Colloidal crystals formed by polymer brush-afforded fine particles
Kohji
Ohno
*
Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: ohno@scl.kyoto-u.ac.jp
First published on 6th August 2010
Abstract
This review highlights the recent development of a new family of colloidal crystals. In this system, monodisperse fine particles grafted with a concentrated polymer brush layer are used as a building block for the crystallization. Unlike the previously known (soft and hard) colloidal crystals, the driving force of crystallization in the new system is a long-range repulsive (non-interpenetrating) interaction between the highly swollen concentrated brush layers. The crystallization concentration is situated between those of typical soft and hard systems. Hence this is termed a semisoft system. Advantages of semisoft colloidal crystal include controllability of interparticle distance and crystal structure by controlling the graft chain length and density, applicability of various particles, applicability of various monomers, and usability of various solvents and solvent mixtures.
 Kohji Ohno | Kohji Ohno received his PhD from Kyoto University in 1999, under the supervision of Professor Takeshi Fukuda. From 1999 to 2001, he worked as a postdoctoral researcher in the group of Professor David M. Haddleton at the University of Warwick in the United Kingdom. He has been working at the Institute for Chemical Research of Kyoto University as an assistant professor (2001–2009) and as an associate professor (2010–present). His current research interests include living radical polymerizations, polymer brushes, synthesis and self-assembly of fine particles, and biomedical applications of fine particles. |
1. Introduction
Colloidal crystals have attracted much attention for fundamental understanding of condensed matter crystallization1–13 as well as for their potential applications, in particular, as photonic crystals which are characterized by alternating domains of higher and lower refractive indices with a periodicity on the order of the wavelengths of visible lights.14–16 Typical colloidal crystals are formed by spherical particles suspended in a liquid. At low particle volume concentration ϕ, such a suspension is a (disordered) fluid, while above a crystallization concentration ϕf, crystallites separate out from the fluid, and crystal and fluid phases coexist in thermodynamic equilibrium until the concentration reaches another critical value ϕm, above which the system is totally crystalline. Driving forces for the formation of colloidal crystals are believed to be repulsive potentials working between colloidal particles, of which essentially two types have been known so far. One is the hard-sphere potential, which is steric and short-range in nature, inducing crystallization of rigid uncharged particles (hard colloidal crystal).3–6,9,10 The other is the electrostatic potential, which can be of extremely long range depending on the ionic strength of the system, inducing crystallization of charged particles (soft colloidal crystal).7,8,17,18We recently developed a third type of colloidal crystals, semisoft colloidal crystals, in which the driving force of crystallization is the excluded-volume interactions between polymer chains densely grafted on spherical particles.19–21 This system is distinguished from any of the previously observed colloidal crystals or similar ordered assemblies formed by e.g., block copolymers and star-shaped polymers. In fact, colloidal crystals of spherical particles with polymer chains terminally grafted on the surface have been known since early 1970s, and it has been speculated that those graft chains would work as a repulsive interparticle potential, a rather “soft” potential qualitatively analogous to the electrostatic one. However, in all of the previously studied systems of this kind,3,4,6,8,12 the graft density was so low, and in many cases, the graft chain length was so small that the graft chains simply played the role of stabilizing the particles, having no dominant effect on the interparticle potential, and thus the system essentially showed the characteristics of a hard colloidal crystal. Using surface-initiated atom transfer radical polymerization (ATRP),22 we succeeded for the first time in preparing silica particles (SiPs) having a shell layer of well-defined polymer chains densely and thickly end-grafted on the particle surface with no aggregation of particles caused and the narrow distribution of particle size maintained throughout the course of preparation.23 The surface density of the grafted poly(methyl methacrylate) (PMMA) chains reached some 0.7 chains nm−2, going deeply into the regime of concentrated polymer brush (CPB). CPBs had not attracted much attention until recently, when ATRP and other techniques of living radical polymerization (LRP) proved to be capable of routinely providing well-defined CPB samples, with which experiments got started disclosing a number of unique properties of CPBs,24,25 essentially different from those of less densely grafted surfaces semidilute polymer brushes (SDPBs). Important properties of CPBs relevant to colloidal crystallization include the extremely large thickness of swollen CPB layer, almost comparable to the fully stretched length of graft chains,26,27 and the non-interpenetrating or immiscible property of confronted CPBs even at extremely high compressing pressures.25 The latter property contrasts to that of confronted SDPBs, which do interpenetrate or mix with each other beyond a critical pressure because of the less extended conformation of SDPB chains. Thus the brush-brush interaction is suggested to be qualitatively different between CPB and SDPB systems.
2. Synthesis of perfectly dispersive particles grafted with a concentrated polymer brush
We succeeded for the first time in preparing perfectly dispersive, monodisperse silica particles (SiPs) grafted with a CPB.23 This success strongly encouraged us to try to fabricate a semisoft colloidal crystal. SiPs are among the most extensively studied particles for the application of surface-initiated ATRP.23,28–35 Various ATRP initiators that can be fixed on silicon oxide surfaces have been synthesized by several groups.28–33,36–47 All of them were mono- or trichlorosilane derivatives, except the monoethoxysilane derivative synthesized by Patten et al.28,29 In search for a better route for the modification of SiPs by surface-initiated ATRP, we synthesized a new triethoxysilane derivative to introduce ATRP initiation sites onto SiP surfaces without causing any aggregation of the particles.23 Even though the chlorosilane group has the advantage of high reactivity, the reaction with it must be carried out in a dried aprotic solvent to prevent an unfavorable side reaction. On the other hand, the reaction with the triethoxysilane group can be carried out in protic solvent, even in the presence of water. This is a clear advantage for achieving a homogeneous modification of the SiP surface because SiPs exhibit higher dispersibility in a protic solvent than in an aprotic one due to the hydrophilic character of the SiP surface with silanol groups. In addition, the reaction with triethoxysilane group forms a stable Si–O–Si network via the trivalent reaction. The triethoxysilane derivative with an initiating site for ATRP was synthesized via the two-step reaction: in short, 5-hexen-1-ol was acylated with 2-bromoisobutyryl bromide to obtain 1-(2-bromo-2-methyl)propionyloxy-5-hexene (BPH), the aryl group of which was subsequently hydrosilylated with triethoxysilane in the presence of Karstedt's catalyst to give the final product (2-bromo-2-methyl)propionyloxyhexyl triethoxysilane (BHE). The ATRP initiator BHE was fixed onto the SiP surface in ethanol solution with ammonia (NH3) added as an alkaline catalyst.23 The NH3 concentration is a key parameter in this reaction: if it is too high, it will cause particle aggregation due to the high ionic strength, while if it is too low, it will make the reaction too slow. The final NH3 concentration was optimized to be 1 M.The initiator-coated SiPs were subsequently used for the copper-mediated ATRP of methyl methacrylate (MMA) in bulk.23 To obtain a satisfactory result, the following points were important. First, the initiator-coated SiPs should never be dried before the polymerization. Once dried, the particles are difficult to homogeneously redisperse in the polymerization medium even with the aid of ultrasonication. Second, the polymerization should be carried out in the presence of the “sacrificial” free initiator EBiB. With free polymers produced by EBiB, many entanglements between the graft and free polymers would work cooperatively and produce an extremely high friction force depending on chain lengths, graft density, and free polymer concentration, which would ensure that the particles hardly diffuse. This would prevent interparticle coupling which would bring about gelation or particle aggregation. The role of the free initiator is also to accumulate an appropriate amount of Cu(II) species via the termination of polymer radicals, thus to control the polymerization by the so-called persistent radical effect.48–50 The polymerization proceeded in a living manner, producing SiPs grafted with well-defined PMMA with a graft density as high as 0.7 chains nm−2. Dynamic light scattering (DLS) measurement shows that the particles have retained their high dispersibility throughout the experimental processes, i.e., before and after the initiator fixation and also before and after the graft polymerization.
3. Formation of semisoft colloidal crystals
In a typical experiment, SiPs with PMMA brushes were used for crystal formation. The hybrid particles had the SiP core and a shell of PMMA chains with weight-average molecular weight Mw = 188,000 and polydispersity index Mw/Mn = 1.19, end-grafted on the SiP surface with a surface density as high as 0.7 chains nm−2. The PMMA-SiPs (sample P3) were suspended in a nearly isorefractive and isobuoyant mixture of solvent. The refractive index matching provides nearly transparent suspensions, which is essential for optical observations, and the density matching reduces the effect of gravity, which is essential for the system to achieve a thermodynamic equilibrium. The PMMA-SiP suspensions in the solvent mixture with different particle volume fractions (ϕ = 0.0785–0.111) were placed in Pyrex glass cells (10 × 10 × 40 mm) connected with a glass tube for sealing, and the systems were sealed off and allowed to stand at 25 °C for 7 days. Fig. 1a shows the photographs of the series of samples illuminated from behind by white light. The numbers shown in Fig. 1a represent the sample codes. In sample 1 with the lowest concentration examined here, the entire body of the suspension remained slightly turbid due to the Tyndall scattering. Sample 9 with the highest particle concentration exhibited heterogeneously iridescent colors within an hour after sample setting, which hardly changed throughout the experimental period of 7 days. In samples 2–8 with intermediate concentrations, tiny iridescent flecks were observed soon after the onset of experiment, indicating the formation of Bragg-reflecting crystallites. In sample 8, the formed crystallites filled the whole volume of the suspension from an early stage of experiment without sedimentation. In samples 2–7, as time elapsed, the crystallites sedimented under the effect of gravity, and the boundary between the iridescent-colored sediment (crystalline phase) and the slightly turbid supernatant (fluid phase) became more and more distinct, ultimately giving a sharp horizontal boundary line. This spontaneous phase separation can be interpreted by the idea of the Kirkwood-Alder transition.51,52 The sedimentation of the crystalline phase occurs because the particle concentration (and hence the density) of the crystalline phase is slightly larger than that of the fluid phase.4,6Fig. 1b shows a close-up of sample 8, which is composed of a pure crystalline phase. Single crystals 0.1 to 1 mm in size are visible, showing different colors depending on their orientations. Upon tumbling the cell, the iridescent colors observed in samples 2–8 instantaneously disappeared, and upon standing they appeared again in almost the same way as before. | ||
| Fig. 1 Photographs of PMMA-SiP suspensions in the mixed solvent (1,2-dichloroethane/chlorobenzene/o-dichlorobenzene = 53/20/27 volume ratio) illuminated from behind by white light; 7 days after sample preparation.19 The weight-average molecular weight of the PMMA grafts is 188,000, and the diameter of the SiP core is 130 nm. (a) Samples with different PMMA-SiP volume fractions (ϕ). The ϕ value increases from 0.0785 (leftmost) to 0.111 (rightmost). (b) A close-up of sample 8. Reproduced by permission of The American Chemical Society from ref. 19: Ohno et al., Macromolecules, 2006, 39, 1245. Copyright 2006 American Chemical Society. | ||
Similar experiments were carried out using another two hybrids with a fixed SiP diameter of 130 nm and varying PMMA graft length of Mw = 88,000 (sample P1) and 518,000 (sample P5). The results were essentially similar to those for sample P3 described above. Namely, the crystallization occurred via the Kirkwood-Alder transition. Fig. 2 shows the phase diagrams of the suspensions of samples P1, P3, and P5, in which the volume percentage of the crystalline phase is plotted as a function of the volume fraction ϕ of the PMMA-SiP particle. In all cases, the plot shows a linear relationship in the fluid-crystal coexistence region, as is demanded for the coexisting two phases to be in a thermodynamic equilibrium. Extrapolation of the linear line to 0% and 100% crystal gives the freezing and melting volume fractions, ϕf and ϕm, respectively. The melting point ϕm decreased from about 0.14 for the smallest sample (P1) to about 0.04 for the largest sample (P5). Note that ϕ is the sum of the volume fractions of the SiP core and grafted chains, and thus these results suggest that the values of ϕm of the present systems (spherical particles afforded with CPB) can possibly cover the volume fraction range throughout from typical soft (ϕm ∼ 0.01) to hard (ϕm = 0.545) systems by simply changing the graft chain length.
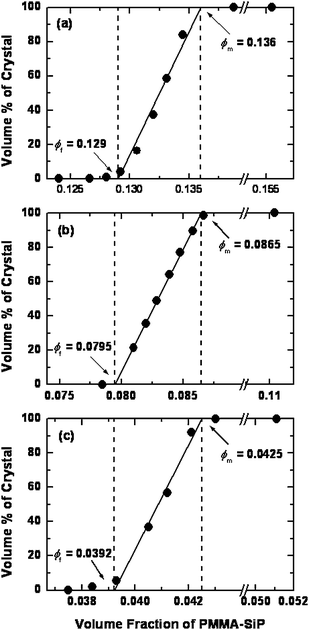 | ||
| Fig. 2 Phase diagrams showing the volume fraction of crystalline phase as a function of the volume fraction of PMMA-SiP hybrid particles P1, P3, and P5 suspended in the mixed solvents.20 Parts (a), (b), and (c) show the data for samples P1, P3, and P5, respectively. Reproduced by permission of The American Chemical Society from ref. 20: Ohno et al., Macromolecules, 2007, 40, 9143. Copyright 2007 American Chemical Society. | ||
4. Structural analysis of semisoft colloidal crystals
Direct observation of the structure of semisoft colloidal crystals was carried out by confocal laser scanning microscopy (CLSM). To achieve a high resolution, we used fluorescence dye, rhodamine-labeled silica particles (RhSiPs) prepared by the Stöber method, then carried out a fluorescence-mode CLSM analysis with a sires of CPB-RhSiP hybrids having a larger core diameter (590 nm) and a wide range of PMMA graft chain lengths (from 75,000 to 1,099,000 in Mw) of their averaged surface density 0.7 chains nm−2. Colloidal crystallization of the rhodamine-labeled samples (R-series) is essentially similar to that of non-labeled samples (P-series) described above.Fig. 3 shows a typical example of the three-dimensional (3-D) image, observed by fluorescence mode CLSM, of the ordered arrays of RhSiPs formed by hybrid particles. The crystalline structure can be analyzed by slicing the 3-D image on desired planes. Fig. 4 collects the CLSM images of the two-dimensional (2-D) close-packed (cp) planes of the crystals formed by the hybrid particles with varying graft chain lengths. In all images, the RhSiP cores can be seen as red circles and the PMMA brushes which should be surrounding the RhSiP cores are invisible, as they are not fluorescence-labeled. Noteworthy are not only the high degree of positional order of the RhSiP cores but also the strong dependence of the interparticle distance on the chain length of PMMA grafts.
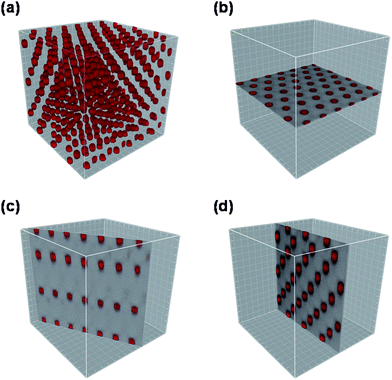 | ||
| Fig. 3 CLSM images of the colloidal crystal formed by PMMA-RhSiPs. The diameter of RhSiP core is 590 nm, and the Mw of the PMMA grafts is 620,000.21 (a) shows a three-dimensional image for a part of a micro crystallite. (b), (c), and (d) show the images of the (001), (100), and (110) planes, respectively, extracted from (a) by the image analysis software Imaris. Note that these planes are indexed according to the Miller description for the hexagonal system, in which the two-dimensional close-packed plane (001) is defined as an x–y plane of a unit lattice. Reproduced by permission of The American Chemical Society from ref. 21: Morinaga et al., Macromolecules, 2008, 41, 3620. Copyright 2008 American Chemical Society. | ||
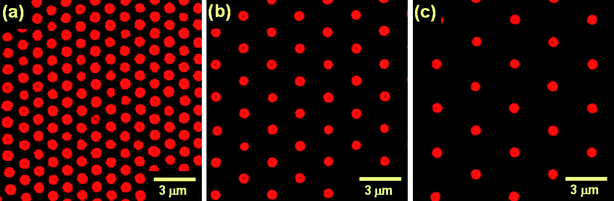 | ||
| Fig. 4 CLSM images of the (001) plane (two-dimensional close-packed plane) of the colloidal crystals formed by PMMA-RhSiP hybrid particles.21 The Mws of the PMMA grafts are (a) 159,000, (b) 620,000, and (c) 1,099,000. The mean nearest-neighbor center-to-center distances in the images measure (a) 1,230 nm, (b) 2,360 nm, and (c) 3,190 nm. Reproduced by permission of The American Chemical Society from ref. 21: Morinaga et al., Macromolecules, 2008, 41, 3620. Copyright 2008 American Chemical Society. | ||
The 2-D cp planes only exist in crystals featuring face-centered cubic (fcc), hexagonal close-packed (hcp) or random hexagonal close-packed (rhcp) structures. The difference between these crystalline structures lies in the sequential stacking of cp planes, which can be made visible by analyzing the plane perpendicular to the cp plane. Of the several crystalline planes perpendicular to the cp plane, the (110) plane most clearly exhibits the difference between fcc and hcp stackings.53Fig. 5 shows an example of the CLSM image of the (110) crystalline plane. As demonstrated in Fig. 5b, the fcc stacking is characterized by a linear arrangement of three consecutive spheres in the (110) plane, and the hcp stacking is given by their zig-zag arrangement, and therefore these two stacking modes are easily distinguished in the image of the (110) plane. The stacking probability α, i.e., the number fraction of fcc stackings is given by
| α = Nfcc/(Nfcc + Nhcp) | (1) |
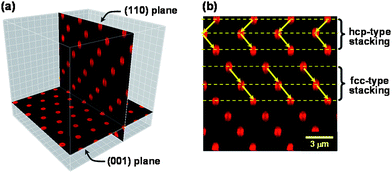 | ||
| Fig. 5 CLSM images of the (001) and (110) crystalline planes of a colloidal crystal.21 (a) shows the spatial relation between the (001) and (110) planes, and (b) demonstrates the difference of hcp and fcc stackings appearing in the (110) plane. Reproduced by permission of The American Chemical Society from ref. 21: Morinaga et al., Macromolecules, 2008, 41, 3620. Copyright 2008 American Chemical Society. | ||
In Fig. 6, α is plotted as a function of the logarithm of Mw (or scaled Mw for the P-series of samples) of the PMMA grafts. The figure suggests the existence of three regions. In the first region of relatively small chain lengths, α is nearly constant and equal to about 0.59. Namely, in this region, fcc and hcp structures coexist in nearly equal proportion, similar to the rhcp structure in which α = 0.5. In the second “transition” region of intermediate chain lengths, α increases rather sharply and approaches 1 with increasing Mw. In the third region of large chain lengths, α is constant and equal to 1.0, i.e., perfect fcc.
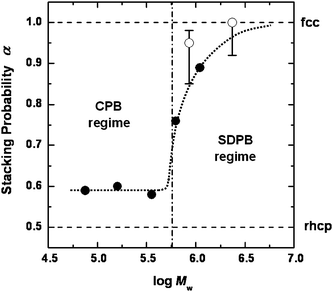 | ||
| Fig. 6 Plot of stacking probability α versus logarithm of Mw of PMMA grafts.21 (●): observed for R-series of samples by CLMS in fluorescence mode; (○): observed for P-series of samples by CLSM in reflection mode. The vertical dot-dash line shows the approximate crossover between CPB and SDPB regimes estimated from the chain length dependence of the hydrodynamic diameter of the P-series of samples (see ref. 20). Reproduced by permission of The American Chemical Society from ref. 21: Morinaga et al., Macromolecules, 2008, 41, 3620. Copyright 2008 American Chemical Society. | ||
The observed chain length dependence of α seems to go in parallel with the chain length dependence of brush layer thickness or graft chain conformation. With the P-series of samples,20 we showed that in a region of small graft chain lengths, the whole graft layer is in a CPB (concentrated polymer brush) regime in which the excluded-volume effect is unimportant, while in a region of large chain lengths, the brush layer is divided into two regimes by a characteristic radius rc. In the inside of this radius (r < rc), the brush layer is in a CPB regime, and in the outside of it (r > rc), it is in a SDPB (semidilute polymer brush) regime, where excluded-volume effect is important. Therefore, the chain conformation and the interparticle potential curve of hybrid particles with short grafts should be very different from those of hybrid particles with long grafts. In fact, Fig. 6 seems to be roughly dividable into CPB and SDPB regimes by the vertical straight line corresponding to the CPB-SDPB boundary.
As noted above, our system with graft chains in the CPB regime (or relatively short graft chains) behaved like a hard sphere system. This is understandable, since chains in CPB regime are highly extended. We now get involved in the controversy regarding the structure of the hard sphere system.54 The fcc structure is favored by the theory54 but earlier experiments10,55–58 indicated a structure close to rhcp. Our results for those samples that are considered to be in the CPB regime indicated that α is about 0.59 with no clear chain length dependence in this region. This will vote for the experimental favor of rhcp in hard or quasi-hard systems.
Our result also indicated that α increased rather sharply as the chain length increased beyond a critical value, and approached 1 for the perfect fcc. We explain this dependence of α on graft chain length in terms of the qualitative change in interparticle potential accompanied by the CPB to SDPB crossover. Systems with long graft chains will be characterized by the rather “soft” interparticle potential of SDPBs. Since graft chains in a SDPB system are less extended on average and can take numerous conformations of differing degrees of chain extension, the interparticle potential between SDPB systems should be, unlike that between CPB systems, necessarily of long range nature.
All these observations and discussion suggest that fcc might be a thermodynamically stable phase in hard as well as semisoft systems. However, during the crystallization process, some hcp arrangements might be frozen in irreversibly. This non-equilibrium process will be more likely to happen for systems with a shorter range of interparticle potential like those of the hard sphere and CPB systems. The entropy difference between fcc and hcp phases theoretically deduced54 may be too small to realize an equilibrium system in usual experimental conditions. On the other hand, as the interparticle potential range becomes longer, not only the nearest-neighbor but also the second-nearest (and higher-order) interactions can play a role in crystallization, giving much larger energetic and entropic differences between fcc and hcp phases. Therefore non-equilibrium processes will be less likely to occur in systems with a longer range of interparticle potential.
5. Conclusions and outlook
Monodisperse SiPs grafted with a concentrated PMMA brush layer and suspended in a good solvent of PMMA formed a colloidal crystal in a certain concentration range. Unlike the previously known (soft and hard) colloidal crystals, the driving force of crystallization in this system is a long-range repulsive (non-interpenetrating) interaction between the highly swollen concentrated brush layers. The crystallization concentration is situated between those of typical soft and hard systems. Hence this is a new type of colloidal crystal, which we term a semisoft system. The internal structure of semisoft colloidal crystals was directly observed by CLSM mainly in fluorescence mode. The crystalline structure was of closed-packed type in all cases, and the nearest-neighbor interparticle distance in the crystal increased with increasing graft chain length. The crystals consisted of two-dimensional hexagonal close-packed planes which are stacked in a statistical sequence of hcp and fcc arrangements. The probability α of finding a fcc stacking was about 0.59 in a region of relatively short graft chains and substantially equal to 1.0 (perfect fcc) in a region of relatively long graft chains, exhibiting a rather narrow intermediate transition region, where α steeply increased with increasing chain length. This transition of crystalline structure from a nearly random stacking to an fcc arrangement was ascribed to a qualitative change in graft chain conformation and hence interparticle potential curve accompanying the CPB-to-SDPB crossover of the effective graft density. Thus semisoft colloidal crystals of CPB-afforded hybrid particles possibly cover, by simply changing the graft chain length, a wide range of crystallization concentrations and interparticle potentials between those of typical hard and soft colloidal crystals. Ordering of particle-like polymeric architectures such as star-shaped polymers and block copolymer micelles in solution has been found by both experiment and computer-simulation.59–62 In some cases, structural transformation from fcc to bcc lattice occurred as the interparticle potential became softer.60,61 In view of these results, our semisoft systems may, in the limit of large chain length relative to core diameter, exhibit a fcc to bcc transition. Thus we may be able to cover the whole range of possible crystalline structures including rhcp, fcc, and bcc structures with a single CPB-core particle system by simply changing the graft chain length. This remains to be studied as a future target. Thanks to the usability of various solvents in the semisoft colloidal system, we are trying to use a non-volatile ionic liquid solvent which has a good advantage for future applications such as the constructions of laser devices and light wave guides.Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (Grant-in-Aid 17002007 and 17685010) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan and by Industrial Technology Research Grant Program in 2004 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan.References
- P. Habdas and E. R. Weeks, Curr. Opin. Colloid Interface Sci., 2002, 7, 196 CrossRef CAS.
- A. Yethiraj and A. van Blaaderen, Nature, 2003, 421, 513 CrossRef CAS.
- W. K. Kegel and A. van Blaaderen, Science, 2000, 287, 290 CrossRef CAS.
- P. N. Pusey and W. van Megen, Nature, 1986, 320, 340 CrossRef CAS.
- W. van Megen and S. M. Underwood, Nature, 1993, 362, 616 CrossRef CAS.
- A. Kose and S. Hachisu, J. Colloid Interface Sci., 1974, 46, 460 CrossRef CAS.
- S. Hachisu, Y. Kobayashi and A. Kose, J. Colloid Interface Sci., 1973, 42, 342 CrossRef CAS.
- S. Hachisu and K. Takano, Adv. Colloid Interface Sci., 1982, 16, 233 CrossRef CAS.
- Z. Cheng, W. B. Russel and P. M. Chaikin, Nature, 1999, 401, 893 CrossRef CAS.
- J. Zhu, M. Li, R. Rogers, W. Meyer, R. H. Ottewill, STS-73 Space Shuttle Crew, W. B. Russel and P. M. Chaikin, Nature, 1997, 387, 883 CrossRef CAS.
- S. Auer and D. Frenkel, Nature, 2001, 409, 1020 CrossRef CAS.
- U. Gasser, E. R. Weeks, A. Schofield, P. N. Pusey and D. A. Weitz, Science, 2001, 292, 258 CrossRef CAS.
- A. P. Gast and Y. Monovoukas, Nature, 1991, 351, 553 CrossRef CAS.
- E. Yablonovitch, Nature, 1999, 401, 539 CrossRef CAS.
- A. Arsenault, S. Fournier-Bidoz, B. Hatton, H. Míguez, N. Tétreault, E. Vekris, S. Wong, S. M. Yang, V. Kieaev and G. A. Ozin, J. Mater. Chem., 2004, 14, 781 RSC.
- V. L. Colvin, MRS Bull., 2001, 26, 637 CAS.
- P. A. Hiltner and I. M. Krieger, J. Phys. Chem., 1969, 73, 2386 CrossRef CAS.
- T. Okubo, Prog. Polym. Sci., 1993, 18, 481 CrossRef CAS.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2006, 39, 1245 CrossRef CAS.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143 CrossRef CAS.
- T. Morinaga, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2008, 41, 3620 CrossRef CAS.
- (a) J.-B. Kim, M. L. Bruening and G. L. Baker, J. Am. Chem. Soc., 2000, 122, 7616 CrossRef CAS; (b) R. A. Sedjo, B. K. Mirous and W. J. Brittain, Macromolecules, 2000, 33, 1492 CrossRef CAS; (c) C. Perruchot, M. A. Khan, A. Kamitsi, S. P. Armes, T. von Werne and T. E. Patten, Langmuir, 2001, 17, 4479 CrossRef CAS; (d) X. Huang and M. J. Wirth, Anal. Chem., 1997, 69, 4577 CrossRef CAS; (e) D. M. Jones, A. A. Brown and W. T. Huck, Langmuir, 2002, 18, 1265 CrossRef CAS.
- K. Ohno, T. Morinaga, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2005, 38, 2137 CrossRef CAS.
- Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Adv. Polym. Sci., 2006, 197, 1 CAS.
- T. Fukuda, Y. Tsujii and K. Ohno, Macromolecular Engineering. Precise Synthesis, Materials Properties, Applications, ed. K. Matjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007, pp. 1137–1178 Search PubMed.
- S. Yamamoto, M. Ejaz, Y. Tsujii, M. Matsumoto and T. Fukuda, Macromolecules, 2000, 33, 5602 CrossRef CAS.
- S. Yamamoto, M. Ejaz, Y. Tsujii and T. Fukuda, Macromolecules, 2000, 33, 5608 CrossRef CAS.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409 CrossRef CAS.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497 CrossRef CAS.
- J. Pyun, K. Matyjaszewski, T. Kowalewski, D. Savin, G. Patterson, G. Kickelbick and N. Huesing, J. Am. Chem. Soc., 2001, 123, 9445 CrossRef CAS.
- J. Pyun, S. Jia, T. Kowalewski, G. D. Patterson and K. Matyjaszewski, Macromolecules, 2003, 36, 5094 CrossRef CAS.
- G. Carrot, S. Diamanti, M. Manuszak, B. Charleux and J.-P. Vairon, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4294 CrossRef CAS.
- H. Mori, D. C. Seng, M. Zhang and A. H. E. Müller, Langmuir, 2002, 18, 3682 CrossRef CAS.
- X. Y. Chen and S. P. Armes, Adv. Mater., 2003, 15, 1558 CrossRef CAS.
- X. Y. Chen, S. P. Armes, S. J. Greaves and J. F. Watts, Langmuir, 2004, 20, 587 CrossRef CAS.
- R. R. Shah, D. Merreceyes, M. Husemann, I. Rees, N. L. Abbott, C. J. Hawker and J. L. Hedrick, Macromolecules, 2000, 33, 597 CrossRef CAS.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424 CrossRef.
- K. Matyjaszewski, P. J. Miller, N. Shukla, B. Immaraporn, A. Gelman, B. B. Luokala, T. M. Siclovan, G. Kickelbick, T. Vallant, H. Hoffmann and T. Pakula, Macromolecules, 1999, 32, 8716 CrossRef CAS.
- J. D. Jeyaprakash, S. Samuel, R. Dhamodharan and J. Rühe, Macromol. Rapid Commun., 2002, 23, 277 CrossRef CAS.
- H. Mori, A. Boker, G. Krausch and A. H. E. Müller, Macromolecules, 2001, 34, 6871 CrossRef CAS.
- D. A. Savin, J. Pyun, G. D. Patterson, T. Kowalewski and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2667 CAS.
- Y.-P. Wang, X.-W. Pei and K. Yuan, Mater. Lett., 2005, 59, 520 CrossRef CAS.
- A. El Harrak, G. Carrot, J. Oberdisse, C. Eychenne-Baron and F. Boué, Macromolecules, 2004, 37, 6376 CrossRef.
- P. Liu, J. Tian, W. Liu and Q. Xue, Polym. Int., 2004, 53, 127 CrossRef CAS.
- J. Parvole, G. Laruelle, C. Guimon, J. Francois and L. Billon, Macromol. Rapid Commun., 2003, 24, 1074 CrossRef CAS.
- X. Huang and M. J. Wirth, Anal. Chem., 1997, 69, 4577 CrossRef CAS.
- X. Huang, L. J. Doneski and M. J. Wirth, Anal. Chem., 1998, 70, 4023 CrossRef CAS.
- M. Ejaz, S. Yamamoto, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 1998, 31, 5934 CrossRef CAS.
- M. Ejaz, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2000, 33, 2870 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS.
- W. G. Hoover and F. H. Ree, J. Chem. Phys., 1968, 49, 3609 CrossRef CAS.
- B. J. Alder, W. G. Hoover and D. A. Young, J. Chem. Phys., 1968, 49, 3688 CrossRef CAS.
- (a) M. S. Elliot, B. T. F. Bristol and W. C. K. Poon, Phys. A, 1997, 235, 216 CrossRef CAS; (b) M. S. Elliot and W. C. K. Poon, Adv. Colloid Interface Sci., 2001, 92, 133 CrossRef CAS.
- L. V. Woodcock, Nature, 1997, 385, 141 CrossRef CAS.
- P. N. Pusey, W. van Megen, P. Bartlett and B. J. Ackerson, Phys. Rev. Lett., 1989, 63, 2753 CrossRef CAS.
- N. A. M. Verhaegh, J. S. van Duijneveldt, A. van Blaaderen and H. N. W. Lekkerkerker, J. Chem. Phys., 1995, 102, 1416 CrossRef CAS.
- D. Frenkel and A. J. C. Ladd, J. Chem. Phys., 1984, 81, 3188 CrossRef CAS.
- P. G. Bolhuis, D. Frenkel, S.-C. Mau and D. A. Huse, Nature, 1997, 385, 235 CrossRef.
- T. A. Witten, P. A. Pincus and M. E. Cates, Europhys. Lett., 1986, 2, 137 CrossRef CAS.
- L. Willner, O. Jucknischke, D. Richter, B. Farago, L. J. Fetters and J. S. Huang, Europhys. Lett., 1992, 19, 297 CrossRef CAS.
- (a) G. A. McConnell, A. P. Gast, J. S. Huang and S. D. Smith, Phys. Rev. Lett., 1993, 71, 2102 CrossRef CAS; (b) G. A. McConnell and A. P. Gast, Macromolecules, 1997, 30, 435 CrossRef CAS.
- M. Watzlawek, C. N. Likos and H. Löwen, Phys. Rev. Lett., 1999, 82, 5289 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |