Hyper/highly-branched polymers by radical polymerisations
Richard Mark
England
and
Stephen
Rimmer
*
The Polymer and Biomaterials Chemistry Laboratories, Dept. Chemistry, University of Sheffield, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.rimmer@sheffield.ac.uk; Tel: +44 114 222 0000
First published on 30th July 2010
Abstract
Hyper branched and highly branched polymers (HB polymers) can be prepared by a variety of radical polymerisations. Here we describe the various techniques that can be used and include hybrid polymerisations that involve both radical and ring opening polymerisations. Adaptations of established techniques that use transfer to monomer are described. Then controlled radical polymerisations are outlined. Finally, some of emergent applications are reviewed with a special emphasis on applications in the life sciences.
 Richard Mark England | Richard England was a PhD student in the department of chemistry from 2006 to 2009 he is currently a post-doctoral worker at The Prince Felipe Research Centre, Valencia, Spain. |
 Stephen Rimmer | Stephen Rimmer is a Reader in Polymer and Biomaterials Chemistry at the University of Sheffield. His research interests revolve around the synthesis and properties of functional polymers with a particular emphasis on stimulus responsive polymers and polymers designed for applications in cell biology and regenerative medicine. |
1. Introduction
Branched polymers, which include dendritic, highly- and hyper-branched (HB), or multibranched systems exhibit interesting and versatile properties that drive synthetic efforts to their production. This is not surprising if we consider that natural macromolecular systems contain many branched structures. In order to produce synthetic polymers that mimic these evolved and therefore almost optimised structures, it is necessary to develop controlled precision syntheses. Potentially, changes in architectures produce different physical and biological properties with a wide range of possible applications. Some of the different branched architectures are shown in Fig. 1.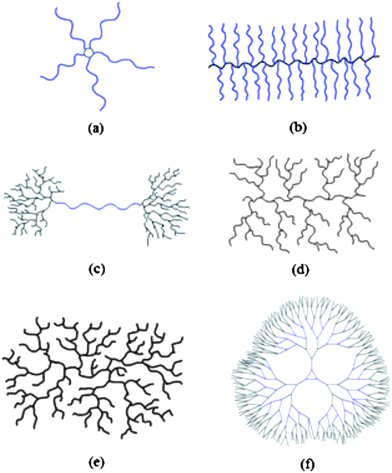 | ||
| Fig. 1 Examples of polymer architectures. (a) Star (b) Brush (c) Pom-Pom (d) Dendrigraft (e) Highly/hyper- Branched (f) Dendrimer. | ||
Dendrimers are often considered as perfect branched architectures, that is they can be produced as single molecular species and they possess high degrees of symmetry. They have high numbers of functional groups on their surface, excellent solubility and very low solution viscosities making them particularly good candidates for a wide range of applications from catalyst supports to drug carriers. However, despite many years of synthetic effort, the production of dendrimers still requires multistep syntheses with time consuming purifications. These labour intensive syntheses make their production prohibitively costly processes that are not well suited to scale-up. For this reason HB polymers are a more attractive architecture as they are far easier to produce but they still have many of the desirable properties of dendrimers whilst providing other properties that are not features of dendrimers.
Interest in HB macromolecules has grown due to several unique properties (compared to linear analogues), including low solution and melt viscosities, improved solubilities, tunable solution behaviour, critical phase behaviour and the presence of large numbers of functional end groups. These functional end groups offer the possibility for further modification for various high performance emerging applications. There are however some drawbacks associated with these polymers that include: broad molecular weight distributions; irregular branching and an uneven distribution of functional groups throughout the macromolecule.1 Also, characterisation of polydisperse branched, often copolymeric, systems can be very difficult and full characterisation can involve time consuming fractionation steps with the application of multiple techniques. In this review we will describe the current state-of-the-art techniques for the synthesis of HB polymers using radical polymerisation, which is by far the most versatile and scalable chain growth polymerisation available for the synthesis of functional polymers. HB polymers are defined by three main features, degree of branching, chain end functionality and repeat unit structure, and we examine some of the applications that are beginning to emerge from the synthetic efforts in this area.
2. Transfer to monomer/polymer
The simplest method of producing branched architecture in radical polymerisations is in conditions where transfer to either monomer or polymer is significant. In general this occurs in polymerisations of monomers that propagate by non-stabilised radicals such as ethylene, vinyl acetate, etc. Of course HB polyethylene produced by transfer to polymer is a very important commodity material, which has been studied for many years.2,3 Also, transfer to monomer has been shown to be significant in acrylate polymerisations4 but in controlled radical polymerisations the frequency of branching is much diminished.5 Branching, by transfer in polymerisations with low rates of transfer to polymer or monomer, would require the addition of comonomers with functionality that is more susceptible to transfer. In the case of acrylate, methacrylates and acrylamides these are generally thiol functionalities. With this in mind, Liu et al. synthesised HB polystryene by copolymerisation with vinyl benzyl thiol.6 However, thiol comonomers are often unstable because they are susceptible to self-condensation by Michael addition with more electron deficient alkenes. On the other hand increasing degrees of branching by transfer to comonomers during the polymerisation of vinyl acetate or N-vinyl pyrrilidinone is a much more realistic possibility because the propagating radicals have significant rates of transfer with a wide variety of stable functionalities. For example we used comonomer, 1 (Scheme 1), to produce HB poly(vinyl acetate),7 HB poly(N-vinyl pyrollidinone)8 and HB poly(1,1-bis(ethoxycarbonyl) vinyl cyclopropane).9 Clearly, transfer to monomer or polymer can be an economical and effective way to produce HB polymers. However, the method provides little control of end group structure, which is becoming increasingly important in many of the emergent applications.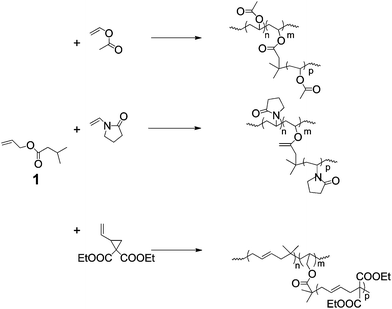 | ||
| Scheme 1 Copolymerisation with a branching comonomer, 1, which transfers by abstraction of the 2-propyl hydrogen during the polymerisation of monomers that propagate via chain radicals that are not stabilised. | ||
3. Self condensing vinyl polymerisation
Self Condensing Vinyl Polymerisation (SCVP) was first reported by Fréchet et al. in 1995.10 In this process a vinyl monomer presents a second functional group capable of initiating the polymerisation of other vinyl groups. The monomer unit acts as an initiator, either activated thermally for radical polymerisation or by an activating group. Fréchet's method involved the polymerisation of 3-(1-chloroethyl)ethenyl benzene in the presence of SnCl4 to produce hyperbranched polystyrene. The monomer satisfies the AB2 requirements for formation of hyperbranched polymers as the vinyl group acts as the difunctional B group and an additional alkyl halide functional group acts as the A group. The activation of the A group using the Lewis acid meant polymerisation through the double bond occurred cationically. Since the discovery of SCVP, the method has been expanded for use with radical polymerisation, in particular with living techniques to control the molecular weight distribution of the materials and prevent gelation.Nitroxide SCVP
In 1995 Hawker et al.11 prepared hyperbranched and star polymers using SCVP with nitroxide mediated polymerisation (NMP). Their method involved polymerisation of a monomer unit containing a polymerisable styryl group and an initiating/propagating moiety consisting of a nitroxide linked to a substituted benzylic carbon atom, 1, as shown in Fig. 2. Because of the low bond dissociation energy of the carbon-oxygen bond linking to the nitroxide group, thermal activation of the self-condensing living free radical polymerisation was possible. This technique yielded branched material with weight average molecular weights ranging from 65,000 to 300,000 g mol.−1 and with polydispersities from 1.6 to 4.4 showing that the living nature of the reaction led to control over the molecular weight distribution.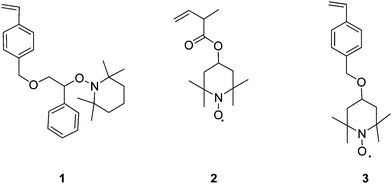 | ||
| Fig. 2 Examples of nitroxides used for SCVP to create hyperbranched polymers. | ||
A number of other polymerizable nitroxide monomers have been synthesised to produce branched materials. Tao et al.12 synthesised two polymerizable nitroxides, 2 and 3, for creating branched polystyrene (Fig. 2). When polymerised, the presence of the nitroxides at the branch points rather than at the chain ends resulted in “weak-links” in the material. The thermal homolysis/recombination of the nitroxide allows for branches to be severed from the main chain. Tao's group exploited this behaviour by irreversibly cleaving the branches using the reducing agent phenylhydrazine thus terminating the nitroxyl and carbon-centered radicals. This termination effect can also be achieved by adding other quenching agents such as ascorbic acid and heating the polymer in solution.13 The advantage of having these cleavable branches lies in that the materials can be used as branched macroinitiators allowing for further propagation of an another monomer near to the branching points. The thermally reversible decomposition of these polymeric clusters has also been studied in depth by Niu et al.14
ATRP SCVP
In 1996, Matyjaszewski et al.15 first presented a route to branched and hyperbranched polystyrene using Atom Transfer Radical Polymerisation (ATRP) in the presence of Cu(I). This method varied from Fréchet's cationic system in that the resulting functional groups in the macromolecule were described as AnB2, where n is the number of repeat units in the polymer. Secondly there were no heteroatoms in the backbone resulting from condensation reactions i.e. aliphatic chains and no “weak links”. The method involved the monomer, p-(chloromethyl) styrene using normal ATRP conditions (Cu(I) and 2,2′-bipyridyl). The p-(chloromethyl)styrene acted as an initiator and thus the result was a chain with a double bond at one chain end and a chlorine atom at the other. The double bond could be incorporated into the growing polymer chain and therefore acted as a branch point (Scheme 2).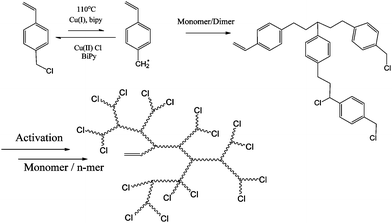
Weimer, Fréchet and Gitsov had found little evidence from Matyjaszewski's work to prove that hyperbranched material with a high degree of branching had been obtained. They published on a set of experiments to optimise the extent of branching in these materials. What was discovered was that due to an unequal reactivity of initiating and propagating species the reaction did not follow an ideal SCVP. Thus, high conversion was required to generate high molecular weight and branched polymer. They also found that low catalyst to monomer ratio led to largely linear polymer formation and little in the way of HB polymer.16 For SCVP using ATRP conditions, there has to be a high initiator (monomer/initiator) concentration in order to favour a shift in equilibrium towards the active radicals. This usually results in an initial large concentration of radicals which terminate efficiently, leaving an excess of X-Cu(II). This process consumes large proportions of the Cu(I) and prevents the activation or reactivation of alkyl halides. In order to produce hyperbranched polymers to high conversion using SCVP with inimers such as those shown in Fig. 3, there is a need for a sufficient concentration of Cu(I) to be maintained throughout the reaction. Matyjaszewski's group accomplished this by using copper in its zero valence state as it reduces the X-Cu(II) complex to generate two equivalents of Cu(I).17 Although 4 was polymerised using Cu(I) and 4,4′-ditert-butyl-2,2′-bipyridine,18 the polymerisation of 5, 6 and 7 required the addition of Cu(0).
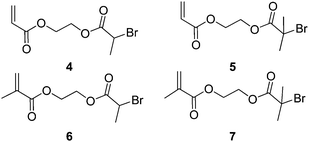
Fluorinated branched polymers are interesting because they can be used to provide low surface energy surfaces. With this in mind ATRP has been used to copolymerise either 4-chloromethyl styrene, 4-bromomethyl sytrene or 4-[oxy(tri(ethyleneglycol)) bromo-isobutyryl -2,3,5,6-tetrafluorostyrene (a poly(ethylene glycol containing inimer) with 2,3,4,5,6-penta-fluorostyrene19,20
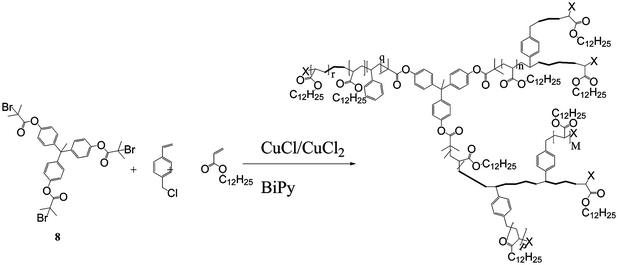
The halide functional monomers act as both an initiator and a monomer (an inimer), to produce highly fluorinated branched polymers with approximately 30% of the repeat units containing a branch point. In another variant, Wooley et al. used a trifunctional initiator, 8, to copolymerise chloromethyl styrene and lauryl acrylate, as shown in Scheme 3. Further ATRP from the active chain ends produced a HB block copolymer with a hydrophobic core and a shell composed of segments of poly(1,1,1-trifluoroethyl methacrylate-stat-acrylic acid) that formed micelle-like particles in aqueous media.21 Similar procedures have been used to prepare HB polyelectyrolytes22 and HB polymers with sugar functionality.23,24
The control of the size distribution is difficult with SCVP. However, if a sequential approach is adopted in which segments are synthesised then attached using protect-deprotection protocols much narrower distributions are possible. For example, Percec et al25 reported a sequential synthesis of HB poly(methyl methacrylate) (HB-PMMA) by first capping PMMA with a halide end group derived from ATRP with a silyl enol ether containing also two dialkyl thiocarbamate groups, which act as latent initiating groups. Then conversion of the dialkyl thiocarbamate groups to aryl sulfonyl chloride groups that reinitiate ATRP. Iteration of the process produced sucessive branching.
RAFT SCVP
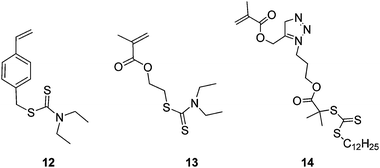
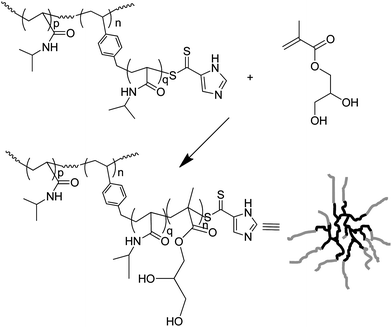
In 2003, Yang et al.26 produced branched polymers by incorporating a polymerisable dithioester (RAFT agent) into a styrene polymerisation. However, although this work showed the possibility of using RAFT polymerisation to produce branched polymers the branching agent, 9, (Fig. 4) placed a weak link in the form of the dithioester in the main chain. This aspect severely hampered the use of this technique until Carter et al. modified their method to place the dithioester at the chain ends using the branching monomer 10 or 11 (Fig. 5) in polymerisations of N-isopropyl acrylamide.27,28 This route to HB polymers appears to be extremely promising and we were able to use the route to produce one of the few examples of block copolymer HB polymers: HB poly(N-isopropyl acrylamide-block-glycerol monomethacrylate)29 (Scheme 4). As with all polymerisations aimed at block copolymer syntheses that follow the RAFT mechanism the production of homopolymer, from the initiation of the second monomer by the primary radical is inevitable.30 However, the branched polymers produced by RAFT SCVP are generally of high molecular weight whereas the homopolymers tend to be of much lower molecular weight. This large difference in size facilitates removal of the homopolymer.![The RAFT branching monomer 9 designed by Yang et al. [11] and the monomers that place the dithioate ester at the chain ends, 10 and 11, reported by Carter et al.27,28](/image/article/2010/PY/c0py00154f/c0py00154f-f4.gif) | ||
| Fig. 4 The RAFT branching monomer 9 designed by Yang et al. [11] and the monomers that place the dithioate ester at the chain ends, 10 and 11, reported by Carter et al.27,28 | ||
 | ||
| Fig. 5 Polymerizable dithiocarbamates used for the synthesis of hyperbranched block copolymers31,32 and a dithiocarbonate branching monomer.33 | ||
 | ||
| Scheme 4 The synthesis of block HB polymers: HB poly(N-isopropyl acrylamide-block-glycerol monomethacrylate) | ||
A method for the synthesis of block copolymers that does not produce homopolymer uses the generation of radicals by photolysis of dithiocarbamates. Photoinitiation via homolytic cleavage of the dithiocarbamate group has for many years been known to provide polymer products that are almost identical to those produced by RAFT and prior to our work Ishizu et al. used the dithiocarbamate chain ends of HB polymers to produce block copolymer hyperbranched polymers by using the branching monomers 12 and 13 shown in Fig. 5.31,32
Further reports of the branching RAFT monomers are currently appearing in the literature including monomers such as 14, which can be easily produced using a “click” strategy.33
In 2005, Perrier et al.34 reported a method to (a) completely remove all of the thiocarbonyl-thio end groups, (b) introduce a wide range of chain-end functionalities, and (c) recover the chain transfer agent (CTA). This method involved the reaction of the polymer with an excess of a radical initiator. The initiator decomposes to form radicals, which if they are generated in concentrations in excess of the chain end radicals react in bimolecular terminations to functionalise the chain ends. The chain end group structures depend on the radical initiator used and this can be utilised to provide terminal chain end functionality. Rimmer et al.28 were able to utilise this method using the azo-initiator, 4,4′-azobis(4-cyanopentanoic acid) to convert pyrrole dithioate chain end groups into carboxylic acids. This material proved to be a key precursor for attaching peptide sequences, e.g. arginine-glycine-aspartic acid, that bind to cell surface receptors.
A technique that appears to have recieved surprisingly little coverage is the use of chain stopping reactions in the branching step of SCVPs. This is best regarded as a convergent SCVP synthesis. The work of Yamada et al.35 is one of the few examples in this area. They use addition-fragmentation with simultaneous copolymerisation using 15 (Scheme 5), to produce HB-PMMA. Addition of the propagating radical to the propenyl sulfide group results in non-reversible termination of the primary chain and release of a new initiating species in a classical non-reversible transfer reaction. Thus, unlike the RAFT SCVP the primary chain does not continue to grow as monomer conversion progresses. Limiting the chain length in this way should be expected to produce important differences in properties between similar polymers prepared by RAFT SCVP and non-reversible addition-fragmentation SCVP. An extension of this process would involve the supression of reinitiation by the benzyl thiolyl radical, which is liberated in the fragmentation part of the transfer reaction. If this could be achieved the process could limit the functionality of the end groups to that provided by a functional initiator.
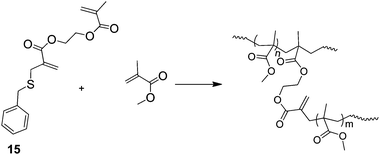 | ||
| Scheme 5 The synthesis of HB PMMA by addition-fragmentation during radical polymerisation with a propenyl sulfide-functional comonomer.35 | ||
4. Control of primary chain length in polymerisations with difunctional monomers
Multifunctional monomers (MFMs) are used to make crosslinked polymers. In free-radical polymerisation the inclusion of only small amounts of MFMs are required to produce a crosslinked network and gelation occurs when the number of crosslinks per chain exceeds unity. Clearly, for a given conversion of monomer to polymer the number of chains increase with decreasing degrees of polymerisation and gelation occurs at lower conversion as the degree of polymerisation increases. It has been shown that even in very dilute solution polymerisation (≪10% monomer), the gel point is reached at substantially less than 20% conversion of monomer to polymer.36 Not surprisingly, the synthesis of soluble branched polymers without crosslinking had seemed impossible in the free-radical batch copolymerisation of MFMs polymerised to high conversion. However, in 2000, Sherrington and co-workers used an approach based on the suppression of gel formation by extensive chain transfer for synthesising a new class of HB polymer.37 This method has become known as the ‘Strathclyde route’, as it was developed at the Univeristy of Strathclyde in the UK. The process is a facile, one step and cost-effective way to produce branched vinyl polymers, employing conventional free radical polymerisation (Fig. 6).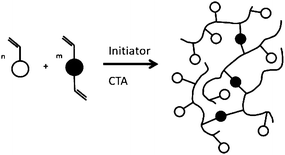 | ||
| Fig. 6 Synthesis of branched vinyl polymer using a combination of cross-linker and radical transfer agent. | ||
In this process gelation is delayed because transfer limits the degree of polymerisation of the primary kinetic chain so that the average number of branch points per chain is reduced when compared to the analogous process in the absence of transfer. The synthesis strategy consisted of using sufficiently concentrated solutions of comonomers but with a low concentration of branching monomer and equimolar conentrations of a CTA, typically a thiol. Using this method it is possible to achieve HB polymer with good yields in short time periods. The architectures, yields and molecular weights of the polymers can be tailored to a degree depending on a number of factors e.g. functionality of the comonomer38,39 and choice of CTA.40 Liu et al. have recently applied this approach to the synthesis of HB poly(butyl methacrylate) in emulsion by using a hydrophobic CTA that mainly partitions to the monomer/polymer particles during emulsion polymerisation.41 However, the process is limited by the availability of suitable transfer agents and this imposes restrictions on the degrees of functionality that can be achieved. This is because most of the available transfer processes generally produce two types of end group: from the transferred atom or group and the initiating radical produced by transfer. Thus, only in exceptional circumstances is it possible to obtain polymers with more than half of the end groups with a target functionality. Also, whilst thiol transfer agents are suitable transfer agents for the polymerisation of methacrylates and acrylamides, they quench the polymerisations of other monomers, such as vinyl acetate and N-vinyl pyrrolidinone, which propagate by non-resonance stabilised radicals. For example, in the polymerisation of vinyl acetate in the presence a thiol transfer agent and a MFM the thiol transfer agent was exhausted at low conversion and gelation could only be prevented by carrying out the polymerisation in 2-propoxy ethanol.42 In this system gelation was almost certainly prevented by transfer to the solvent and because of this effect the authors reported no effects on the polymerisation when the concentration of thiol was changed.
Recently, ATRP, rather than transfer, was used to regulate chain length and thus prevent gelation.43 Even ATRP homopolymerisation of MFMs has been reported to produce soluble polymer provided the conversions are limited.44 The use of controlled radical polymerisation can also potentially provide better control of end group functionality. Isaure et al. were able to produce soluble HB PMMA using Cu-based ATRP in a one-pot reaction and the later work from the same laboratories further expanded this approach by polymerizing hydroxypropyl methacrylate by ATRP in the presence of ethylene glycol dimethacrylate45 or a cleavable disulfide dimethacrylate.46 Monte Carlo simulation of the statistics of branching was simpler in these ATRP systems than in similar non-controlled polymerisations and Bannister et al. were able to use this technique to predict that at critical fractions of MFM the proportions of small loops in the system was low so that HB structure could be formed.47
Perrier et al. were the first to use a dithionate ester as the CTA, in a RAFT polymerisation, to limit gelation.48 Dong et al.49 synthesised branched polystyrene with many pendant vinyl groups by using an asymmetric divinyl monomer, 16, and RAFT polymerisation.
The asymmetrical vinyl monomer had a high reactivity styryl group and a low reactivity butenyl group (Fig. 7).The pendant butenyl vinyl groups allowed for further modification of the polymers. Modifications included epoxidation, amination and hydroxylation. The authors considered that the formation of branched material without gelation is possible because only a small number of the butenyl vinyl groups react resulting in low quantities of cross-linking. The low reactivity of the butenyl group in RAFT polymerisation would undoubtedly allow for the synthesis of butenyl-functionalised polymers. However, recent work on polymerisation in the presence of MFMs using the RAFT technique has shown how the use of high monomer concentration (above C* (the critical entanglement concentration) for an equivalent linear polymer) can prevent the production of intramolecular cyclic structures without the need to use MFMs with alkenes of different reactivity.50 Recently, the NMP process has also been used to limit chain lengths in the presence of MFMs.51
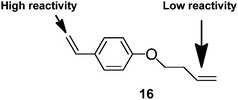 | ||
| Fig. 7 Asymmetric divinyl monomer for branching polymerisation.49 | ||
5. Hyperbranched polymers from living radical and ring opening polymerisation (ROP)
The combination of ROP with RAFT, ATRP or NMP techniques provides a route to novel (co)polymers with narrow molecular weight distributions and well defined architectures. Zou et al. used the difunctional (for ATRP and ROP) initiator, 17, in combination with the ATRP inimer, 18, to produce HB polymers with a linear poly(-caprolactone) “tail”52 This was followed by Kakwere and Perrier, who used RAFT to produce low molecular weight linear chains of ethyl acrylate and hydroxyethyl acrylate, which were used as initiators for ROP of L-lactide to produce star-shaped polymers.53 In the same publication they report the chain extension of the linear chains by utilising RAFT to polymerise styrene onto chains before being used for the ROP of L-lactide. This method produces miktoarm star polymers, which are so termed because of their varying chemical composition with varying molecular weights and/or chemically different arms. Due to their unique chemical compositions there has been a lot of interest in the synthesis of Miktoarm polymers using controlled radical polymerisation techniques.53–57 Hedrick et al.58 also produced hyperbranched ε-caprolactone using ROP to create AB2 macromonomers, such as 19. Structures of 17, 18 and 19 are shown in Fig. 8.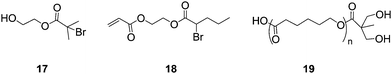 | ||
| Fig. 8 Branching monomers for use in ROP and ATRP. | ||
ROP is a useful synthetic technique due to the control of molecular weights leading to narrow polydispersities. The technique can be applied in the synthesis of dendronised polymers as shown by Hedrick et al.59,60 who demonstrated the use of living ROP of ε-caprolactone initiated from a bis(hydroxymethyl) central core to synthesise dendrimer-like, star block copolymers. They were able to enhance the molecules by capping the chain ends with dendrons containing activated bromide moieties to produce “macro-initiators” for further polymerisation via ATRP. The controlled synthesis from both living conditions for ROP, followed by ATRP led to high molecular weight material with low polydispersities (<1.2). Additionally, the design of these polymers, utilising the build up of different generations in separate reactions allowed for the incorporation of layers with amphiphilic character.
Morandi et al.61 recently reported a method for producing brush polymers using a “grafting from” approach using the combination of ROP and ATRP. This method is based on an inimer, 20, which can be ring opened (Scheme 6) whilst the presence of two initiating sites for ATRP leads to well controlled graft sites.
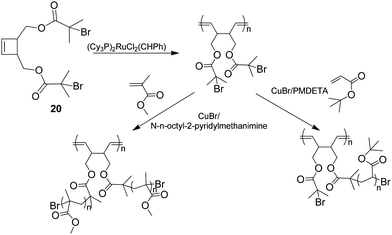 | ||
| Scheme 6 Cis-3,4-disubstituted cyclobutene derivative (inimer) containing active sites for ATRP after a ROP polymerisation.56 | ||
The presence of these initiating sites allows for the synthesis of a number of different monomers from the main chain. In the same publication, styrene, methyl methacrylate and tert-butyl acrylate were initiated from the active backbones to give a variety of graft copolymers. Ring-opening metathesis polymerisation was used by Fontanille et al.,62–64 who designed a novel route to branched materials using macromonomers of polystyrene and poly(ethylene oxide) with norbornene chain end groups. These end groups were then susceptible to ring opening polymerisation to produce branched and star-like material.
Barriau et al.65 devised a three-step strategy for developing well-defined amphiphilic, linear hyperbranched block copolymers by hypergrafting. This method involved the synthesis of a linear AB block copolymer of styrene and butadiene by anionic polymerisation followed by hydroxylation of the vinyl groups from the butadiene. These groups provided sites for the ring opening polymerisation of a functional epoxide to give hyperbranched grafts (hypergrafts) (See Scheme 7).
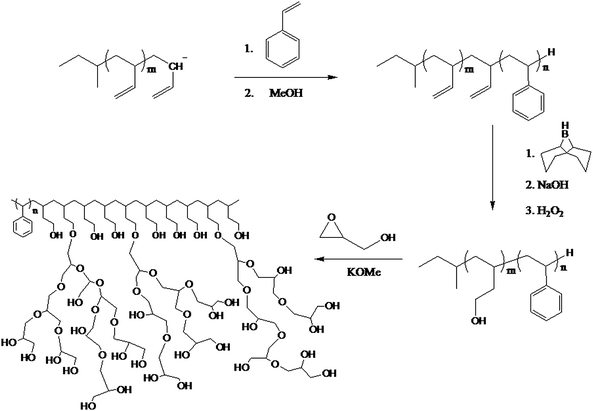 | ||
| Scheme 7 Synthesis of an amphiphilic linear-hyperbranched block copolymer.65 | ||
6. Controlled radical graft polymerisation onto linear polymer backbones
Grafting polymer chains onto linear backbones makes it possible to make branched materials in a step by step method to give greater control over the branch lengths. This involves synthesising a polymer backbone with functional monomers along the chain which, either directly or after chemical modification are capable of participating in initiating a polymerisation reaction. These functional monomers act as sites from which branches can either be (a) “grafted-to” where linear polymer chains with functional end groups can be directly attached to the backbone or (b) “grafted-from” so that addition of monomer (and an appropriate initiator, if required) will create branches (Scheme 8).66–70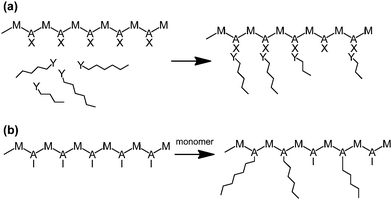 | ||
| Scheme 8 Schematic for grafting polymerisation, (a) “Grafting-to” approach, (b) “Grafting-from” approach. | ||
Gilbert et al.71 used RAFT and an activated ester comonomer in order to synthesise comb-like poly(butyl methacrylate). Butyl methacrylate and N-acryloxysuccinimide (NAS) were copolymerised using free radical polymerisation before the NAS groups were modified with a trithiocarbonate group thus providing a RAFT motif attached to the linear chain, capable of controlling the polymerisation of grafts from the backbone. This grafting point was used to polymerise butyl methacrylate homopolymer in a grafting from approach to create comb-like structures. After grafting, the polymers exhibited increased molecular weight because of the new graft material. Control over the reaction from the RAFT process kept the polydispersities relatively low (< 2.5).
Davis and co-workers also reported the synthesis of comb-like polymers using styrene and a comonomer, 4-vinylbenzyl chloride, which after polymerisation was substituted for a dithiobenzoate group (see Scheme 9).68 This allowed for the synthesis of HB polystyrene (also in a grafted-from approach) via RAFT polymerisation from the vinylbenzyl dithiobenzoate groups located along the backbone. Unlike the work by Gilbert et al., the molecular weights were found to increase dramatically after an induction period of 2–5 h. At 24 h many of the polymers were found to gel due to radical-radical coupling between comb structures. A similar approach was used by us to prepare PNIPAM graft polymer.30
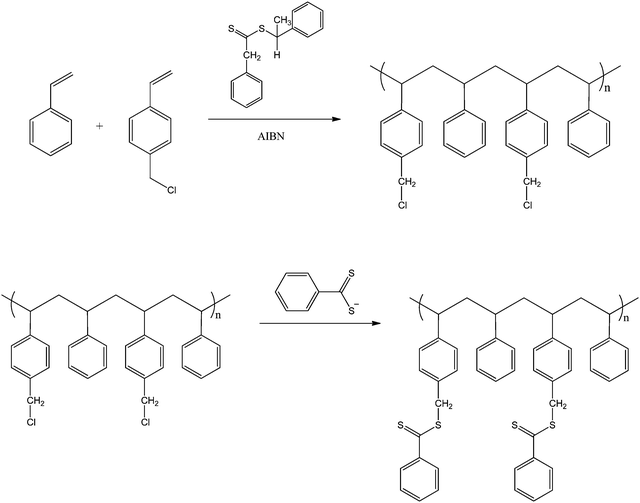 | ||
| Scheme 9 Synthesis of the comb precursor from poly(Sty-co-VBC).68 | ||
7. Applications
Even at this early stage of the development of this class of polymer we are already seeing the emergence of a number of interesting applications that are dependant on their unusual properties. Although modification of rheology compared to linear analogues is frequently discussed as a potential application of HB polymers at the current time there appeared to be few studies, available in the open literature, using HB polymers prepared by radical polymerizsation. Chisholm et al.72 produced HB P(MMA-co-EA-co-EDMA) by copolymerising MMA and ethyl acrylate with ethandiol dimethcarylate (EDMA) in combination with a thiol CTA, by suspension polymerisations. Plots of complex shear viscosity against molecular weight showed clear differences between the rheological behaviour of the HB P(MMA-co-EA-co-EDMA) and linear analogues. The results showed the expected decrease in viscosity for the HB polymers compared to the linear polymers and the data reflected a decrease in chain entanglement for the HB polymers. Chisholm et al.72 showed the possibility of producing HB polymer beads and earlier work from by Baurdry and Sherrington had illustrared the use of emulsion polymerisation to produce sub-micron particles using the MFM plus CTA route.73 Much of the recent work reported on new polymer particles is directed at their response to stimuli, usually a change in polymer conformation due to changing solvation. All aqueous polymer solutions should display lower critical solution temperature (LCST) behaviour, which produces a switch from an open coil and highly solvated state to a more compact globular and less solvated state: a coil-to-gobule transition. Above the LCST, linear polymers tend to form flocculated masses of material unless they are at very low concentrations. However, branched polymers behave differently and it is possible to produce colloidally stable particulate dispersion above the LCST that form homogeneous solutions below the LCST. Fig. 9 shows the two modes of providing colloidal stability above the LCST that are possible for dispersed polymer globules and aggregates of globules. Stable nano and micro dispersions can be formed from HB polymers by either functionalisation of the end groups with hydrophilic/polar species or increasing the swelling of the globules by adding hydrophilic/polar comonomers. We reported the formation of stable sub-micron dispersions of HB-PNIPAM that were provided with colloidal stability by virtue of charged end groups; either charged peptides or carboxylate groups.28b Stable HB PNIPAM particles were also formed by us by copolymerising 1,2-propandiol-3- monomethacrylate (GM) with NIPAM and 9.74 The equivalent polymers produced by polymerisation of NIPAM with 9, in the absence of GM produced flocculated masses of polymer above the LCST with little stability provided by the relatively hydrophobic imidazole end groups. In Fig. 9 we illustrate these two modes for producing colloidally stable dispersions. At least qualitively, an important consequence of these results is that we expect the globule to be less swollen for an end group-stabilised particle than for comonomer-stabilised particles. However, this hypothesis has not yet been fully tested. Weaver et al. have used the comonomer-stabilised mode to prepare pH responsive particles.75 In their procedure a MFM is copolymerised with 2-(diethylamino)ethyl methacrylate and poly(ethylene glycol) monomethacrylate in the presence of a thiol CTA.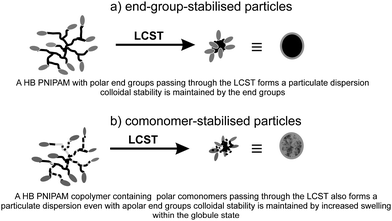 | ||
| Fig. 9 The two modes of colloidal stability that can be provided by HB polymers in dispersion. | ||
Clearly, there are several possibilities for using this class of branched polymer particles for controlled release and delivery and with this in mind we have shown that both comonomer-stabilised and end group stabilised particles enter human cells in a temperature dependant manner: entry occurs above the LCST in the particle form but the polymers are excluded from the cells in the open chain form below the LCST.74,76 End-group stabilised particles formed from HB-PNIPAM with the GRGDS end groups, which bind to the cell surface fibronectin and vitronectin receptors (integrins α5β1 and αvβ3), do not enter cells. Instead the cells are lifted from their previous culture substrate and they can be transported and released by cooling to below the LCST.76 This lifting/transport process and controlled entry and delivery, both of which are illustrated in Fig. 10, have enormous potential as new tools in cell biology and regenerative medicine.
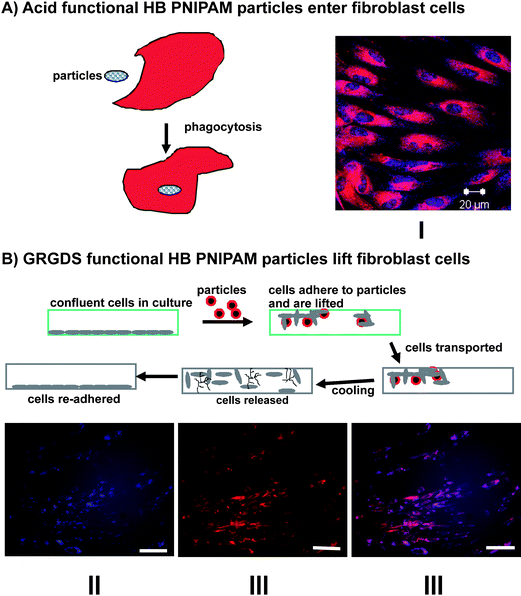 | ||
| Fig. 10 Application of HB PNIPAM in cell biology: (A) schematic of entry of HB PNIPAM particles into cells and I a micrograph of the particles inside human dermal fibroblasts (blue-polymer, red-actin cytosketon); (B) schematic cell lifting and reseeding technology. II, III and IV micrographs of cells and GRGDS-functional HB PNIPAM. The images show the co-location of the cells and bound polymer-II polymers labeled blue, III fibroblasts stained red and IV images II and III overlayed.76 | ||
A potential tool for use as a scaffold in regenerative medicine uses the discovery that disufide containing polymers can be degraded by the action of glutathione. Wang et al. used this concept to prepare electrospun non-woven scaffolds from HB-poly(hydroxy ethyl methacrylate) using an ATRP process with a disulfide functional dimethacrylate MFM. The presence of the disulfide in the branching units ensured that degradation produced oligomeric soluble fragments with known good cytocompatibility.77
Another attractive concept is the possibility of inducing the LCST by binding to a cell surface. This should be possible because many of the binding interactions in nature are electrostatic, polar interactions involving the desolvation of highly solvated ligands. Binding of such ligands at the multitude of chain ends present in a HB polymer then produces a large perturbation of the overall solvency of the polymer and this perturbation can produce significant decreases in the LCST. The concept is illustrated in Fig. 11 and exemplified in ref. 78 for interactions between vancomycin-functionalised HB-PNIPAM and Staphyloccus aureus. This change in the LCST drives the polymer collapse process if binding is sufficient to decrease the LCST to below the application temperature. Availability of the end groups for binding both above and below the coil-globule transition is essential in this process and the HB architecture ensures that this occurs because the end groups do not penetrate easily into the coil of a HB polymer. On the other hand functionalised linear polymers shield the ligand-receptor interactions in the globule state. Therefore, the coil-to-globule transition will involve dissociation of the complex. For this reason the transition is rarely observed on binding of linear polymers. Another important application of HB polymers is their use as multi-functional surfaces. A good example of this is the HB glycoacrylates prepared using ATRP SCVP by Muthukrishnan et al.79 The polymers can be spin coated and then cross-linked with an argon plasma to provide stable glyco-functional coatings. A important advantage of their use of ATRP is the fine control of end group structure that can be achieved. Recently, HB poly(acrylic acid-co-poly(ethylene glycol) methacrylate)s and HB poly(2-(diethylamino)ethyl methacrylate-co-poly(ethylene glycol) methacrylate) with various mixed end groups80,81 were used as pH responsive surfactants. The polymers clearly show important colloidal stability and aggregation effects associated with the nature of the end groups.
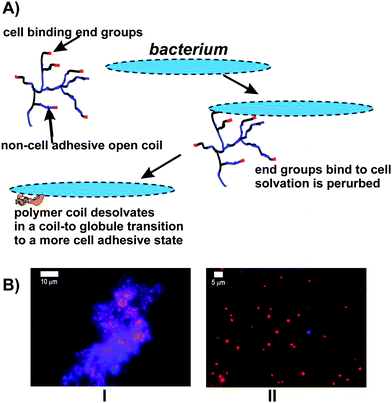 | ||
| Fig. 11 (A) Schematic of HB PNIPAM with end groups that bind to bacteria illustrating the coil-to-globule transition induced by a perturbation of solvation on binding. (B) Staphyloccus aureus (red) bound to HB-PNIPAM with vancomycin end groups: I aggregates of bacteria and polymer at 37 °C are disassembled by cooling to 4 °C, II, as the polymer passes from the globular state to the open coil on cooling.78 | ||
Galeav et al. reported the use of PNIPAM functionalised with ligands to remove various proteins from solution by complexation and temperature induced precipitation.82 Inspired by these reports we also used the multi-functionality of HB PNIPAM with imidazole end groups to purify recombinant BBRCA 1: a protein implicated in breast cancer.83,84 The tracking of polymers in vivo is difficult but the use 19F MRI has been suggested as a useful tool. The technique requires polymers with high fluorine contents. With this in mind Thurecht et al. prepared a HB branched copolymer from a RAFT MFM copolymerisation of dimethylaminoethylacrylate, and trifluoroethyl acrylate and ethylen glycol dimethacrylate.85 They functionalised the chain ends with first with a PEG-methacrylate block followed by the addition of a mannose ligand and were able to observe the polymers by 19F MRI in an in vivo mouse model.
Summary and prospects
The recent advances in the synthesis of HB polymers using radical polymerisations are now providing new materials with many potential applications. Applications such as anti-biofouling and interfacial properties are only just beginning to be explored but other applications in the life sciences are now emerging. Clearly, the use of either transfer to monomer/polymer, SCVP, or the MFM (Strathclyde) route can be scaled to commercially viable production strategies but there are still several unanswered aspects that will need to be addressed.However, SCVP provides improved control of end group structure compared to the addition of transfer agents in MFM strategies. Adopting a controlled radical polymetrisation MFM approach provides a large improvement in this regard. It should also be noted that both the controlled radical MFM and SCVP routes both involve continuous growth of the primary chain. These routes produce different architectures, which most likely provide different properties, to strategies that involves non-reversible termination. Both the transfer to monomer/polymer and the MFM routes are limited by gelation phenomena and it is only recently that efforts to examine the fractions of cyclic structures have been embarked upon. Considerations of the effects of monomer concentration will clearly be important in these studies50 as will strategies that combine a range of simulation and experimental techniques. The importance of controlling the production of intramolecular cyclic structures is perhaps best illustrated by considering that both branching,27,28,86,87,33 cyclisation88,89 and end group structure28,90 have very significant effects on critical solution behaviour. This property and associated solution properties, will be key to many of the future applications of these polymers. Control of cyclisation and end group structure will also be essential for any rheological applications. Characterisation of these HB polymers remains problematic and will always require application of multiple techniques perhaps in the future involving more use of fractionation prior to analysis.
Although, many applications of the current materials are possible, the most difficult synthetic challenge to address is providing more control of the molecular weight (hydrodynamic volume) distributions from these techniques. However, at the current time there appears to be few prospects in this area. One surprising aspect of this survey of the work in this area is that were few41 reports attempting to control the molecular weight distributions, gelation and functionality by using semi-continuous and continuous feed procedures. This perhaps reflects the paucity of kinetic simulations of these polymerisations, which may in turn reflect a lack of suitable rate parameters.
References
- S. Unal, I. Yilgor, E. Yilgor, J. P. Sheth, G. L. Wilkes and T. E. Long, Macromolecules, 2004, 37, 7081 CrossRef CAS.
- D. J. Cutler, P. J. Hendra, M. E. A. Cudby and H. A. Willis, Polymer, 1977, 18, 1005 CrossRef CAS.
- W.-M. Chan, P. E. Gloor and A. E. Hamielec, Polymer, 1992, 11, 2243.
- P. A. Lovell, T. H. Shah and F. Heatley, Polym. Comm., 1991, 32, 98 CAS.
- N. M. Ahmad, B. Charleux, C. Farcet, C. J. Ferguson, S. G. Gaynor, B. S. Hawkett, F. Heatley, B. Klumperman, D. Konkolewicz, P. A. Lovell, K. Matyjaszewski and R. Venkatesh, Macromol. Rapid Commun., 2009, 30, 2002 CrossRef CAS.
- J. Liu, Y. Wang, Q. Fu, X. Zhu and W. Shi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1449 CrossRef CAS.
- S. Rimmer, S. Collins and P. Sarker, Chem. Commun., 2005, 6029 RSC.
- L. E. Smith, S. Collins, Z. Liu, S. MacNeil, R. Williams and S. Rimmer, ACS Symp. Ser., 2007, 977, 197.
- P. Sarker, J. R. Ebdon and S. Rimmer, Macromol. Rapid Commun., 2006, 27, 2007 CrossRef CAS.
- J. M. J. Frechet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 CrossRef CAS.
- C. J. Hawker, J. M. J. Frechet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763 CrossRef CAS.
- Y. Tao, J. He, Z. Wang, J. Pan, H. Jiang, S. Chen and Y. Yang, Macromolecules, 2001, 34, 4742 CrossRef CAS.
- C. Li, J. He, L. Li, J. Cao and Y. Yang, Macromolecules, 1999, 32, 7012 CrossRef CAS.
- A. Niu, C. Li, Y. Zhao, J. He, Y. Yang and C. Wu, Macromolecules, 2001, 34, 460 CrossRef CAS.
- S. Gaynor, D. Greszta, J. Wang and K. Matyjaszewski, Macromolecules, 1996, 29, 1079 CrossRef CAS.
- W. M. Weimer, J. M. Frechet and I. Gitsov, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 955 CrossRef CAS.
- K. Matyjaszewski, J. Pyun and S. Gaynor, Macromol. Rapid Commun., 1998, 19, 665 CrossRef CAS.
- K. Matyjaszewski, S. Gaynor, A. Kulfan and M. Podwika, Macromolecules, 1997, 30, 5192 CrossRef CAS.
- C. Cheng, K. L. Wooley and E. Khoshdel, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4754 CrossRef CAS.
- K. T. Powell, C. Cheng and K. L. Wooley, Macromolecules, 2007, 40, 4509 CrossRef CAS.
- W. Du, A. M. Nystrom, L. Zhang, K. T. Powell, Y. Li, C. Cheng, Samuel A. Wickline and K. L. Wooley, Biomacromolecules, 2008, 9, 2826 CrossRef CAS.
- H. Mori, A. Walther, X. André, M. G. Lanzendörfer and A. H. E. Müller, Macromolecules, 2004, 37, 2054 CrossRef CAS.
- S. Muthukrishnan, G. Jutz, X. André, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 9 CrossRef CAS.
- S. Muthukrishnan, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 3108 CrossRef CAS.
- V. Percec, B. Barboiu, C. Grigoras and T. K. Bera, J. Am. Chem. Soc., 2003, 125, 6503 CrossRef CAS.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and H. Y. Yang, Macromolecules, 2003, 36, 7446 CrossRef CAS.
- S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595 CrossRef CAS.
- (a) S. R. Carter, S. Rimmer, R. Rutkaite, L. Swanson and J. Haycock, Polym. Mater. Sci. Eng., 2005, 93, 503 CAS; (b) S. Rimmer, S. R. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971 RSC.
- S. Carter, S. Rimmer, A. Sturdy and M. Webb, Macromol. Biosci., 2005, 5, 373 CrossRef CAS.
- S. R. Carter, R. M. England, B. Hunt and S. Rimmer, Macromol. Biosci., 2007, 7, 975 CrossRef CAS.
- K. Ishizu, T. Kojima, O. Takaaki, S. Yoshihiro and T. Shibuya, J. Colloid Interface Sci., 2004, 272, 76 CrossRef CAS.
- K. Ishizu, R. A. Khan, T. Furukawa and M. Furo, J. Appl. Polym. Sci., 2004, 91, 3233 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033 CrossRef CAS.
- B. Yamada, O. Konosu, K. Tanaka and F. Oku, Polymer, 2000, 41, 5625 CrossRef CAS.
- A. Matsumoto, Adv. Polym. Sci., 1995, 123, 41 CAS.
- (a) N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027 CrossRef CAS; (b) P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245 CrossRef CAS.
- A. T. Slark, D. C. Sherrington, A. Titterton and I. K. Martin, J. Mater. Chem., 2003, 13, 2711 RSC.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2004, 37, 2096 CrossRef CAS.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 1455 CrossRef CAS.
- Y. Liu, J. C. Haley, K. Deng, W. Lau and M. A. Winnik, Macromolecules, 2008, 41, 4220 CrossRef CAS.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 5230 CrossRef CAS.
- F. Isaure, P. A. G. Cormack, S. Graham, D. C. Sherrington, S. Armes and V. Bütün, Chem. Commun., 2004, 1138 RSC.
- W. Wang, Y. Zheng, E. Roberts, C. J. Duxbury, L. Ding, D. J. Irvine and S. M. Howdle, Macromolecules, 2007, 40, 7184 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155 CrossRef CAS.
- I. Bannister, N. C. Billingham and S. P. Armes, Soft Matter, 2009, 5, 3495 RSC.
- (a) B. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 2131 CrossRef; (b) B. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Polymer, 2005, 46, 6293 CrossRef CAS.
- Z. M. Dong, X. H. Liu, Y. Lin and Y. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6023 CrossRef CAS.
- (a) J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919 CrossRef CAS; (b) J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145 CrossRef CAS.
- A. Khan, M. Malkoch, M. F. Montague and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6238 CrossRef CAS.
- P. Zou, L.-P. Yang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7628 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6396 CrossRef.
- A. Vora, K. Singh and D. C. Webster, Polymer, 2009, 50, 2768 CrossRef CAS.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2007, 40, 4154 CrossRef CAS.
- O. Atltintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci. A Polym. Chem., 2008, 46, 3588.
- D. Mecerreyes, M. K. Georges, P. Dubois, R. Jerome, J. L. Hedrick, C. J. Hawker, E. E. Malmstom and M. Trollsas, Angew. Chem., Int. Ed., 1998, 37, 1274 CrossRef.
- J. L. Hedrick, M. Trollsas, C. J. Hawker, B. Atthoff, H. Claesson, A. Heise, R. D. Miller, D. Mecerreyes, R. Jerome and P. Dubois, Macromolecules, 1998, 31, 8691 CrossRef CAS.
- M. Trollsås, B. Atthoff, H. Claesson and H. J. L. Hedrick, Macromolecules, 1998, 31, 3439 CrossRef.
- G. Morandi, S. Pascual, V. Montembault, S. Legoupy, N. Delorme and L. Fontaine, Macromolecules, 2009, 42, 6927 CrossRef CAS.
- V. Heroguez, Y. Gnanou and D. Fontanille, Macromol. Rapid Commun., 1996, 17, 137 CrossRef.
- V. Heroguez, Y. Gnanou and D. Fontanille, Macromolecules, 1997, 30, 4791 CrossRef CAS.
- V. Heroguez, S. Breunig, Y. Gnanou and D. Fontanille, Macromolecules, 1996, 29, 4459 CrossRef CAS.
- E. Barriau, A. Garcia-Marcos, H. Kautz and H. Frey, Macromol. Rapid Commun., 2005, 26, 862 CrossRef CAS.
- H. G. Borner and K. Matyjaszewski, Macromol. Symp., 2002, 177, 1 CrossRef CAS.
- X. P. Guo, Z. A. Li, Q. G. Du, Y. L. Yang and M. D. Lin, J. Appl. Polym. Sci., 2002, 84, 2318 CrossRef CAS.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956 CrossRef CAS.
- Z. Li, Q. C. Du, Y. L. Yang and M. D. Lin, Macromol. Chem. Phys., 2001, 202, 2314 CrossRef CAS.
- G. Martinez, E. de Santos and J. L. Millan, Macromol. Chem. Phys., 2001, 202, 2377 CrossRef CAS.
- J. L. Vosloo, M. P. Tonge, C. M. Fellows, F. D'Agosto, R. D. Sanderson and R. G. Gilbert, Macromolecules, 2004, 37, 2371 CrossRef CAS.
- M. Chisholm, N. Hudson, N. Kirtley, F. Vilela and D. C. Sherrington, Macromolecules, 2009, 42, 7745 CrossRef CAS.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 1455 CrossRef CAS.
- S. Hopkins, S. Carter, L. Swanson, S. MacNeil and S. Rimmer, J. Mater. Chem., 2007, 17, 4022 RSC.
- J. V. M. Weaver, R. T. Williams, B. J. L. Royles, P. H. Findlay, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985 RSC.
- S. Hopkins, S. R. Carter, J. W. Haycock, N. J. Fullwood, S. MacNeil and S. Rimmer, Soft Matter, 2009, 5, 4928 RSC.
- L. Wang, C. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566 CrossRef CAS.
- J. Shepherd, P. Sarker, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, J. Am. Chem. Soc., 2010, 132, 1736 CrossRef CAS.
- S. Muthukrishnan, M. Nitschke, S. Gramm, Z. Özyürek, B. Voit, Carsten Werner and A. H. E. Müller, Macromol. Biosci., 2006, 6, 658 CrossRef CAS.
- J. V. M. Weaver, S. P. Rannard and A. I. Cooper, Angew. Chem., Int. Ed., 2009, 48, 2131 CrossRef CAS.
- R. T. Woodward, R. A. Slater, S. Higgins, S. P. Rannard, A. I. Cooper, B. J. L. Royles, P. H. Findlay and J. V. M. Weaver, Chem. Commun., 2009, 3554 RSC.
- A. Kumar, A. Srivastava, I. Yu Galaev and B. Mattiasson, Prog. Polym. Sci., 2007, 32, 1205 CrossRef CAS.
- S. Carter, S. Rimmer, A. Sturdy and M. Webb, Macromol. Biosci., 2005, 5, 373 CrossRef CAS.
- S. Carter, S. Rimmer, R. Rutkaite, L. Swanson, J. P. A. Fairclough, A. Sturdy and M. Webb, Biomacromolecules, 2006, 7, 1124 CrossRef CAS.
- K. J. Thurecht, I. Blakey, H. Peng, O. Squires, S. Hsu, C. Alexander and A. K. Whittaker, J. Am. Chem. Soc., 2010, 132, 5336 CrossRef CAS.
- C. K. Chee, B. J. Hunt, S. Rimmer, R. Rutkaite, I. Soutar and L. Swanson, Soft Matter, 2009, 5, 3701 RSC.
- M. Luzon, C. Boyer, C. Peinado, T. Corrales, M. Whittaker, L. Tao and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2783 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Symp., 2009, 278, 10 CrossRef CAS.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069 CrossRef CAS.
- S. Furyk, Y. Zhang, D. Oritiz-Acosta, P. S. Cremer and D. E. Bergreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |