Characterization of network structure in radiation-cured resins of polyfunctional acrylic ester and N-vinylpyrrolidone by MALDI-MS combined with supercritical methanolysis
Hideki
Matsubara
*a,
Hiroyasu
Kataoka
b and
Hajime
Ohtani
b
aAichi Industrial Technology Institute, Kariya, 448-0013, Japan. E-mail: hideki_matsubara@pref.aichi.lg.jp
bDepartment of Materials Science and Engineering, Graduate School of Engineering, Nagoya Institute of Technology, Nagoya, 466-8555, Japan
First published on 19th May 2010
Abstract
The cross-linking structures of the ultraviolet and electron beam cured resins prepared from pentaerithritol triacrylate and N-vinylpyrrolidone (VP) mixtures were characterized by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) combined with supercritical methanolysis. The MALDI-mass spectra of the methanolysis products contained a series of peaks of sodium-cationized methyl acrylate/VP co-oligomers formed through selective cleavage and methylation at ester linkages in the radiation-cured resins, which reflected the cross-linking sequences. The composition distributions of the methanolysis products were interpreted in terms of curing conditions such as energy source, photoinitiators, dosage of radiation and formulations.
Introduction
Polymerization induced by ultraviolet (UV) or electron beam (EB) radiation has been widely employed in industrial materials such as inks, paints, coatings, and adhesives because of its distinct advantages such as no need of solvents, low energy consumption and low temperature requirements.1–3 For both UV and EB-curing process, formulations consisting of acrylated monomers and prepolymers having various molecular structures are mainly utilized. Although both the curing processes are based upon free radical polymerization, there are some fundamental differences between them. In the UV-curing process, free radical polymerization proceeds in the presence of photoinitiators, whereas, in the EB-curing process, polymerization occurs normally in their absence. Although UV-curing is generally carried out under an air atmosphere with high productivity, EB-curing requires an inert atmosphere such as nitrogen gas. In addition, the properties of resulting resins by UV and EB irradiations are often considerably different even in the same formulations.Characterization of the cross-linking network structure of the radiation-cured resins has been requested to correlate it with various properties of the resins. However, it is not an easy task to analyze cured resins even using advanced spectroscopic method such as high-resolution NMR due to their insoluble nature. Although solid-state NMR has been successfully utilized to study the heterogeneity of network structures in photo-cured resins,4,5 this technique is not necessarily suitable for characterizing microstructures in the rigidly cross-linked resins mainly because of insufficient resolution.
On the other hand, pyrolysis-gas chromatography (Py-GC) has been utilized as a practical tool even for the characterization of insoluble cross-linked polymers such as vulcanized rubbers,6 polystyrene gels,7 and epoxy resins.8,9 Moreover, Py-GC in the presence of tetramethylammonium hydroxide (TMAH) has brought us valuable information on detailed chemical structures in the intractable polymeric materials including UV-cured acrylic resins.10–13 In this technique, the ester linkages in the cured resin samples selectively decomposed into methyl esters and methyl ethers which provide various information such as conversion, copolymer composition and average molar mass of prepolymer.
The authors characterized the chain length distribution of network junctions in UV-cured poly(ethylene glycol) diacrylate (PEDA) using Py-GC in the presence of TMAH.11 The chain length distribution of the network junctions composed of up to six acryloyl units in the UV-cured PEDA could be estimated from the peak intensities of the characteristic methyl acrylate (MA) oligomers reflecting cross-linking sequence structure in the resins. However, the network junctions containing longer sequence comprised of more than seven acryloyl units were not elucidated because the corresponding higher MA oligomers were not observed in the pyrogram due to their lower volatility.
Recently, in order to overcome this limitation, the authors evaluated the kinetic chain length of the network junctions of UV-cured acrylic ester resins prepared from dipentaerythritol hexacrylate (DPHA) by matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) combined with supercritical methanolysis.14 In this case, UV-cured DPHA was selectively decomposed at the ester linkages to give poly(methyl acrylate)s (PMAs) reflecting cross-linking sequences, which were observed in the MALDI-mass spectrum of the decomposition products as their sodium-cationized molecules. Furthermore, collecting a size exclusion chromatographic fraction of the products followed by MALDI-MS analysis enabled us to evaluate almost the whole range of molar mass distribution of PMA reflecting the widely distributed sequences of the network junctions.
In general, radiation-curable materials consist of several kinds of monomers and prepolymers to satisfy various properties such as rheology of the formulations, curing-speed and mechanical strength of the resulting resins. Among many kinds of comonomers currently available for radiation-curable materials, N-vinylpyrrolidone (VP) is frequently utilized because of its significant effect to provide increasing cure rate and excellent adhesion to plastics. Although such important characteristics are closely related to the resulting chemical structures in the radiation-cured resins, the detailed network structure in the resins consisting of plural curable ingredients has not been clarified up until now.
In this work, MALDI-MS combined with supercritical methanolysis was extended and applied to study the network structures in radiation-cured resins prepared with pentaerithritol triacylate (PETA) and VP mixtures. The observed MALDI-mass spectra of the supercritical methanolysis products were mainly interpreted in terms of the composition distribution of the network junctions in the resins cured under the various conditions in energy sources, irradiation dosages and monomer compositions.
Experimental
Samples
PETA was obtained from Kyoeisha Chemical Co., Ltd, Japan. VP was purchased from Wako Pure Chemical Industries, Ltd, Japan. Two types of photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPA; Wako Pure Chemical Industries, Ltd., Japan) and benzophenone (BP; Tokyokasei Kogyo Co., Ltd, Japan), were utilized for UV-curing without further purification.The UV and EB-cured resin samples are listed in Table 1 together with the compositions and curing condition. Scheme 1 shows typical UV and EB-curing pathways of the PETA/VP resins. In the UV-curing process, active free radicals are generated upon UV exposure in the presence of suitable photoinitiators, either by homolytic C–C bond scission of DMPA, or by hydrogen abstraction from H-donor molecules to BP.15,16 These active species initiate photopolymerization of PETA and VP, which generates the cross-linked polymers shown in Scheme 1.
| Samples | Molar ratio of functionality | Radiation source | Composition of photoinitiator (parts by weight)a | Irradiation dose/J g−1 | |
|---|---|---|---|---|---|
| Acryloyl/Vinyl | DMPA | BP | |||
| a The total amount of PETA and VP is defined to be 100. | |||||
| U1–A | 2/1 | UV | 3 | — | — |
| U2–A | 1/1 | UV | 3 | — | — |
| U3–A | 1/2 | UV | 3 | — | — |
| U1–B | 2/1 | UV | 3 | 1 | — |
| U2–B | 1/1 | UV | 3 | 1 | — |
| U3–B | 1/2 | UV | 3 | 1 | — |
| E1–20 | 2/1 | EB | — | — | 20 |
| E2–20 | 1/1 | EB | — | — | 20 |
| E3–20 | 1/2 | EB | — | — | 20 |
| E1–40 | 2/1 | EB | — | — | 40 |
| E2–40 | 1/1 | EB | — | — | 40 |
| E3–40 | 1/2 | EB | — | — | 40 |
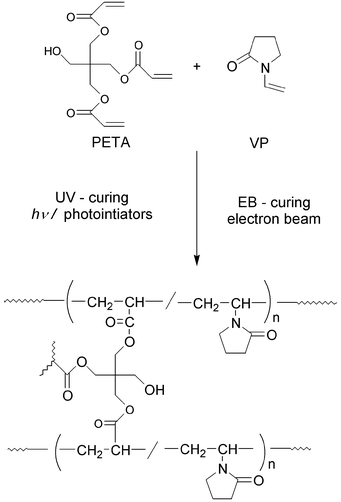 | ||
| Scheme 1 A typical UV and EB-curing pathway for PETA/VP resin. | ||
Meanwhile, in the EB-curing process, free radicals are produced by the impact of accelerated electrons at the energy levels sufficient to break chemical bonds. Although polymerization proceeds in a similar manner to that of the UV-curing process, photoinitiator are not necessary because of initiation induced by high energy electrons.
UV-curing process
The procedure for preparing UV-cured resin samples is basically the same as that described in our previous papers.11,14 Prepolymer was compounded by dissolving the photoinitiators into the mixtures of PETA and VP at 70 °C. Then, the viscous prepolymer solution was applied to a stainless steel plate to form a wet film with 100–200 μm thickness by using a bar applicator. UV-curing was carried out in air by passing the UV-curable prepolymer on the plate through the UV-curing equipment consisting of a conveyor and a medium-pressure mercury lamp (80 W cm−1 and total output 2 kW) mounted in a dichroic mirror. The dosage for preparing UV-cured resins was determined by a UV-integrator (UVPF-36; EYE Graphics, Japan, wavelength rage = 300–360 nm).EB-curing process
Similarly to the UV-curing, the EB-curable prepolymer solution without any photoinitiator was applied to the stainless steel plate. EB-curing was carried out in a nitrogen atmosphere at room temperature by using scanning type electron beam processing system (EPST-3001-1H; NHV Corporation, Japan). The accelerating voltage used was 300 kV, and the irradiation dose was adjusted by changing filament current. The films applied on the plate were irradiated at dose of either 20 or 40 J g−1.Supercritical methanolysis procedure
The apparatus and procedure for supercritical methanolysis are almost the same as those in our previous paper.14 The autoclave was made of a stainless steel (SUS 316) tube (1/4 inch Φ, ca. 7 cm long) with the inner volume of 1 ml, of which both ends were sealed with Swagelok caps. Prior to supercritical methanolysis, the cured resin sample was cryomilled into a fine powder by a cryogenic sample crusher (JFC-300; Japan Analytical Industry Co., LTD., Japan) at liquid nitrogen temperature in order to improve the efficiency of methanolysis. About 2 mg of the ground resin sample was put into an autoclave together with 0.7 ml of methanol. The sealed autoclave was heated at 290 °C for 2 h in a gas-chromatograph oven. The reaction temperature and time were empirically optimized by using size exclusion chromatographic measurements to attain higher decomposition efficiency with minimizing pyrolytic cleavage of C–C bonds in the polymer chains.14 Under these conditions, methanol in the reaction tube was in supercritical state over the critical point. After heating for the prescribed period, the autoclave was cooled down in a water bath, the decomposed products were taken out of the autoclave and the inner wall of the tube was washed out using tetrahydrofuran. The products in methanol were mixed with the rinsing solutions and dried in vacuo at 40 °C, and then the obtained solid products were dissolved again in 0.5 ml of chloroform to be subjected to MALDI-MS measurement.MALDI-MS conditions
From several matrix reagents examined, 4-hydroxybenzylidene malononitrile (HBMN; Sigma, USA) was empirically selected as the optimum matrix and used without further purification. In addition, sodium iodide (Wako Pure Chemical Industries, Ltd., Japan) was used as the cationization salt. First, 0.5 μl of 0.01M sodium iodide solution in tetrahydrofuran was spotted on a stainless steel sample plate. Next, 10 μl of 4 mg mL−1 methanolysis products solution in chloroform was mixed with 10 μl of 0.1M HBMN solution in tetrahydrofuran. Then, 0.5 μl of the sample/matrix mixed solution was spotted onto the sodium iodide layer on the sample plate. MALDI-mass spectra were acquired in linear mode using a Shimadzu/Kratos AXIMA CFR Plus time-of-flight mass spectrometer (Shimadzu Corporation, Japan) equipped with a pulsed nitrogen laser (λ = 337 nm, 3 ns pulse width) and a delayed extraction ion source. Ions generated by the laser desorption were introduced into the flight tube with an accelerating voltage of 20 kV. The delay time was set at 90 ns. All mass spectra were acquired by averaging 100 individual laser shots. Mass calibration was accomplished using both CHIMASSORB 119FL (Ciba Japan K. K., Japan) (MW = 2285) and TINUVIN 144 (Ciba Japan K. K.) (MW = 684) as external standards.Results and discussion
MALDI-mass spectrum of decomposed products of the radiation-cured resins
In the case of supercritical methanolysis of UV-cured DPHA, methyl acrylate (MA) oligomers and/or polymers were mainly formed through selective cleavage and methylation at ester linkages in the cured resin.14 Provided that supercritical methanolysis of the radiation-cured PETA/VP resins proceeds in the same manner as that of UV-cured DPHA, selective cleavage and methylation should occurr at ester linkages in the resins. Scheme 2 illustrates the most probable decomposition pathway of the radiation-cured PETA/VP resins in supercritical methanol. It is considered that co-oligomers and/or copolymers containing MA and VP units are formed through selective cleavage and methylation at ester linkages in the radiation-cured resins. Meanwhile, pentaerithritol moiety should be converted to corresponding alcohol. Unless the pyrolytic cleavages at C–C bonds in the sequence of acryloyl and vinyl groups take place, the MA/VP co-oligomers and/or co-polymers should reflect the sequences of network junctions in the cured resins.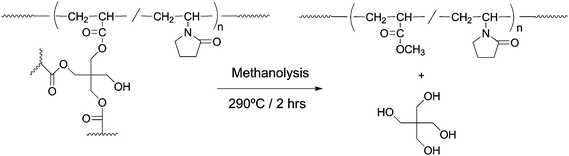 | ||
| Scheme 2 Most probable decomposition pathway of the radiation-cured PETA/VP resin in supercritical methanol. | ||
Fig. 1 shows the MALDI-mass spectra of the supercritical methanolysis products of the UV-cured resins (U1-A, U2-A and U3-A) initiated by DMPA alone with varying composition between PETA (acryloyl) and VP (vinyl). In the mass spectra, many peaks were commonly observed in the mass range up to more than m/z ca. 1500, although the distributions were somewhat different among them. In the mass spectrum of U1-A (acryloyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :vinyl = 2:1 in mole ratio), the peaks of co-oligomers having possible monomer combinations were observed in every n-mer region. For example, as shown in the expanded mass spectrum of U1-A, one of the representative peaks a1 at m/z = 872.5 was assigned to be sodium-cationized MA/VP co-oligomer having one vinylidene terminal, six MA monomer and three VP units. The peak a2 at m/z = 886.5 was identified to be the molecule having one vinylidene terminal, one MA monomer unit and seven VP units.
:vinyl = 2:1 in mole ratio), the peaks of co-oligomers having possible monomer combinations were observed in every n-mer region. For example, as shown in the expanded mass spectrum of U1-A, one of the representative peaks a1 at m/z = 872.5 was assigned to be sodium-cationized MA/VP co-oligomer having one vinylidene terminal, six MA monomer and three VP units. The peak a2 at m/z = 886.5 was identified to be the molecule having one vinylidene terminal, one MA monomer unit and seven VP units.
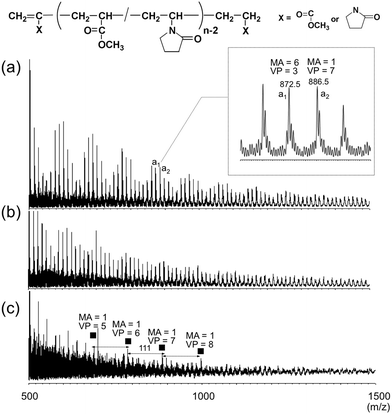 | ||
| Fig. 1 MALDI-mass spectra of supercritical methanolysis products of the UV-cured resins prepared with DMPA alone: (a) U1-A, (b) U2-A, (c) U3-A. | ||
Similarly to U1-A, the peaks of MA/VP co-oligomers were clearly observed in the spectrum of U2-A (Fig. 1(b); stoichiometric composition, acryloyl:vinyl = 1:1 in mole ratio). On the other hand, in the mass spectrum of U3-A with higher VP content (Fig. 1(c); acryloyl:vinyl = 1:2 in mole ratio), a series of peaks (labeled by solid squares) with the intervals of m/z = 111 were specifically observed. The intervals of m/z = 111 are consistent with a VP monomer unit and the observed peaks were assigned to sodium cationized MA/VP co-oligomers containing only one MA unit. These results suggests the network junctions in the UV-cured resins were mostly decomposed into corresponding MA/VP copolymer (co-oligomer) in the supercritical methanol under the optimized conditions.
Fig. 2 shows the MADI-mass spectra of supercritical methanolysis products of the EB-cured resins with 20 J g−1 irradiation dose prepared with varying compositions. In the mass spectra of the EB-cured resins, a series of peaks corresponding to possible MA/VP co-oligomers were observed. Additionally, the spectrum of E3-20 (Fig. 2(c); acryloyl:vinyl = 1:2, VP rich formulation) also contained the remarkable peaks (labeled by ■) of the co-oligomers containing only one MA unit similarly to U3-A. Meanwhile, in the spectrum of E1-20, the peaks of MA homo-oligomers (labeled by ●) with the intervals of m/z = 86 corresponding MA unit were prominently observed.
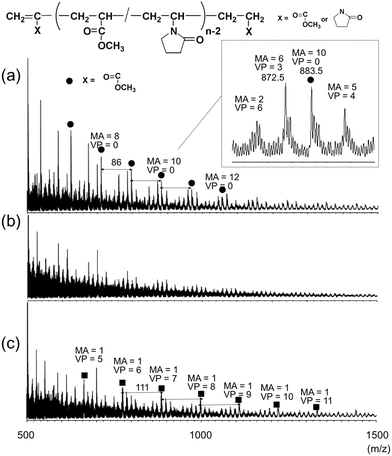 | ||
| Fig. 2 MALDI-mass spectra of supercritical methanolysis products of the EB-cured resins prepared with 20 J g−1 dosage: (a) E1-20, (b) E2-20, (c) E3-20. | ||
Evaluation of composition distributions in the network junctions
In order to estimate composition distribution of the polymer chains in the network junctions of the radiation-cured resins, MA/VP ratio in the decomposition products was evaluated from the intensities of characteristic peaks in the MALDI-mass spectra. Although the peaks of the decomposition products containing up to ca. 20 monomer units were observed in the spectra, the calculation on the basis of longer sequences lacked in accuracy because of poor S/N ratio in the high m/z region. In contrast, that on the basis of shorter chains was less reliable due to the restricted combination of MA and VP units. Therefore, 10-mers were representatively selected for the calculation of the composition distribution.Fig. 3 shows composition distributions of 10-mer products estimated from the specific peaks in the MALDI-mass spectra of the decomposition products of the UV-cured resins. As a whole, the products were widely distributed with the peak shifts corresponding to the copolymer composition, which indicates PETA and VP copolymerized in a random manner during the cure, and the distributions shifted to the co-oligomer region containing more VP units with increase in the VP content in feed. However, comparing the profile for U1-A prepared with DMPA alone as the photoinitiator with that for U1-B obtained with a combination of DMPA and BP, MA homo-decamer was produced more abundantly in the latter. It has been reported that the conversions of acrylates in the acrylate/VP mixture cured in air with VP contents lower than 50% were only 20–50%, while those in the absence of oxygen reached nearly 90%.15 Therefore, higher yield of MA homo-oligomers in U1-B might be concerned with increasing conversion of acrylic double bonds caused by decreased oxygen inhibition15–17 due to oxygen scavenging in the presence of VP and BP.15
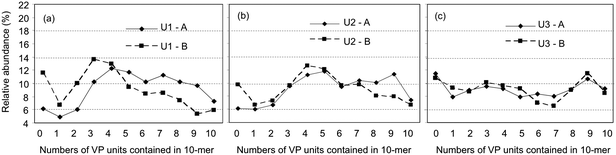 | ||
| Fig. 3 Composition distributions of co-oligomers composed of ten monomer units observed in the MALDI-mass spectra of the methanolysis products of the UV-cured resins: (a) U1-A and B, (b) U2-A and B, (c) U3-A and B. | ||
Meanwhile, as for U3-A and B, the co-oligomer containing only one MA unit and nine VP units, and even the VP homo-decamer were considerably yielded regardless of the kind of photoinitiator used. Theoretically, charge-transfer interaction might occur between PETA and VP, having electron poor acryloyl group and electron rich N-vinyl group, respectively. Although alternating copolymerization would be supposed to proceed preferentially in such a charge-transfer complex system, this observation suggests VP could homo-polymerize substantially in its higher content formulations. This consideration agrees with a lack of the specific UV-visible absorption spectra due to the formation of charge-transfer complex between acrylates and VP reported previously18 and enhanced photopolymerization rate of VP in the presence of oxygen.18,19
Fig. 4 shows composition distributions of 10-mer products estimated for the EB-cured resins from the corresponding peak intensities in the MALDI-mass spectra of the decomposition products. Similarly to those of the UV-cured resins, the observed distributions shifted to the regions of VP rich oligomers with increasing VP contents in formulations. In E1-20 and E1-40 (acryloyl:vinyl = 2:1), and E2-20 and E2-40 (acryloyl:vinyl = 1:1), MA homo-decamer generated more largely than that in the UV-cured resins of the corresponding composition, especially for curing with the lower dosage (20 J g−1). Although this higher yield of MA oligomer cannot be explained conclusively at present, its dependency on EB dosage might be related to the difference in the amount of initiating species generated by EB irradiation. Moreover, as shown in Fig. 4 (c) for E3-20 and E3-40 (acryloyl:vinyl = 2:1), relatively large amount of co-oligomers containing only one MA unit and nine VP units, and even VP homo-decamer were produced for VP rich formulation similarly to U3-A and B. To the best of our knowledge, no experimental data have been available on the detailed polymerization process of EB-curing of VP. Therefore, it is noteworthy that relatively long sequences of VP units were found in the network junctions for the acrylate/VP curing system.
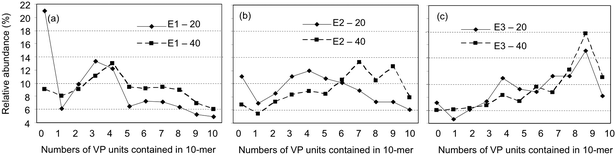 | ||
| Fig. 4 Composition distributions of co-oligomers composed of ten monomer units observed in the MALDI-mass spectra of the methanolysis products of the EB-cured resins: (a) E1-20 and 40, (b) E2-20 and 40, (c) E3-20 and 40. | ||
Conclusions
The MALDI-mass spectra of the supercritical methanolysis products of the radiation-cured PETA/VP resins contained a series of peaks of sodium-cationized MA/VP co-oligomers. This observation suggests the network junctions in the UV-cured resins were mostly decomposed into the co-oligomers under the optimized conditions through selective cleavage and methylation at ester linkages in the cured resins, reflecting the cross-linking sequences. For instance, the composition distributions of the 10-mer products estimated from the obtained MALDI-mass spectra were somewhat different among the curing conditons such as energy source, photoinitiators, dosage of radiation and formulations. In conclusion, the results obtained in this work demonstrate MALDI-MS combined with supercritical methanolysis make it possible to elucidate the cross-linking structure of radiation-cured resins in detail, even for the composition distribution in copolymer-type, which cannot be measured by any other conventional analytical methods.Acknowledgements
The authors are grateful to Mr Hironori Hata (Artec Co., Ltd) for his help in EB irradiation. This work was supported in part by the research foundation of Frontier Laboratory Inc., and Grant-in-Aid for Scientific Research (B) (19350037) of Japan Society for the Promotion of Science, Japan.References
- J. L. Garnett, Radiat. Phys. Chem., 1995, 46, 925 CrossRef CAS.
- A. V. Rao, D. S. Kanitar and A. K. Parab, Prog. Org. Coat., 1995, 25, 221 CrossRef CAS.
- S. C. Lapin, RadTech Report, 2008, September/October, 27 Search PubMed.
- A. Lungu and D. C. Neckers, Macromolecules, 1995, 28, 8147 CrossRef CAS.
- V. M. Litvinov and A. A. Dias, Macromolecules, 2001, 34, 4051 CrossRef CAS.
- H. Nakagawa, S. Tsuge and K. Murakami, J. Anal. Appl. Pyrolysis, 1986, 10, 31 CrossRef CAS.
- H. Nakagawa and S. Tsuge, Macromolecules, 1985, 18, 2068 CrossRef CAS.
- H. Nakagawa, S. Wakatsuka, S. Tsuge and T. Koyama, Polym. J., 1988, 20, 9 CrossRef CAS.
- H. Nakagawa, S. Wakatsuka, S. Tsuge and T. Koyama, Polymer, 1992, 33, 4556 CrossRef CAS.
- H. Matsubara, Y. Yoshida, H. Ohtani and S. Tsuge, J. Anal. Appl. Pyrolysis, 2002, 64, 159 CrossRef CAS.
- H. Matsubara, A. Yoshida, Y. Kondo, S. Tsuge and H. Ohtani, Macromolecules, 2003, 36, 4750 CrossRef CAS.
- H. Matsubara and H. Ohtani, J. Anal. Appl. Pyrolysis, 2006, 75, 226 CrossRef CAS.
- H. Matsubara and H. Ohtani, Anal. Sci., 2007, 23, 513 CrossRef CAS.
- H. Matsubara, S. Hata, Y. Kondo, Y. Ishida, H. Takigawa and H. Ohtani, Anal. Sci., 2006, 22, 1403 CrossRef CAS.
- C. W. Miller, C. E. Hoyle, S. Jonsson, C. Nason, T. Y. Lee, W. F. Kuang, and K. Viswanathan, in Photoinitiated Polymerization, ed. K. D. Belfield and J. V. Crivello, American Chemical Society, Washington, D.C., 2003, p. 2 Search PubMed.
- J. G. Kloosterboer, Adv. Polym. Sci., 1988, 84, 1 CAS.
- H. F. Gruber, Prog. Polym. Sci., 1992, 17, 953 CrossRef CAS.
- M. Takeishi and G. Tao, J. Polym. Sci., Part C: Polym. Lett., 1989, 27, 301 CrossRef CAS.
- N. Davidenko, C. Peniche, R. Sastre and J. S. Roman, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1753 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |