in the mid-MHz range was not highly affected by conformational differences, although systematically the T1 relaxation times of the collapsed structures were lower than those observed for the extended structures. The T2 relaxation times, which are sensitive to lower frequency motions, were much more sensitive to changes in conformation.
The observed differences in the relaxation times, along with varying fluorine contents, resulted in different SNR in 19F MRI. In particular the T2 relaxation times were found to be highly indicative of the sample performance, and the T2 results are therefore discussed with special attention in the following sections. The detailed T2 results, including the exponential fitting of the data, are presented in ESI†.
Sample C-12 contained only 12 mol% TFEMA, and was soluble in water even at pH 8, where the extent of DMAEMA protonation was low. At this pH both relaxation rates of 19F were relatively short, T1 = 310 ms and T2 = 12 ms (0.25 M NaCl solution, Table 2). When the pH was decreased, the relaxation rates increased substantially, especially T2, which at pH 2 increased one order of magnitude to T2 = 160 ms (0.25 M NaCl solution). This change is expected to be due to a globule-to-coil transformation in the chain conformation, as drawn in Scheme 1 and tabulated in Table 2. When the pH was decreased, the repulsion between cationic monomer units increased, overcoming the attraction between the hydrophobic –CF3 and –CH3 groups in the copolymer. This resulted in increased segmental mobility around the –CF3 groups, which increased the 19F T2 relaxation time.
C-24, which contained 24 mol% of TFEMA, was no longer soluble in water at pH 8. Otherwise, the behavior of the sample as a function of pH was similar to C-12. Most notably at pH 2, when the extent of DMAEMA protonation was high, the relaxation results of C-24 and C-12 were similar, e.g. at 0.25 M NaCl solutions T2 of C-12 and C-24 were 160 ms and 120 ms, respectively (Table 2). C-51, which contained 51 mol% of TFEMA groups was not soluble in water even at pH 6.5, and was measured only at pH 2. The relaxation rates of C-51 at pH 2 were significantly decreased from the other two samples at low pH, down to values T1 = 320 ms and T2 = 8.4 ms (0.25 M NaCl solution), similar to the collapsed form of C-12 at pH 8. This indicates that at this highest fluorine content, the electrostatic repulsion was not sufficiently strong to overcome the attraction between the numerous –CF3 groups in the copolymer chain. These trends in the aqueous conformations of hydrophobic polyelectrolytes have been predicted theoretically.13 The quaternized sample QC-51 was soluble in water without adjustment of pH. The relaxation times measured for QC-51 were very close to values measured for C-51 at pH 2, which is not surprising, as both samples had equal charge and TFEMA densities.
When comparing the relaxation rate results of salt-free and 0.25 M NaCl solutions, it can be seen from Table 2 that the T2 relaxation time of C-12 at pH 6.5 in salt-free solution (T2 = 140 ms) is almost twice as large as the corresponding T2 of C-12 at pH 7.3 in 0.25 M NaCl solution (T2 = 76 ms). However, in general it is difficult to draw conclusions from the differences between these two samples as it is difficult to estimate the charge density of the polymers. The samples were in the pH range where relatively significant HCl additions resulted in minor changes in the solution pH, but in major changes in the charge density of the polymer. This area often appears in the titration curves of hydrophobic polyelectrolytes before the neutralization point, and has been argued to derive from the phase transition between the collapsed and extended states.13
The reason for the different pH values in the intermediate pH C-12 samples (6.5 for salt-free solution and 7.3 for 0.25 M NaCl solution) was that both the solutions were prepared with approximately equal amounts of added HCl. DMAEMA units were more readily protonated in 0.25 M NaCl solutions than in salt-free solutions, due to the screening of the polymer chain charges by the salt.
Otherwise, if the pH values 6.5–7.3 were not taken into consideration, the comparison of relaxation rate results of salt-free and 0.25 M NaCl solutions revealed only small differences. The general trend was that in most cases the salt-free solutions had slightly longer relaxation times, but the differences were not large. In theory, increasing the ionic strength of the solution should have a significant impact on the solution conformation of hydrophobic polyelectrolytes.11,13,17,18 However, due to the high polymer concentration of the solutions, the ionic strength of the salt-free solutions was already rather high. This could explain why no differences in the relaxation rates were observed between the salt free and 0.25 M NaCl solutions.
When comparing these relaxation rate results to the performance of the samples in imaging experiments, it can be seen that increasing T2 indeed increased the image intensity, as suggested by eqn (1). C-12 could not be imaged at pH 8 (T2 = 13 ms, salt-free solution) but at pH 2 (T2 = 140 ms, salt-free solution) it gave a weak image (SNR = 3.9). However, the samples C-24 and C-51 at pH 2 resulted in roughly equal image intensities. The one-magnitude shorter T2 of C-51 (T2 = 8.4 ms, salt-free solution) when compared to C-24 (T2 = 150 ms, salt-free solution) was completely compensated by the increase in the fluorine nuclei present in the polymer chain. The samples C-51 at pH 2 and QC-51 had approximately equal relaxation times. The higher intensity of the C-51 image most likely resulted from the higher fluorine content of the sample (Table 1); the fluorine content of the quaternized QC-51 was significantly reduced by the incorporation of heavy iodine ions into METAI units.
In summary, there was a clear balance in behaviour: increasing fluorine content on one hand increased the image intensity by adding more 19F nuclei to the image, but on the other hand decreased the T2 relaxation time, potentially reducing the image intensity, because of increased association between the more abundant fluorinated groups. From the results of the C-12, C-24, C-51 and QC-51 series it can be concluded that optimal images would be obtained if extended coil conformation could be retained at high fluorine contents. On the other hand, it can be seen that if the T2 remains at a reasonably high level, as in C-51 at pH 2 and in QC-51, images can be obtained even if mobility is restricted to some extent.
Large variations were again observed in the T2 relaxation rates. Two different T2 values could be separated from the T2 relaxation curves of all the nanoparticle solutions (Table 2, for the relaxation decays see ESI†). The presence of two separate T2 values indicates the presence of separate 19F populations, at least on the time scale of the relaxation time measurements. It can be assumed that the shorter T2 derives from the TFEMA units within the core blocks of the particles, while the longer T2 is due to the TFEMA units within the statistical copolymer blocks forming the coronas of the particles. The relative populations of short and long 19F T2 values could be estimated from the relaxation decays (listed in Table 2) and correspond to the relative TFEMA populations in the core and corona blocks reasonably well. For example the B-47 block copolymer had 17% of its TFEMA groups in the statistical corona block, and correspondingly the relative size of the 19F population with long T2 was measured to be 14%.
The major component in the T2 relaxation times of the block copolymer particles was short, <1 ms relaxation time arising from the particle cores. These values were so short that in practice the core 19F nuclei did not contribute to the MRI images with the pulse sequence we used (with echo time of 4.8 ms). Thus, only the nuclei present within the corona blocks were detected in the images.
The corona blocks of the block copolymer samples were statistical copolymers, and their 19F NMR properties were found to follow similar trends as the statistical copolymers discussed above. The TFEMA unit density of the corona blocks was either 6 mol% (B-20) or 13 mol% (B-47 and QB-47). The environment for the fluorine atoms in the coronas of the B-20 and B-47 particles was therefore similar to the environment for fluorine in C-12 chains at pH 8 as the chain charge densities were in all cases low (the block copolymers were measured at unadjusted pH) and the fluorine contents similar. Correspondingly, the environment for fluorine in the coronas of QB-47 particles was similar to the environment for fluorine in C-12 chains at pH 2, because the charge densities were now high in both cases, and the fluorine contents again similar.
The long T2 component of both B-20 and B-47 was T2 = 11 ms, which was almost the same as the value measured for C-12 at pH 8, T2 = 13 ms (0 M NaCl in both measurements, Table 2). The long T2 component of QB-47 was T2 = 81 ms. This was somewhat lower than the value for the C-12 at pH 2, T2 = 140 ms (0 M NaCl in both measurements, Table 2). However, the trend of increasing T2 relaxation time with increasing charge density was evident both in the case of block copolymer coronas and in the case of statistical copolymers.
The similarities in the results of the C-12 statistical copolymers with the corresponding corona blocks of the nanoparticles indicate that the results we obtained with statistical copolymers can be used to qualitatively estimate the behavior of corresponding nanoparticle coronas. However, the systems are not exactly comparable due to the extra confinement effects provided by the particle cores.
The DLS results for the B-47 particles as a function of solution pH (Table 3) indicated that the conformation of the block copolymer particle coronas was highly dependent on the charge density of the corona block. The relaxation time results in Table 2 strongly support this conclusion. The conformations of the B-20 and B-47 particles are therefore expected to be compact (low charge density, short T2) whereas the conformation of the QB-47 particles is expected to be extended (high charge density, long T2), as depicted in Scheme 1 and tabulated in Table 2. The DLS measurements in Table 3 gave indication only of the behavior of the block copolymers, but since the relaxation rates followed similar trends in both the block copolymer and statistical copolymer samples, it can be expected that such collapse and extension phenomena were present in the statistical copolymers as well.
None of the nanoparticle solutions gave distinguishable MRI signals at 1 w/v% concentration. When the QB-47 sample was concentrated to 2.5 w/v% concentration, a weak image was obtained. However, the weaker imaging performance of the nanoparticle solutions when compared to statistical copolymers is understandable—because the core block formed the majority of the block copolymer chains, the concentration of the detectable corona fluorine nuclei was low in the nanoparticle solutions. Nanoparticles constructed from such block copolymers, where the corona block forms the majority of the chain, are expected to perform significantly better in 19F MRI, if the overall concentration is kept constant.
Conclusions
A series of partly fluorinated, electrostatically charged statistical and block copolymers were synthesized and studied in aqueous solutions. The aim of the study was to evaluate the performance of the polyelectrolytes in 19F MRI, especially as corona components in 19F-detectable nanoparticles. The copolymers had TFEMA as fluorine-containing monomer unit copolymerized with cationic DMAEMA or METAI units. It was noticed that maintaining sufficient mobility of the 19F nuclei was important for obtaining images of high intensity. Mobility of the fluorinated groups could be increased by preventing their aggregation in water by exploiting electrostatic repulsion between monomer units. The 19F NMR T2 relaxation times were found to be highly indicative of the sample 19F imaging performance. Conversely, it was noticed that if sufficient 19F mobility was achieved having a high fluorine density in the polymer was important to obtain an image of high intensity. Block copolymers assembled into nanoparticles were found to perform in the 19F NMR experiments in a similar manner to statistical copolymers, provided that the fluorine groups remained within the particle coronas. The imaging performance of the fluorinated rigid particle cores, consisted of PTFEMA homopolymer, was poor.In order to achieve superior imaging performance it is necessary to retain the fluorine group mobility with higher fluorine loadings. If the 19F nuclei are incorporated in the coronas of the nanoparticles, as in this study, one solution could be to increase the hydrophilicity of the corona blocks, and especially incorporate hydrophilic functionalities as close to the 19F nuclei as possible. Another way of achieving high 19F detectability could be the synthesis of nanoparticles with liquid-like fluorophilic cores, where large amounts of fluorinated compounds could be incorporated.
Acknowledgements
The authors acknowledge financial support from the Graduate School in Chemical Engineering (Finland). Financial support from the Queensland Government for funding under the NIRAP scheme for the International Biomaterials Research Alliance is also gratefully acknowledged. Part of this work utilized equipment funded under the NCRIS Australian National Fabrication Facility.Notes and references
- J. Yu, V. D. Kodibagkar, W. Cui and R. P. Mason, Curr. Med. Chem., 2005, 12, 819–848 CrossRef CAS.
- J. M. Janjic, M. Srinivas, D. K. K. Kadayakkara and E. T. Ahrens, J. Am. Chem. Soc., 2008, 130, 2832–2841 CrossRef CAS.
- W. Du, A. M. Nyström, L. Zhang, K. T. Powell, Y. Li, C. Cheng, S. A. Wickline and K. L. Wooley, Biomacromolecules, 2008, 9, 2826–2833 CrossRef CAS.
- W. Du, Z. Xu, A. M. Nyström, K. Zhang, J. R. Leonard and K. L. Wooley, Bioconjugate Chem., 2008, 19, 2492–2498 CrossRef CAS.
- H. Peng, I. Blakey, B. Dargaville, F. Rasoul, S. Rose and A. K. Whittaker, Biomacromolecules, 2009, 10, 374–381 CrossRef CAS.
- A. M. Nyström, J. W. Bartels, W. Du and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1023–1037 CrossRef CAS.
- J. M. Criscione, B. L. Le, E. Stern, M. Brennan, C. Rahner, X. Papademetris and T. M. Fahmy, Biomaterials, 2009, 30, 3946–3955 CrossRef CAS.
- M. Higuchi, N. Iwata, Y. Matsuba, K. Sato, K. Sasamoto and T. C. Saido, Nat. Neurosci., 2005, 8, 527–533 CrossRef CAS.
- P. Mirau, Practical Guide to Understanding the NMR of Polymers, John Wiley & Sons, Inc., Hoboken, New Jersey, 2005 Search PubMed.
- A. V. Dobrynin, Curr. Opin. Colloid Interface Sci., 2008, 13, 376–388 CrossRef CAS.
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- J. Kötz, S. Kosmella and T. Beitz, Prog. Polym. Sci., 2001, 26, 1199–1232 CrossRef CAS.
- S. Ulrich, A. Laguecir and S. Stoll, J. Chem. Phys., 2005, 122, 094911 CrossRef.
- O. V. Borisov, F. Hakem, T. A. Vilgis, J. F. Joanny and A. Johner, Eur. Phys. J. E, 2001, 6, 37–47 CrossRef CAS.
- Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Yuan, M. Ballauff and A. H. E. Müller, Polymer, 2008, 49, 3957–3964 CrossRef CAS.
- L. J. Kirwan, G. Papastavrou, M. Borkovec and S. H. Behrens, Nano Lett., 2004, 4, 149–152 CrossRef CAS.
- S. Minko, A. Kiriy, G. Gorodyska and M. Stamm, J. Am. Chem. Soc., 2002, 124, 3218–3219 CrossRef CAS.
- A. Kiriy, G. Gorodyska, S. Minko, W. Jaeger, P. Stepanek and M. Stamm, J. Am. Chem. Soc., 2002, 124, 13454–13462 CrossRef CAS.
- J. Preuschen, S. Menchen, M. A. Winnik, A. Heuer and H. W. Spiess, Macromolecules, 1999, 32, 2690–2695 CrossRef CAS.
- I. Furo, I. Iliopoulos and P. Stilbs, J. Phys. Chem. B, 2000, 104, 485–494 CrossRef CAS.
- G. D. Poe, W. L. Jarrett, C. W. Scales and C. L. McCormick, Macromolecules, 2004, 37, 2603–2612 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- J. Pietrasik, B. S. Sumerlin, H. Lee, R. R. Gil and K. Matyjaszewski, Polymer, 2007, 48, 496–501 CrossRef CAS.
- R. N. Keller and H. D. Wycoff, Inorg. Synth., 1946, 2, 1–4.
- D. M. Haddleton, C. B. Jasieczek, M. J. Hannon and A. J. Shooter, Macromolecules, 1997, 30, 2190–2193 CrossRef CAS.
- N. J. Hovestad, G. van Koten, S. A. F. Bon and D. M. Haddleton, Macromolecules, 2000, 33, 4048–4052 CrossRef CAS.
- V. Jaacks, Makromol. Chem., 1972, 161, 161–172 CrossRef CAS.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Muller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS.
- N. M. L. Hansen, K. Jankova and S. Hvilsted, Eur. Polym. J., 2007, 43, 255–293 CrossRef CAS.
- N. M. L. Hansen, M. Gerstenberg, D. M. Haddleton and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8097–8111 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- O. Théodoly, M. Jacquin, P. Muller and S. Chhun, Langmuir, 2009, 25, 781–793 CrossRef CAS.
- R. E. Hendrick, Magn. Reson. Imaging, 1987, 5, 31–37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Experimental details for the synthesis of block copolymers of TFEMA and DMAEMA. (2) Experimental details for the synthesis of statistical copolymers of TFEMA and DMAEMA. (3) Detailed polymerization results for TFEMA/DMAEMA block and statistical copolymers. (4) Reactivity ratio measurements of the DMAEMA/TFEMA monomer pair. (5) The exponential decay curves of the spin–spin (T2) relaxation time measurements. See DOI: 10.1039/c0py00035c |
| This journal is © The Royal Society of Chemistry 2010 |