Single step reductive polymerization of functional 3,4-propylenedioxythiophenes via direct C–H arylation catalyzed by palladium acetate†
Anshu
Kumar
and
Anil
Kumar
*
Department of Chemistry and Center of Excellence in Nanoelectronics, Indian Institute of Technology Bombay, Mumbai 400076, India. E-mail: anilkumar@iitb.ac.in; Fax: +91-22-25767152; Tel: +91-22-25767153
First published on 21st December 2009
Abstract
In this Communication we report a single step reductive polymerization of ProDOT derivatives which is compatible with functional side chains and is amenable for scale-up.
This Communication reports, for the first time, syntheses and characterization of functional polymers based on 3,4-propylenedioxythiophenes prepared in a single step reductive polymerization catalyzed by palladium acetate. Conjugated polymers based on 3,4-alkylenedioxythiophenes have been of great interest for scientists working in academic as well as industrial laboratories. These polymers exhibit unique characteristics due to their low oxidation potential, high thermal and chemical stability in doped state and high transparency in the doped state. These properties make them ideally suited for various applications such as hole transport layer in light emitting devices, design and fabrication of chemical and biological sensors, antistatic coatings, transparent electrodes, and electrochromic displays etc.1–7 Various grades of aqueous dispersions of poly(3,4-ethylenedioxythiophene), PEDOT, are commercially available under the trade name of Clevios. Since the PEDOT is insoluble, various substituted EDOTs and 3,4-propylenedioxythiophenes, ProDOTs, have been developed in order to introduce solubility and functionality.8–14
Polymers based on ProDOT derivatives show improved electrochemical properties as compared to EDOT derivatives due to the regiosymmetric nature of the final polymer.14–18 Two commonly used methods for the chemical polymerization of ProDOT derivatives are (a) oxidative and (b) reductive polymerization. In oxidative polymerization, ProDOT derivatives are subjected to oxidizing agents such as iron(III) salts and persulfate etc.9–11,14,19–22 In case of reductive polymerization, Grignard metathesis (GRIM) polymerization is the method of choice.16,17,22,23 Oxidative polymerization is a simple one step process and has advantages such as relatively less stringent polymerization conditions (both organic and aqueous medium can be used) and tolerance to functional groups. On the other hand, reductive polymerization via GRIM method requires more stringent polymerization conditions, sensitive to functional groups and economically more expensive compared to oxidative polymerization. The other main disadvantage of reductive GRIM polymerization method is that the polymerization is irreversible, i.e., in case of a failed reaction, one can not recover the monomers. Though the reductive polymerization is more tedious and expensive compared to oxidative polymerization, it gives much better quality materials in terms of their electrochemical properties and therefore is the preferred method.
Hence there is a need to develop a new reductive polymerization method for the synthesis of these polymers which is economically viable, inert to the presence of functional groups and uses less stringent polymerization conditions. In this direction, we came across two reports on the direct C–H arylation of thiophenes substituted with an electron rich side chain.24,25 Since ProDOT is an electron rich thiophene derivative due to the presence of dialkoxy groups at the 3 and 4 positions, we presumed it would be highly reactive towards this direct C–H arylation. If this is possible, then it should lead to the formation of high quality polymers by the suitable design of monomers based on ProDOTs. In this direction, we report a new synthesis method for the reductive polymerization of ProDOT derivatives which has many advantages over the known GRIM polymerization method such as functional group tolerance, moisture tolerance, being economically more attractive, amenable for scale-up and its ability to control molecular architectures such as in the synthesis of alternate copolymers.
In order to study the polymerization of ProDOT derivatives via direct C–H arylation using palladium acetate as the catalyst, dihexyl-ProDOT (1) was used as the model monomer along with 2,5-dibromo EDOT (2). The selection of these model monomers was based on the fact that the resulting alternating copolymer of dihexyl substituted ProDOT and EDOT was found to be soluble in common organic solvents and hence it is easy to characterize the final polymer. Dihexyl-ProDOT and 2,5-dibromo EDOT were synthesized following the reported procedure and were fully characterized before polymerization.17,26 1 mmol of Dihexyl-ProDOT along with 1 mmol of tetrabutylammonium bromide (TBAB), 4 mmols of sodium acetate 80 ml of dry DMF were taken in a three neck round bottom flask equipped with a reflux condenser and addition funnel. The reaction mixture was stirred at room temperature for 15 min followed by the addition of 1 mmol of 2,5-dibromo EDOT and 10 mol% of palladium acetate. The reaction mixture was then heated to 70 °C under nitrogen atmosphere for 48 h. The color of the polymerization mixture turned to dark red within 20 min of the addition of the catalyst and finally to purple indicating the formation of polymer. The polymer was isolated by precipitating the reaction mixture in methanol. The resulting polymer was centrifuged and washed three times with fresh methanol and then was subjected to soxhlet extraction first using methanol then acetone and finally with chloroform. The final chloroform soluble fraction was used for further characterization and the polymer P1-a was isolated as dark purple solid in 60% yield. We also carried out a preliminary analysis of the effect of varying the amount of catalyst on the polymerization by carrying out polymerization reactions at 1 mol% of the catalyst to obtained polymer P1-b. It was interesting to note that it took around 5 h for the color to change to dark red on the addition of catalyst in case of as compared to around 20 min when 10 mol% of the catalyst was used indicating slower rate of polymerization in case of 1 mol% catalyst. We also tried carrying out the polymerization using 0.01 mol% of the catalyst but the polymerization was unsuccessful. UV-vis along with GPC studies on P1-a,b confirmed the formation of reasonably high molecular weight polymers (Table 1) indicating that increasing the amount of catalyst from 1 mol% to 10 mol% increases the rate of polymerization and not the molecular weight. In order to further investigate the polymerization conditions, we explored the effect of water on the polymerization by adding 1 and 2 wt% of water to dry DMF and then carrying out the polymerization using this wet DMF to get P1-c and P1-d, respectivelly. It was interesting to note that the polymerization proceeded in usual fashion as was observed in cased of dry DMF. GPC and UV-vis studies indicated the formation of reasonably high molecular weight polymers indicating that the polymerization conditions are much more benign and do not need dry solvents, which are a must in the case of the GRIM method. The final polymers P1-a–d were found to be soluble in common organic solvents like chloroform, methylene chloride, tetrahydrofuran and toluene and was characterized by NMR, UV-vis spectroscopy and GPC. 1H NMR of P1a in CDCl3 showed no peak at 6.25 ppm corresponding to the chain ends indicating the formation of reasonably high molecular weight polymer. GPC studies carried out in chloroform using polystyrene as standard further confirmed the polymeric nature of the sample with weight average molecular weight of 11.4 kD and polydispersity of 2.5 UV-vis spectra also gave an absorption maximum of 557 nm which is typical of the dialkyl substituted high molecular weight polymers.17,26 Therefore, this confirmed that it is possible to carry out reductive polymerization of ProDOT derivatives by the direct C–H arylation using palladium acetate as the catalyst.
| Polymer | Catalyst (Mol %) | Yield (%) | GPC Mw(PD)a | UV-vis (λmax) |
|---|---|---|---|---|
| a In chloroform using polystyrene as standard. b This unusually low molecular weight could be due to absortion on the column because of pendant hydroxyl groups. | ||||
| P1-a | 10 | 60 | 11400(2.5) | 557 |
| P1-b | 1 | 55 | 8200(2.2) | 557 |
| P1-c | 1 | 50 | 11000(2.4) | 556 |
| P1-d | 1 | 55 | 7000(3.0) | 551 |
| P3a | 10 | 46 | 1300(1.2)b | 567 |
| P3b | 10 | 51 | 13500(3.2) | 543 |
| P3c | 10 | 51 | 13700(3.2) | 544 |
| P3d | 10 | 53 | 6200(2.0) | 542 |
| P3e | 10 | 52 | 9300(2.1) | 541 |
Encouraged by the success of the direct C–H arylation of ProDOT using palladium acetate as the catalyst, we further explored the functional group tolerance of the polymerization conditions. In order to do this, we synthesized functional ProDOT derivatives (Scheme 1) using ProDOT-OH as the starting point following the reported procedure.12 Various functional side chains such as hydroxyl (3a), nitrile (3b), ester (3c), bromide (3d) and azide (3e) were explored. All these functional ProDOT derivatives were fully characterized by various spectroscopic techniques before polymerization. These functional ProDOT derivatives were then copolymerized with 2,5-dibromo dihexyl-ProDOT (4) under similar conditions to obtained final polymers P3a–e and were fully characterized (Table 1). The successful polymerization of these functional side chains clearly indicates the versatility of the present reductive polymerization approach over the reported GRIM method.
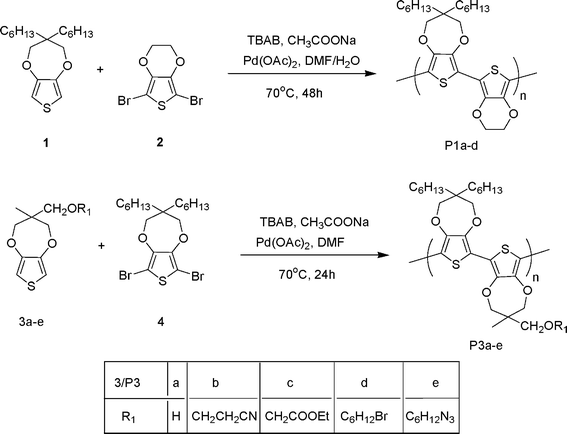 | ||
| Scheme 1 Reductive polymerization of monomers based on ProDOTs using palladium acetate. | ||
We also explored if some homocoupling is also happening along with the heterocoupling during polymerization. For this, we carried out the polymerization of dihexyl-ProDOT in the same manner as explained above but without 2,5-dibromo dihexyl-ProDOT. No color change was observed during the reaction and we could recover the unreacted monomer even after carrying out the polymerization for two days. The same was observed when 2,5-dibromo dihexyl-ProDOT was subjected to polymerization conditions without the addition of dihexyl-ProDOT. This confirms that there is no homocoupling during polymerization. The final composition of all the polymers was found be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as confirmed by 1H NMR spectra. The absence of homocoupling and 1:1 incorporation of the two monomers in the final polymer confirms that the present method gives perfectly alternate structures. Therefore, it opens the possibilities of synthesizing alternate donor–acceptor type polymers based on EDOTs, which is not possible using GRIM method. All these polymers were further studied for solution doping with SbCl5 and exhibited typical doping spectra of substituted ProDOTs.
:1 as confirmed by 1H NMR spectra. The absence of homocoupling and 1:1 incorporation of the two monomers in the final polymer confirms that the present method gives perfectly alternate structures. Therefore, it opens the possibilities of synthesizing alternate donor–acceptor type polymers based on EDOTs, which is not possible using GRIM method. All these polymers were further studied for solution doping with SbCl5 and exhibited typical doping spectra of substituted ProDOTs.
In order to get some insight into if the polymerization follows chain growth mechanism or step growth mechanism, copolymerzation of monomer 1 was carried out with two equivalents of monomer 2 instead of the normal 1:1 ratio. To our surprise we found that this also resulted in the formation of high molecular weight polymer confirming the chain growth mechanism of the polymerization.
Acknowledgements
We would like to acknowledge Department of Science and Technology (DST), India and Ministry of Communication and Information Technology (MCIT), India, for financial support. Anshu Kumar thanks Council of Scientific and Industrial Research, India for Senior Research Fellowship. We also thank Sycon Polymers India Pvt. Ltd for a gift of 3,4-dimethoxythiophene.Notes and references
- J. Roncali, P. Blanchard and P. Frere, J. Mater. Chem., 2005, 15, 1589 RSC.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481 CrossRef CAS.
- C. B. Nielsen, A. Angerhofer, K. A. Abboud and J. R. Reynolds, J. Am. Chem. Soc., 2008, 130, 9734 CrossRef CAS.
- I. Schwendeman, C. L. Gaupp, J. M. Hancock, L. Groenendaal and J. R. Reynolds, Adv. Funct. Mater., 2003, 13, 541 CrossRef CAS.
- K. Krishnamoorthy, R. S. Gokhale, A. Q. Contractor and A. Kumar, Chem. Commun., 2004, 820 RSC.
- R. B. Bazaco, R. Gomez, C. Seoane, P. Baeuerle and J. L. Segura, Tetrahedron Lett., 2009, 50, 4154 CrossRef CAS.
- B. Winther-Jensen, O. Winther-Jensen, M. Forsyth and D. Macfarlane, Science, 2008, 321, 671 CrossRef CAS.
- A. E. Daugaard, S. Hvilsted, T. S. Hansen and N. B. Larsen, Macromolecules, 2008, 41, 4321 CrossRef.
- H. Bu, G. Gotz, E. Reinold, A. Vogt, S. Schmid, R. Blanco, J. L. Segura and P. Bauerle, Chem. Commun., 2008, 1320 RSC.
- J. Sinha, R. Sahoo and A. Kumar, Macromolecules, 2009, 42, 2015 CrossRef CAS.
- E. M. Galand, J. K. Mwaura, A. A. Argun, K. A. Abboud, T. D. McCarley and J. R. Reynolds, Macromolecules, 2006, 39, 7286 CrossRef CAS.
- S. P. Mishra, R. Sahoo, A. V. Ambade, A. Q. Contractor and A. Kumar, J. Mater. Chem., 2004, 14, 1896 RSC.
- B. Sankaran and J. R. Reynolds, Macromolecules, 1997, 30, 2582 CrossRef CAS.
- V. Jain, R. Sahoo, S. P. Mishra, J. Sinha, R. Montazami, H. M. Yochum, J. R. Heflin and A. Kumar, Macromolecules, 2009, 42, 135 CrossRef CAS.
- K. Krishnamoorthy, A. V. Ambade, M. Kanungo, A. Q. Contractor and A. Kumar, J. Mater. Chem., 2001, 11, 2909 RSC.
- C. R. G. Grenier, W. Pisula, T. J. Joncheray, K. Muellen and J. R. Reynolds, Angew. Chem., Int. Ed., 2007, 46, 714 CrossRef CAS.
- B. D. Reeves, C. R. G. Grenier, A. A. Argun, A. Cirpan, T. D. McCarley and J. R. Reynolds, Macromolecules, 2004, 37, 7559 CrossRef CAS.
- D. M. Welsh, A. Kumar, E. W. Meijer and J. R. Reynolds, Adv. Mater., 1999, 11, 1379 CrossRef CAS.
- V. Seshadri, L. Wu and G. A. Sotzing, Langmuir, 2003, 19, 9479 CrossRef CAS.
- J. G. Bokria, A. Kumar, V. Seshadri, A. Tran and G. A. Sotzing, Adv. Mater., 2008, 20, 1175 CrossRef CAS.
- B. D. Reeves, E. Unur, N. Ananthakrishnan and J. R. Reynolds, Macromolecules, 2007, 40, 5344 CrossRef CAS.
- C. B. Nielsen and T. Bjornholm, Macromolecules, 2005, 38, 10379 CrossRef CAS.
- C. R. G. Grenier, S. J. George, T. J. Joncheray, E. W. Meijer and J. R. Reynolds, J. Am. Chem. Soc., 2007, 129, 10694 CrossRef CAS.
- A. Borghese, G. Geldhof and L. Antoine, Tetrahedron Lett., 2006, 47, 9249 CrossRef CAS.
- A. K. Mohanakrishnan, P. Amaladass and J. A. Clement, Tetrahedron Lett., 2007, 48, 539 CrossRef CAS.
- S. P. Mishra, K. Krishnamoorthy, R. Sahoo and A. Kumar, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra along with the UV-vis solution doping studies of the resulting polymers. See DOI: 10.1039/b9py00265k |
| This journal is © The Royal Society of Chemistry 2010 |