Biopolymer templating as synthetic route to functional metal oxide nanoparticles and porous sponges
Yi-Yeoun
Kim
c,
Carolina
Neudeck
ab and
Dominic
Walsh
*a
aSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS. E-mail: d.walsh@bristol.ac.uk; Tel: +44 (0)1225 3316797
bDepartment of Chemistry, Technische Universität München, Lichtenbergstr. 4, 85748, Garching, Germany
cSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS2 9JT
First published on 26th January 2010
Abstract
The biopolymers dextran and xyloglucan were used to attenuate structural development of a range of metal oxide materials. Dextran was used to prepare catalytic iron, iron–gold, and cerium–copper oxide porous sponges composed of joined nanoparticles. Zinc oxide sponges and nanoparticles coated with <5 nm gold particles were similarly prepared using xyloglucan which was shown to have potential advantages in terms of synthesis properties and cost.
Dextran is a waste material in the sugar industry and is used here as an example of a soluble hydroxylated polymer waste product that can be applied for attenuated calcinations of metal nitrates. Dextran is a biocompatible polysaccharide composed of a majority of α1-6 and a minority of α1-3 or α1-4 glycosidic linked glucose units and is readily soluble in aqueous solutions to form mildly to highly viscous mixtures depending on concentration and molecular weight.1 The dextran biopolymer component can also be readily generated from sucrose solution using bioreactors inoculated with Leuconostoc mesenteroides bacteria or using dextran synthase enzyme directly.2 Other example biopolymer materials that could be similarly applied include pullulans, xylans and xyloglucans. Tamarind xyloglucan was here employed as a means to control formation of zinc oxide nanoparticles that could also be readily coated with gold nanoparticles to form a catalytic material. Tamarind xlyoglucan, a waste product from tamarind seed kernels, is a soluble hemicellulose polysaccharide composed of 1,4-linked β-D-glucopyranose residues with many α(1,6)-linked xylopyranose branches, which themselves may be further branched. The large stiff and branched molecule means that a viscous aqueous solution results from dissolving xyloglucan at a few wt%.3
Nanoparticles are becoming increasingly important for the preparation of new materials for a range of applications, for example particular metal oxide nanoparticles, such as iron, zinc and cerium oxides, have important medical applications.4 Also these materials have shown high efficiency for heterogeneous catalysis, including CO oxidation in gas streams.5 An important recent development is the discovery that nanoparticles of gold (<10 nm), when laid onto metal oxide supports, show remarkably enhanced catalytic efficiency.6 Furthermore mixed metal oxide systems have been prepared that show improved catalytic properties. For example cerium–copper oxide mixtures have recently shown increased importance for oxidation catalysis.7 In view of the dramatic increase in demand for these types of materials, especially with the bringing to marketplace of fuel cell powered vehicles, an efficient nanoparticle synthesis process that does not require organic solvents or surfactants has clear advantages.
Synthesis of nanoparticles often involves use of organic solvents, sol–gel condensations and emulsion systems using a wide range of surfactants.8 Nanoparticles have also been prepared using a variety of polymers as stabilizing or capping agents.9 In our study oxide nanoparticles of iron, zinc and mixed cerium–copper were prepared as examples by a simple green and general approach using the mixing of aqueous nitrate solutions with the natural biopolymers dextran and xyloglucan. These mixtures were either freeze-dried or room temperature (RT) dried before heating. It was found that xyloglucan nitrate mixtures could successfully form porous structures without freeze drying.
For the synthesis of iron oxide (Fe2O3) nanoparticles, 5 ml of deionized water containing 0.01 M of Fe(NO3)2·9H2O were mixed with 10 g of commercially available dextran of Mr 70k, the viscous mixtures were then freeze-dried to form porous monoliths. Similarly cerium–copper oxide microsponges of nanoparticles were prepared using 5 ml water containing 0.01 M Ce(NO3)3·6H2O mixed with Cu(NO3)2.2·5H2O (to give copper at 6 and 10 wt%) and 10 g of dextran of either Mr 70k or 500k followed by freeze drying. All freeze-dried metal oxide–dextran mixtures were then heated to 600 °C (10 °C min−1 ramp rate) for 2 h and allowed to cool to RT in the furnace to form monoliths of porous sponges or foams.
For comparison zinc oxide (ZnO) microsponges were prepared from both dextran and xyloglucan. Thus 0.01 M Zn(NO3)2·6H2O was dissolved in 5 ml of distilled water to which 10 g of dextran were added. For the xyloglucan system 0.01 M Zn(NO3)2·6H2O was dissolved in 50 ml of water in which 2 g of tamarind xyloglucan were dissolved (i.e. at 4 wt% of the H2O content). The xyloglucan mixture was stirred for 1 h to form a viscous clear solution which was freeze-dried and also dried at RT before heating under the same conditions as the dextran systems.
Fe2O3 sponges were red-orange, ZnO were white and CeO2–CuO brown-grey with a darkened colour obtained at higher CuO content. Control experiments where the nitrate salts were heated using the same conditions but in the absence of biopolymer produced heterogenous loose powders of macroscopic metal oxide crystals in all cases (not shown).
Significantly, heating of RT air dried dextran–metal nitrate mixtures was less successful in producing open framework microsponges of metal oxide nanoparticles. Here prolonged or heating to higher temperatures was necessary to completely remove the dextran templating agent. As a result much greater sintering of the nanoparticles and weak and dense frameworks of microcrystals were formed (not shown). Conversely, freeze-dried metal oxide–dextrans had porous open structures that aided calcination and removal of the biopolymer agent with minimal heating and consequently much less sintering of the oxide components. For the ZnO xyloglucan preparation, however, open framework sponges were readily obtained without freeze drying.
Fig. 1 shows SEM micrographs of the metal oxide microsponges obtained after complete removal of the dextran or xyloglucan template. Fig. 1a shows the prepared Fe2O3 in the form of sponges with filaments around 1 µm in diameter. Fig. 1b shows ZnO microsponges formed from heating mixtures dried at RT. The product is composed of a highly porous flake-like structure with pores ranging from tens of nanometres up to approximately one micron in diameter. Freeze-dried xyloglucan–ZnO mixtures were near identical in structure (not shown). Fig. 1c shows a foam structure of micron sized cells of the CeO–CuO (6 wt% Cu) prepared using dextran of Mr 70k, which differed quite markedly from the same oxide mixture prepared using dextran of Mr 500k. Here the macrostructure was of flakes of oxide containing occasional pores and tears (Fig. 1d). This was presumably due to the much higher viscosity of the high Mr dextran mixture that restricted the formation of a large carbon/oxide nanoparticle open framework foam during heating and resulted in a denser final oxide structure.
 | ||
| Fig. 1 SEM micrographs of heated microsponges composed of (a) Fe2O3 prepared using 70k dextran; (b) ZnO prepared using xyloglucan; (c) CeO2–CuO (6 wt% Cu) using 70k dextran as template directing agent; (d) CeO2–CuO (6 wt% Cu) using 500k dextran as a template. (Dextran–nitrate mixtures were freeze-dried, xyloglucan–ZnO mixtures were dried at RT before heating). | ||
Samples were removed from the furnace at intervals during heating to elucidate the transformation process. It was observed that heating of the freeze-dried dextran–metal nitrate mixtures resulted in transformation of the dextran into a viscous phase which decomposed, sometimes rapidly, into an expanded carbon foam containing dispersed nanoparticles. Continued heating brought the constituents together producing open framework microsponges composed of connected oxide nanoparticles. The porous products obtained were reasonably robust and could be handled with care and placed into solution without collapse of the structure.
Gold was also incorporated onto metal oxides to form gold nanoparticle decorated structures which are efficient and reusable oxidation catalyst materials. Gold was incorporated into the Fe2O3 and ZnO materials by addition of Au as HAuCl4 salt with Au at 4 wt% of the Fe2O3 and at 1.5 wt% of the ZnO. This was achieved by stirring the oxide mixture with gold solution pH adjusted to 7 and held at 70 °C overnight, followed by drying and heating.10 The prepared Fe2O3–AuOH was heated to 300 °C and ZnO–AuOH heated to 240 °C for 2 h to convert the AuOH into bonded Au nanoparticles decorated upon the oxide surfaces. Fe2O3–Au was darkened and ZnO transformed to purple in colour.
Fig. 2 shows the X-ray diffraction data (XRD) of all the prepared heated materials which corresponded to Fe2O3 (hematite) (JCPDS no. 00-033-0664) (Fig. 2a) and ZnO (zincite) (JCPDS no. 00-036-1451) (Fig. 2c). Small XRD peaks at d 2.36 [111] and 2.04 [200] Å due to the presence of Au (JCPDS no. 004-0784) were present in the composites of these materials but are only just barely discernible with the lower Au content in ZnO (Fig. 2b and d). CeO2–CuO prepared with 500k dextran with CuO present at 6 (Fig. 2e) and 10% (Fig. 2f) is shown (JCPDS no. for CeO2 (ceria) 00-034-0394 and CuO (tenorite) 00-048-1548), CuO content at 10 wt% Cu with weak d 2.52 Å (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and d 2.32 Å (111) reflections is visible. CuO content was only clearly discernible at 10 wt% or above as has been reported.7 X-Ray reflections from CeO2–CuO systems at the two CuO levels using dextran of Mr 70k (not shown) were near identical to their counterparts' prepared using Mr 500k dextran.
) and d 2.32 Å (111) reflections is visible. CuO content was only clearly discernible at 10 wt% or above as has been reported.7 X-Ray reflections from CeO2–CuO systems at the two CuO levels using dextran of Mr 70k (not shown) were near identical to their counterparts' prepared using Mr 500k dextran.
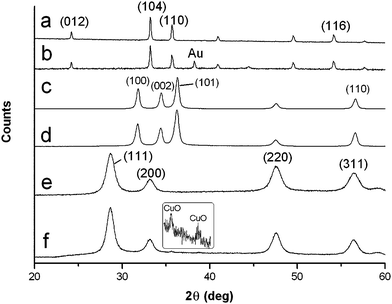 | ||
| Fig. 2 Powder X-ray diffraction of (a) Fe2O3; (b) Fe2O3 and 4 wt% Au nanoparticles; (c) ZnO; (d) ZnO and 1.5 wt% Au nanoparticles; (e) CeO2–CuO with 6 wt% CuO; (f) CeO2–CuO with 10 wt% CuO (inset). | ||
Fig. 3 presents TEM microscopy of the gold decorated metal oxides and mixed oxides, Fig. 3a and b show that the Fe2O3–Au was formed as porous plates composed of interconnected fused Fe2O3 crystals of 50–100 nm dimension decorated by gold nanoparticles of 4–10 nm in size. ZnO–Au prepared by use of xyloglucan consisted of 20–50 nm ZnO crystals decorated by gold nanoparticles of 3–5 nm in size, in this case the tendency of the ZnO crystals to fuse together was less and they were largely discrete (Fig. 3c). Significantly experiments using dextran–zinc nitrate produced larger ZnO nanoparticles of 50–80 nm in size (not shown). CeO2–CuO (6 wt% Cu) mixtures are shown in Fig. 3d, here fused nanocrystal plates were obtained which showed little porosity at the nanoscale. A lower magnification TEM is shown in Fig. 3e of the open framework macrostructure of the product. Using dextran of Mr 70k average crystal dimension was 12 nm, when dextran of Mr 500k was employed average crystal dimension decreased to 9 nm. This is likely due to the higher viscosity of the environment during calcinations with the higher Mr dextran that resulted in enhanced inhibition of crystal growth and sintering. The dimensions of these ceria oxide mixtures compare favourably with commercially available ceria which is reported to be ∼46 nm in size.7
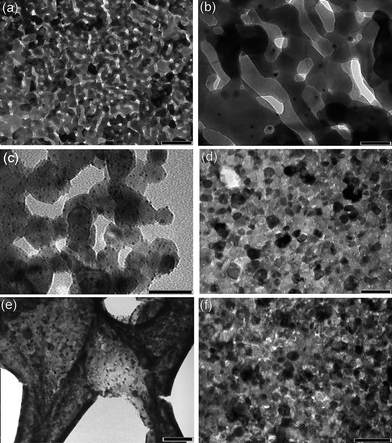 | ||
| Fig. 3 TEM micrographs showing (a) nanoporous Fe2O3 crystals prepared using dextran and decorated with Au nanoparticles (scale bar = 200 nm); (b) enlargement of (a) (scale bar = 50 nm); (c) ZnO nanocrystals prepared using xyloglucan and decorated with Au nanoparticles (scale bar = 50 nm); (d) CeO2–CuO (6 wt% Cu) using 70k dextran as template directing agent (scale bar = 50 nm); (e) as (d) at lower magnification showing open framework of embedded nanocrystals (scale bar = 200 nm); (f) CeO2–CuO (6 wt% Cu) using 500k dextran as template directing agent (scale bar = 50 nm). | ||
In order to elucidate the possible mechanism for the varied porosity obtained the thermal behaviour in air of the metal nitrate with dextran or xyloglucan samples were investigated using TGA (Fig. 4). Samples for TGA were prepared by rapid freezing of the dried polymer–nitrate solids in N2(l) to enable grinding into a fine powder. Pure polymers were also dissolved in water and freeze-dried or RT dried before grinding and TGA measurement. Overall from the TGA data it was clear that the polymer–nitrate mixtures decomposed at temperatures well below that of the pure polymer counterparts, this was due to the evolution of oxygen and nitrogen oxide gases from the nitrates that accelerated combustion of the organic component. TGA of Fe(NO3)2·9H2O and dextran produced a curve that differed from the other metal nitrate systems as onset of decomposition occurred at a significantly lower temperature of about 110 °C. Fe(NO3)2·9H2O is known to decompose at a relatively low temperature and transforms to Fe2O3via dehydroxylation of iron hydroxides below 200 °C.11 This compares to Zn(NO3)2·6H2O for example which does not decompose fully to ZnO until around 310 °C.11 Thus it is possible that the observed nanopores present in the Fe2O3 crystals are due to partially trapped steam from the phase changing crystals within the rubbery dextran phase. Decomposition of the dextran produced the overall macroporous sponge structures, with carbon removed by around 340 °C. For the other dextran nitrate systems decomposition occurs at higher temperatures with minimal steam evolution and a more rapid combustion step occurring, thus discrete whole crystals were obtained in these cases.

For CeO2–CuO (6 wt% Cu) 70k dextran samples the combustion step occurred at 180 °C, for the corresponding 500k dextran sample it occurred more gradually at around 10 °C higher, the remaining carbon/oxide foams then underwent decomposition of the carbon component. For the 70k sample this had fully occurred at 280 °C and for the 500k sample this end point was at the higher temperature of 340 °C. The differences in decomposition and final removal of the dextran templating agent were reflected in the smaller size of the nanocrystals obtained at higher Mr dextran. BET measurements supported this finding as surface area of the 70k sample was measured as 9 m2 g−1 and the corresponding 500k sample as more than twice this at 19 m2 g−1. RT dried ZnO–xyloglucan systems showed a combustion step at 120 °C followed by a relatively gradual decline of remaining zinc nitrate and carbon content until around 400 °C when ZnO at approximately 20 wt% of original dry weight content remained. The sharp weight loss on combustion is most likely due to the small amount of xyloglucan present relative to the dextran systems. Here partial decomposition of the zinc nitrate with consequent release of combustion accelerant gases was sufficient to promote decomposition of the majority of the xyloglucan component at a relatively low temperature.
Yields of final oxides product from the dried nitrate–dextran composites after heating in the furnace varied from approximately 2 wt% for Fe2O3 up to approximately 8.5 wt% for CeO2–CuO and 20 wt% for ZnO systems.
Conclusions
In conclusion we have demonstrated a simple green and general method for the synthesis of a range of metal oxides as nanoparticles arranged in the form of microsponges by the use of the natural biopolymers dextran and xyloglucan as templating agents. To form microsponges of nanoparticles freeze drying of the metal oxide–dextran mixture to form porous monoliths was necessary to aid combustion and minimize crystal sintering during the heating step. The prepared oxides nanoparticles could be readily decorated with gold nanoparticles for applications as high efficiency oxidation catalysis. The results suggest that oxide crystal structure was effected by the decomposition pathway of the nitrate, Fe(NO3)2·9H2O had a low decomposition temperature accompanied by steam evolution that gave rise to nanoporous crystals. For all systems, decomposition of the metal nitrate occurred via a combustion step with onset between ∼120 and 190 °C depending on the particular nitrate and biopolymer used. Decomposition of the biopolymer component produced the macroporous sponges composed of the joined metal oxide nanocrystals. Particle size was affected by Mr of the dextran with higher chain length resulting in smaller nanocrystals due to the higher glass transition temperature and higher thermal stability of the polymer template, this therefore attenuated crystal growth to a greater extent. Xyloglucan is not readily available with differing Mr but for ZnO systems successfully generated open framework sponges without freeze drying being required. With this biopolymer much lower concentrations could be employed that controlled the crystal development on heating.Acknowledgements
We thank Wataru Ogasawara, Department of Bioengineering Nagaoka University of Technology, Nagaoka, Japan for generous donation of Tamarind xyloglucan. We thank the EPSRC ARF (EP/C544803/1) (DW) for financial support of this study, Jack Butterfield for technical support and Dr Barbara Palazzo, School of Chemistry, University of Bologna for BET measurements.Notes and references
- T. Khan, J. K. Park and J.-H. Kwon, Korean J. Chem. Eng., 2007, 24, 816 Search PubMed.
- S. A. U. Qader, A. Abid Azhar, L. Iqbal, A. Aman and E. Shireen, Turk. J. Biochem., 2005, 31, 21 Search PubMed.
- A. Mishra and A. V. Malhotra, J. Mater. Chem., 2009, 19, 8528 RSC.
- (a) T. Kawaguchi, T. Hanaichi, M. Hasegawa and S. Maruno, J. Mater. Sci.: Mater. Med., 2001, 12, 121 CrossRef CAS; (b) M. R. Kongara, A. Punnoose, D. Wingett and K. R. Madhusudan, Patent Numbers WO2009039508-A2; US2009136580–A1, 2009; (c) J. Chen, S. Patil, S. Seal and J. F. McGinnis, Nat. Nanotechnol., 2006, 1, 142 CrossRef CAS.
- (a) Y. F. Shen, R. P. Zerger, R. N. DeGuzman, S. L. Suib, L. McCurdy, D. I. Potter and C. L. Young, Science, 1993, 260, 511 CAS; (b) H. K. Lin, H. C. Chiu, H. C. Tsai, S. H. Chien and C. B. Wang, Catal. Lett., 2003, 88, 169 CrossRef CAS.
- (a) G. Y. Wang, W. X. Zhang, H. L. Lian, D. Z. Jiang and H. W. Tong, Appl. Catal., A, 2003, 239, 1 CrossRef CAS; (b) M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175 CrossRef CAS; (c) M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS; (d) J. A. von Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651 CrossRef CAS.
- X. C. Zheng, S. H. Wu, S. P. Wang, S. R. Wang, S. M. Zhang and W. P. Huang, Appl. Catal., A, 2005, 283, 217 CrossRef CAS.
- (a) J. Eastoe, M. J. Hollamby and L. Hudson, Adv. Colloid Interface Sci., 2006, 128, 5 CrossRef; (b) G. Schmid, Nanoparticles, Wiley-VCH, Weinheim, 2003 Search PubMed.
- (a) R. B. Grubbs, Polym. Rev., 2007, 47, 197 Search PubMed; (b) L. Zhang, R. He and H.-G. Gu, Appl. Surf. Sci., 2006, 253, 2611 CrossRef CAS; (c) M. Chen and Y. Xing, Langmuir, 2005, 21, 9334 CrossRef CAS.
- X. Wang, D. R. G. Mitchell, K. Prince, A. J. Atanacio and R. A. Caruso, Chem. Mater., 2008, 20, 3917 CrossRef CAS.
- (a) K. Wieczorek-Ciurowa and A. J. Kozak, J. Therm. Anal. Calorim., 1999, 58, 647 CrossRef CAS; (b) M. Maneva and N. Petrov, J. Therm. Anal., 1989, 35, 2297 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |