Acid catalyzed synthesis of carbonyl-functionalized microporous ladder polymers with high surface area†
Reiner Sebastian
Sprick
a,
Arne
Thomas
*b and
Ullrich
Scherf
*a
aMakromolekulare Chemie, Bergische Universität Wuppertal, Gauss-Str. 20, D-42097, Wuppertal, Germany. E-mail: scherf@uni-wuppertal.de; Fax: +49 202 4393880; Tel: +49 202 3871
bInstitut für Chemie, Technische Universität Berlin, Englische Str. 20, D-10587, Berlin, Germany. E-mail: arne.thomas@tu-berlin.de; Fax: +49 30 31429271; Tel: +49 30 31428876
First published on 26th January 2010
Abstract
The synthesis of a new class of very rigid, microporous ladder polymers in a cyclotrimerization reaction of bifunctional diketo-s-indacene-type monomers is described. The cyclotrimerization reaction is carried out under Lewis-acidic (TiCl4) or, preferentially, acidic conditions (methanesulfonic acid—MSA) and yields ladder-type, aromatic networks with high BET surface areas of up to 1650 m2 g−1. The synthetic scheme towards the truxene-cored ladder networks also allows for the incorporation of carbonyl functions that may be transformed to other functionalities in further derivatization reactions.
Organic microporous polymer networks have gained an enormous and increasing interest due to their potential in attractive and promising application areas such as gas separation/gas storage and heterogeneous catalysis.1–3 However, their synthesis often requires high temperatures (ionothermal synthesis at temperatures >400 °C4,5), the use of transition metal C–C-coupling catalysts (palladium or nickel complexes6–9) or strong Lewis acids as Friedel–Crafts catalysts (FeCl3).10 The complete removal of the catalysts and reagents is hampered by the insolubility of the crosslinked reaction products. Moreover, oxidizing agents (FeCl3) may cause the incorporation of unwanted functionalities by oxidative side reactions. Nevertheless, Brunauer–Emmett–Teller (BET) surface areas of >1500 m2 g−1 have been reported for a couple of products.4,5,10
Despite the tremendous progress in the field there is still a need for improvements, especially in the generation of thermally stable and chemically resistant nanoporous polymers under relatively mild reaction conditions and, preferably, without the use of expensive or toxic transition metal reagents or catalysts. In the most preferred case, the reagents/catalysts used should be easily removable by simple solvent extraction procedures. Moreover, microporous polymer networks containing suitable additional functional groups may allow for a further polymer-analogous derivatization of the solid products and for the design of tailored materials for specific applications, e.g. the generation of catalytic or separation materials with an enhanced selectivity for gas sorption or catalyst binding.
Here, we would like to describe the synthesis and first characterization data of a new class of very rigid, microporous ladder polymers made in a cyclotrimerization reaction of bifunctional diketo-s-indacene-type monomers. The cyclotrimerization reaction is carried out under Lewis-acidic (TiCl4) or, preferentially, acidic conditions (methanesulfonic acid—MSA) and yields ladder-type, aromatic networks with high BET surface areas of up to 1650 m2 g−1. The synthetic scheme towards the truxene-cored ladder networks also allows for the incorporation of additional carbonyl functions that can be transformed to other functionalities in further derivatization reactions.
The synthetic scheme for the generation of the ladder-type networks is based on the well-known synthesis of truxene derivatives via cyclotrimerization of indan-1-one derivatives such as indan-1-one, 3,3-dialkylindan-1-one, or indan-1,3-dione (see Scheme 1).11–13 The corresponding bifunctional s-indacene-based monomers A–C (Fig. 1) should accordingly lead to the corresponding truxene-cored ladder networks as targets of our study.
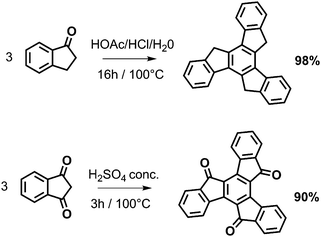 | ||
| Scheme 1 Truxene and truxenone synthesis. Reaction conditions after Dehmlow and Kelle.11 | ||
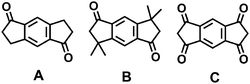 | ||
| Fig. 1 Chemical structures of the cyclotrimerization monomers A–C. | ||
Unsubstituted truxene (10,15-dihydro-5H-diindeno[1,2-a;19,29-c]fluorene) is accessible in the cyclotrimerization of indan-1-one with protic acids (acetic acid–conc. hydrochloric acid,11p-toluenesulfonic acid,12,13 polyphosphoric acid14) or Lewis acids such as titanium tetrachloride,15 boron tribromide,14 silicon tetrachloride,16 or molten zinc chloride.5 In contrast, the condensation of indan-1,3-dione towards the triketo derivative truxenone (diindeno[1,2-a;1′,2′-c]fluorene-5,10,15-trione) was mainly described with protic reagents such as sulfuric acid11,17 or methanesulfonic acid.18 Considering these literature results we have applied related cyclotrimerization conditions in the condensation of the corresponding bifunctional s-indacene monomers A/B (p-toluenesulfonic acid, polyphosphoric acid, titanium tetrachloride, and molten zinc chloride) and C (sulfuric or methanesulfonic acid), see Table 1. The bifunctional monomers A–C have been prepared following literature procedures (A,19B,20C21). First, different reagents and condensation parameters for monomer B (entries 2–5) were tested.† The best yields have been obtained using titanium tetrachloride as condensation reagent in 1,2-dichlorobenzene. These conditions have been subsequently used in the condensation of monomer A (entry 1). For the condensation of monomer C only two protic acids have been applied (entries 6 and 7) according to the literature (truxenone synthesis).17,18
| Entry | Monomer | Reagent–solventd | Temp./°C | Time/h | Yield (%) | S BET/m2 g−1 |
|---|---|---|---|---|---|---|
| a Quantitatively, yields > 100% are caused by non-reacted –CH2–CO– end groups. b Very small SBET value. c Large hysteresis, SBET value not estimated. d MSA: methanesulfonic acid, PPA: polyphosphoric acid, PTSA: p-toluenesulfonic acid, ODCB: 1,2-dichlorobenzene. | ||||||
| 1 | A | TiCl4–ODCB | 180 | 72 | Quant.a | 1165 |
| 2 | B | PTSA–ODCB | 105 | 72 | 31.7 | —b |
| 3 | B | PPA | 160 | 72 | 70.6 | —c |
| 4 | B | Molten ZnCl2 | 400 | 72 | 10.3 | 173 |
| 5 | B | TiCl4–ODCB | 180 | 72 | 84.5 | 395 |
| 6 | C | H2SO4 (20% aqueous) | 100 | 24 | Quant.a | 49 |
| 7 | C | MSA–ODCB | 180 | 3 | Quant.a | 1650 |
Fig. 2 and 3 depict the (idealized) structures of the resulting crosslinked polymer networks. Please note (a) that one would not strictly expect the two-dimensional (2D) structure (Fig. 2) starting from monomers A and B due to the formation of “open”, non-cyclized substructures within a three-dimensional (3D) network, and (b) that the cyclotrimerization of monomer C results in a more irregular structure of the condensation product due to the formation of cis- and trans-configurated s-indacene-dione bridging units between adjacent truxene cores. The ladder-type microporous polymer networks from entries 1–7 showed an excellent thermal stability without thermal degradation up to 330 °C due to thermogravimetric analyses (TGA) in air (Fig. S7 and 8†).‡
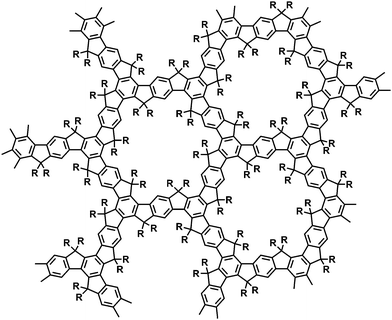 | ||
| Fig. 2 Idealized chemical structure of the condensation products of monomers A and B (A: R = H, B: R = CH3). | ||
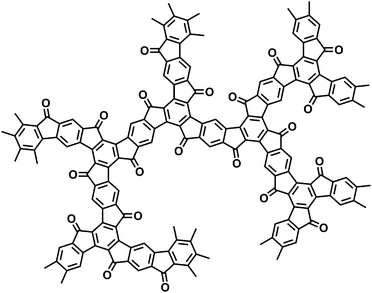 | ||
| Fig. 3 Chemical structure of the condensation product of monomer C. | ||
We have further characterized the resulting condensation products by solid state 13C {1H} CPMAS NMR spectroscopy‡ (Fig. S1–3†). The cyclotrimerization product of entry 1 (monomer A, reagent TiCl4) displays one dominant aliphatic carbon signal centered at 33 ppm that originates from the methylene bridging units. In the aromatic region two signals at 141 (broad)/115 ppm occur for the five non-equivalent carbons of the idealized structure of Fig. 2. In addition, low-intensity carbonyl-related end group signals are detected at chemical shifts of ∼190 ppm. The product from entry 5 (monomer B, reagent TiCl4) shows two signals in the aliphatic region that can be assigned to the methyls (23 ppm) and the aliphatic ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C(CH3)2 carbons bridging adjacent aromatic rings (48 ppm). The aromatic region displays six well-resolved signals (151, 145, 138, 133, 128 and 113 ppm). The idealized structure of Fig. 2 corresponds to only 5 non-equivalent carbons, so the increased number of six signals points to the presence of the already mentioned “open” substructures including end groups. The solid state 13C NMR spectrum of the condensation product from entry 7 (monomer C) displays aromatic carbon signals at 145/118 ppm. Additional peaks at 194/186 ppm can be assigned to the carbonyl functions (related to the truxenone cores and end groups).
C(CH3)2 carbons bridging adjacent aromatic rings (48 ppm). The aromatic region displays six well-resolved signals (151, 145, 138, 133, 128 and 113 ppm). The idealized structure of Fig. 2 corresponds to only 5 non-equivalent carbons, so the increased number of six signals points to the presence of the already mentioned “open” substructures including end groups. The solid state 13C NMR spectrum of the condensation product from entry 7 (monomer C) displays aromatic carbon signals at 145/118 ppm. Additional peaks at 194/186 ppm can be assigned to the carbonyl functions (related to the truxenone cores and end groups).
To test the porosity of the polymeric products nitrogen sorption measurements were carried out and the Brunauer–Emmett–Teller (BET) surface areas extracted (Fig. S4–6†).‡ We could determine considerable SBET values for the condensation products from all three monomers. In the entries 2–5 with monomer B the highest SBET value (395 m2 g−1) was measured for entry 5 with TiCl4 as condensation reagent corresponding to the highest yield (84.5%) of cyclotrimerization product. The TiCl4-condensation product of monomer A (entry 1) displays a largely increased SBET value of 1165 m2 g−1. This should mainly correspond to an increased volume of the pores after replacement of the space-filling methyls by hydrogens. The MSA-condensation product of monomer C (entry 7) leads to the highest SBET value of 1650 m2 g−1. Remarkably, this condensation product is generated in a metal-free synthesis at moderate temperature. The condensation reagent (MSA in 1,2-dichlorobenzene) and, if present, a small amount of low molecular weight by-products can be simply removed by subsequent extraction of the product with water, ethanol, acetone, and chloroform. In contrast, the cyclotrimerization of C in 20% aqueous H2SO4 leads quantitatively to a condensation product with a very low SBET value of only 49 m2 g−1 most probably caused by the insolubility of the monomer in the condensation reagent.
In conclusion, we could describe the generation of a novel class of microporous ladder polymers with high surface areas of up to 1650 m2 g−1 in an acid catalyzed cyclotrimerization scheme of bifunctional s-indacene-type monomers. Moreover, the possibility of an incorporation of carbonyl functions into the resulting 3D networks allows for a subsequent polymer-analogous derivatization and the introduction of specific functional groups or binding sites. Further characterization experiments will be focussed on the determination of pore sizes and pore size distributions as well as the solid state order (by X-ray diffraction experiments) of the microporous organic networks. In addition, we will check the feasibility of a polymer-analogous derivatization of remaining keto functions (especially with the reaction products of monomer C) e.g. by addition of lithium organic or Grignard reagents.
Notes and references
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- J. Germain, J. M. J. Fréchet and F. Svec, Small, 2009, 5, 1098–1111 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- P. Kuhn, A. I. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS.
- E. Stockel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- E. V. Dehmlow and T. Kelle, Synth. Commun., 1997, 27, 2021 CAS.
- T. W. Warmerdam, R. J. M. Nolte, W. Drenth, J. C. van Miltenburg, D. Frenkel and R. J. J. Zijlstra, Liq. Cryst., 1988, 3, 1087–1104 CrossRef CAS.
- A. W. Amick and L. T. Scott, J. Org. Chem., 2007, 72, 3412–3418 CrossRef CAS.
- B. Gómez-Lor, Ó. de Frutos, P. A. Ceballos, T. Granier and A. M. Echavarren, Eur. J. Org. Chem., 2001, 2107–2114 CrossRef CAS.
- R. B. M. Ansems and L. T. Scott, J. Am. Chem. Soc., 2000, 122, 2719–2724 CrossRef.
- S. Kotha, D. Kashinath, K. Lahiri and R. B. Sunoj, Eur. J. Org. Chem., 2004, 4003–4013 CrossRef CAS.
- M. V. Ionescu, Ber. Dtsch. Chem. Ges., 1927, 60, 1228–1235 CrossRef.
- L. Sanguinet, J. C. Williams, Z. Yang, R. J. Twieg, G. Mao, K. D. Singer, G. Wiggers and R. G. Petschek, Chem. Mater., 2006, 18, 4259–4269 CrossRef CAS.
- D. E. Seeger, P. M. Lahti, A. R. Rossi and J. A. Berson, J. Am. Chem. Soc., 1986, 108, 1251–1263 CrossRef CAS.
- H. Sakurai, N. Iwasawa and K. Narasaka, Bull. Chem. Soc. Jpn., 1996, 69, 2585–2594 CAS.
- P. Krief, J. Y. Becker, A. Ellern, V. Khordorkovsky and O. Neilands, Synthesis, 2004, 2509–2514 CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, 13C {1H} CPMAS NMR spectra, BET isotherms, and TGA analysis for the cyclotrimerization products from entries 1, 5 and 7. See DOI: 10.1039/b9py00375d |
| ‡ The 13C NMR MAS spectra were recorded on a Bruker Advance 400 spectrometer in the MAS double resonance technique with a spinning frequency of 10 kHz. The 13C {1H} cross-polarization magic angle spinning (CPMAS) spectra were measured with a contact time of 2 s and referenced to tetramethylsilane (TMS) with adamantane as secondary standard. The thermogravimetric measurements were carried out on a Mettler 1 STARe system (Mettler–Toledo) with a heating rate of 10° min−1. The adsorption isotherms leading to the Brunauer–Emmett–Teller (BET) surface areas (SBET) were measured on a Quantochrome Instruments machine at 77 K. The samples (20–50 mg) were dried at 373 K per 10−3 mbar. The SBET values were calculated in the pressure region P/P0 as described in the literature22 based on a specific surface of 16.2 Å2 per N2 molecule. |
| This journal is © The Royal Society of Chemistry 2010 |