Barnacle repellent nanostructured surfaces formed by the self-assembly of amphiphilic block copolymers
Beng H.
Tan
a,
Hazrat
Hussain
a,
Kuan C.
Chaw
a,
Gary H.
Dickinson
b,
Chakra S.
Gudipati
c,
William R.
Birch
*a,
Serena L. M.
Teo
b,
Chaobin
He
a,
Ye
Liu
*a and
Thomas P.
Davis
*ad
aInstitute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), 3 Research Link, 117602, Singapore. E-mail: ye-liu@imre.a-star.edu.sg; w-birch@imre.a-star.edu.sg
bTropical Marine Science Institute, National University of Singapore, 119223, Singapore
cGE Water and Process Technologies, WS2-M-4, 1 Engineering Drive 1, 117580, Singapore
dCentre for Advanced Macromolecular Design (CAMD), School of Chemical Sciences and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: t.davis@unsw.edu.au
First published on 22nd December 2009
Abstract
Well-defined nanostructured surface with domains of dimension ∼20 nm was formed by the self-assembly of brush-type amphiphilic block copolymers of poly[poly(ethylene glycol)methyl ether methacrylate]-block-poly(2,3,4,5,6-pentafluorostyrene) (P(PEGMA)-b-PPFS) which demonstrate promise in discouraging barnacle settlement as proven using laboratory settlement assays and marine field tests.
It is a formidable challenge to produce anti-fouling surfaces for applications such as bio- and marine anti-fouling without resorting to biocides. Recent findings have revealed that heterogeneous surfaces show promise in reducing the settlement and enhancing the release of fouling organisms. Self-assembly of amphiphilic copolymers is a versatile “bottom up” approach to engineering non-biofouling heterogeneous surfaces.1 Surfaces containing fluoropolymer segregated phases and poly(ethylene glycol) (PEG) domains from hyperbranched fluoropolymers have been shown to reduce the settlement and increase the release of Ulva, a marine algae.2 Nanostructured surfaces from block copolymers possessing polystyrene–polystyrene with amphiphilic PEGylated fluoroalkyl side chains3 and polystyrene–poly(2-vinyl pyridine) together with polystyrene–poly(methyl methacrylate)4 resulted in the weak adhesion and low settlement density of two marine algae, Ulva and Navicula. These earlier findings provide strong evidence that the topological and morphological complexity of heterogeneous surfaces can yield surfaces that are energetically unfavorable for protein and glycoprotein adsorption, thus weakening the adhesive strength of entire organisms to the polymer surface.1–4 Herein we report that nanostructured surfaces formed by the self-assembly of brush-type amphiphilic block copolymers of poly[poly(ethylene glycol)methyl ether methacrylate]-block-poly(2,3,4,5,6-pentafluorostyrene) (P(PEGMA)-b-PPFS) demonstrate promise in discouraging barnacle settlement.
Barnacles are widespread and considered to be a serious pest as they rapidly colonize immersed man-made objects. Their larvae are highly discriminatory during exploration and judge a surface's suitability based on criteria including texture, local hydrodynamics and surface chemistry.5 Barnacles are also fortuitously conducive to laboratory testing of experimental anti-foulants as they breed year-round and settle readily in static water assays.5 Phang et al. observed that barnacle larvae favor settling on hydrophilic over hydrophobic surfaces and speculated that the confined larvae footprint size on hydrophilic surfaces may induce a higher concentration of the protein adhesive.5 Previously, coatings research focused almost exclusively on biocidal technologies and low surface-free-energy ‘fouling-release’ materials. However, novel strategies to inhibit barnacle surface colonization are desirable because barnacle growth increases drag up to 60% thus reducing a vessel's fuel efficiency and increasing green house gas emissions.5
The present work focuses on the surfaces formed by the assembly of P(PEGMA)-b-PPFS and their ability to resist the settlement of barnacles. The atom transfer radical polymerization (ATRP) synthesis and self-assembly behavior of this block copolymer have recently been reported in detail.6 The PPFS imbues typical fluoropolymer characteristics such as low surface energy, high chemical and thermal resistance and distinct self-assembly behavior.7 P(PEGMA) segments can potentially introduce other desirable properties to the copolymer such as water affinity, low toxicity, high biocompatibility and fouling resistance to proteins.8 Therefore the P(PEGMA)-b-PPFS copolymer presents an opportunity for combining the favorable properties of the fluorinated blocks with those of the P(PEGMA) blocks.
The phase separation and morphological features of amphiphilic fluorinated copolymers and blends can be exploited to generate ordered, self-assembled, low surface energy materials.2,3,9 The TEM image and AFM phase image (from tapping mode) of the block copolymer coating as depicted in Fig. 1a and b, respectively, indicate the formation of nanodomains of dimension ∼20 nm resulting from the thermodynamically induced phase segregation of the different components of the coating, driven by the chemical incompatibility of the blocks.3 Two distinct glass transition temperatures (Tg), approx. −50 °C and ∼100 °C corresponding to the Tg of P(PEGMA) and PPFS respectively were also observed using differential scanning calorimetry (DSC). The presence of two distinct transitions confirms that the two polymer blocks (P(PEGMA) and PPFS) are well segregated.
 | ||
| Fig. 1 (a) Transmission electron microscopy (TEM) and (b) atomic force microscopy (AFM) images of P(PEGMA)-b-PPFS diblock copolymer films cast from 10 mg ml−1 of copolymer solution in THF. | ||
Well-defined morphologies with either spherical (20 nm diameter) or lying cylindrical (24–29 nm periodicity) nanodomains were also observed by Martinelli et al. for polystyrene block copolymers carrying an amphiphilic polyoxyethylene–polytetrafluoroethylene chain side group, depending on the amphiphilic copolymer composition. In contrast, no phase segregation or well-defined nanoscale structures were observed for coatings prepared from fluorinated polystyrene homopolymer3 or statistical copolymers formed from a feed containing the same mole fraction of each monomer.4
The surface-free energies of the coatings were calculated from the advancing contact angles, θa, of water, diiodomethane and ethylene glycol using geometric mean (GM)10 and Lifshitz–van der Waals acid–base (LW-AB) models.11 The θas of water on the amphiphilic copolymer, PPFS homopolymer and the amino (–NH2)-group-terminated glass surface were 78.1 ± 0.6°, 102.0 ± 0.4° and 55.8 ± 0.3° respectively confirming the less hydrophobic nature of the P(PEGMA)-b-PPFS and glass surfaces in comparison to the PPFS surface. The amphiphilic copolymer surface also displayed a lower θa to diiodomethane and ethylene glycol when compared to the hydrophobic PPFS surface, indicating an increase in the surface energy of P(PEGMA)-b-PPFS surface in comparison to the hydrophobic PPFS surface. It is noteworthy that the amphiphilic copolymer coating exhibited high values of θa and low values of receding contact angle, θr, leading to significant hystereses (θa − θr). Such phenomena can be attributed to both the chemical heterogeneity and the swelling and reorganization of the PEG domains with the latter being affected by contact with water.1–4 The total surface energies, γs, along with the polar and dispersive components for the three different surfaces were evaluated using advancing contact angles by geometric mean (GM)10 and Lifshitz–van der Waals acid–base (LW-AB)11 approaches. The γs of the amphiphilic P(PEGMA)-b-PPFS copolymer, PPFS homopolymer and the amino-terminated glass surfaces calculated from the GM model are consistent with those obtained from the LW-AB model which are approximately 25.2, 19.5 and 29.8 mJ m−2 respectively. This observation suggests that the incorporation of brush-type polymers based on PEG to fluorinated amphiphilic copolymers increases the surface energy of the coating when compared to hydrophobic surfaces.1–4
The hypothesis of surface reorganization was also confirmed by static contact angle measurements performed after the surfaces were immersed in water for 10 days. It was found that the static contact angles of water, θw, for the amphiphilic copolymer, PPFS homopolymer and the amino-terminated glass surface changed from ∼73° to ∼60°, ∼97° to ∼99° and ∼50° to ∼47° respectively. The γs of the amphiphilic P(PEGMA)-b-PPFS copolymer increased significantly to 29 mJ m−2 while the hydrophobicity of the other two surfaces did not change significantly. Martinelli et al. observed similar findings for polystyrene block copolymers carrying an amphiphilic polyoxyethylene–polytetrafluoroethylene chain side group and concluded that the surface became more enriched in the high surface energy PEG component after contact with water, while the fluorinated segments tended to be segregated in the bulk of the film.3
The coatings were subjected to laboratory bioassays to explore their intrinsic ability to resist the settlement by reducing the adhesive strength of proteins and cyprids. Quartz crystal microbalance with dissipation monitoring (QCM-D) was used to evaluate the adsorption of three different proteins in 10 mM phosphate buffered saline (PBS) i.e. BSA, CFL and LPSE on the P(PEGMA)-b-PPFS polymer surface. Three control surfaces were used in this experiment: (a) PPFS homopolymer, (b) hydrophilic surface comprised of pure gold with an amino-terminated monolayer and without any polymer coating (labelled as hydrophilic gold) and (c) hydrophobic surface consisting of trimethylsilane chloride deposited onto a treated gold surface without any polymer coating (labelled as hydrophobic gold). Trimethylsilane chloride is known to create a very hydrophobic surface on gold.12 The adsorbed mass, Δm, as a function of time when 0.5 mg ml−1 of BSA in 10 mM PBS was passed over the P(PEGMA)-b-PPFS polymer surface is shown in Fig. 2 and compared against the three control surfaces. A small amount of BSA was desorbed from the control and polymer surfaces when the surfaces were rinsed with 10 mM PBS as observed from Fig. 2. The adsorbed mass, Δm, with BSA did not attain equilibrium within 1 h. Similar results were also obtained for the polymer surface with the other two proteins, i.e. CFL and LPSE (data not shown). These data suggest complex structural rearrangements and conformational changes occurring within the polymeric layer consistent with surface reorganization, as QCM-D does not simply measure the mass of polymer or protein but also includes any entrained solvent within the adsorbed layer.13
 | ||
| Fig. 2 Adsorbed mass as a function of time of the amphiphilic copolymer and control surfaces measured by the quartz crystal microbalance with dissipation monitoring (QCM-D) technique when 0.5 mg ml−1 of bovine serum albumin (BSA) was passed through the surfaces. | ||
The control surfaces including the PPFS homopolymer surface adsorbed large amounts of protein when compared to the amphiphilic polymer surface. Spores of marine algae (young plants) have been shown previously to settle (attach) in high numbers on hydrophobic surfaces including fluorinated surfaces.1 The primary thermodynamic interaction between a protein excreted from marine organisms and a polymer upon which it adsorbs is the hydrophobic effect, namely, changes in the interaction of water with the hydrophobic surface and, to a lesser extent, with the hydrophobic amino acid residues on the protein. Water is poorly hydrogen bonded to the polymer surface, and the adsorption of the hydrophilic protein to the surface releases a large amount of entropy in the otherwise ordered water layer. Thus hydrophobic surfaces tend to adsorb large amounts of proteins relative to hydrophilic ones.14
The percentages of the final mass of each protein adsorbed onto the control and polymeric surfaces after rinsing with 10 mM PBS solution are shown in Table 1. The mass of each protein adsorbed onto the surfaces was normalized against the mass of that particular protein adsorbed onto the hydrophobic gold surface (set at 100%). The percentage of BSA and CFL adsorbed onto the P(PEGMA)-b-PPFS polymer surface is below 20% when normalized against the hydrophobic gold surface suggesting that this polymer surface has potential as an anti-protein fouling surface. In contrast, high adsorption of LPSE was observed for this amphiphilic surface when normalized against the hydrophobic gold surface. LPSE is known to adsorb preferentially onto hydrophilic surfaces rather than hydrophobic ones consistent with its high adsorption on the hydrophilic gold surface with the –NH2 terminated monolayer (157%).15 However, the absolute mass of LPSE adsorbed onto the amphiphilic polymer surface is still lower when compared to the control surfaces as shown in Table 1 as the amphiphilic surface can undergo surface reorganization after immersion in water as well as differences in surface nano-roughness, unlike the control surfaces, factors that can influence the adhesion strength of proteins, and attachment of bacteria and animal cells.3,4
| Film | Percentage adsorption of protein (%) | ||
|---|---|---|---|
| BSA | LPSE | CFL | |
| P(PEGMA)-b-PPFS | 16.8 | 30.5 | 18.2 |
| PPFS homopolymer | 68.3 | 44.6 | 63.0 |
| Hydrophilic gold | 47.0 | 157.0 | 45.4 |
| Hydrophobic gold | 100.0 | 100.0 | 100.0 |
The laboratory settlement assays employing barnacle larvae were performed on the amphiphilic copolymer, PPFS homopolymer and the amino-terminated glass surfaces over 24 h. Importantly, no barnacles were observed to settle on the amphiphilic surface in contrast to the control surfaces as depicted in Fig. 3a, b and c. These assays were corroborated by field tests evaluating the performance of the surfaces in a marine environment. The surfaces exposed to seawater for four weeks recruited several macrofouler species: barnacles, tubeworms and bryozoans. From the photographs presented in Fig. 3d and e, it is clear that no barnacles settled on the amphiphilic copolymer surface (Fig. 3e) in contrast to the control surface (Fig. 3d). We believe that we are the first to report an anti-fouling coating capable of resisting the settlement of barnacles for up to four weeks of immersion in seawater. Phang et al. reported that the preferred settlement of barnacles on amino-terminated glass could be explained by the confined larvae footprint size that induces a higher concentration of the settlement inducing protein complex (SIPC) on amino-terminated surfaces.5 The nanoscale patterns on the amphiphilic P(PEGMA)-b-PPFS surface and its ability to undergo surface reorganization in water act as a deterrent to the settlement of barnacles. The size of a typical larvae footprint is ∼30 µm by 20 µm and is expected to settle more readily on surfaces of similar dimensions.5 However, the dynamic nature and complex topology of the amphiphilic P(PEGMA)-b-PPFS surface lower the driving force for the adsorption of SIPC and hence reduce the settlement of barnacles.
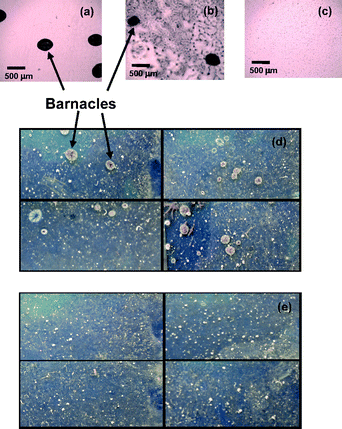 | ||
| Fig. 3 Optical microscope images of laboratory settlement assays employing cyprids on the (a) amino-terminated glass surface, (b) PPFS homopolymer surface and (c) P(PEGMA)-b-PPFS diblock copolymer surface. Photographs of glass slides exposed to marine environment for four weeks containing (d) amino-terminated glass surface (four slides) and (e) P(PEGMA)-b-PPFS diblock copolymer surface (four slides) where the area of each slide shown is 25 mm × 60 mm. | ||
Experimental
Materials
All reagents and proteins (BSA, Mw 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da; LPSE, Mw 10000 Da and CFL, Mw 60000 Da) were purchased from Aldrich.
000 Da; LPSE, Mw 10000 Da and CFL, Mw 60000 Da) were purchased from Aldrich.
Surface preparation
Prior to each QCM-D experiment, the gold sensor surface was cleaned in an UV/ozone chamber for 10 min and subsequently immersed in 1 mM cysteamine prepared in ethanol solution for at least 24 h prior to use. Cysteamine adsorbs spontaneously onto gold surfaces forming a uniform and active amino-group-terminated monolayer.16 The polymers (10 mg ml−1 in THF solution) were then deposited drop-wise onto individual treated gold surfaces and were left to dry overnight under ambient conditions and annealed for 4 h at 110 °C the next day before being mounted in the QCM-D.9 Laboratory assays and field tests were performed on cleaned glass slides of dimension 25 mm × 80 mm × 0.5 mm. The cleaned slides were dipped in aminopropyltrimethoxy silane (APTMS) solution for 20 min, followed by rinsing with DI water and dried under nitrogen. Polymers (10 mg ml−1 in THF solution) were deposited drop-wise onto the functionalized glass surface and annealed at 110 °C for 4 h.Barnacle larvae culture
The culture of Amphibalanus amphitrite has been reported.5Barnacle larvae settlement assay
Cleaned glass slides with amino-terminated surface and polymer surface were suspended vertically in individual centrifuge tubes containing filtered seawater. The area immersed was ∼25 mm × 60 mm and three replicates were used. After 24 h incubation in the dark, the barnacles attached to each slide were counted.Field test by panel immersion
Field test was conducted on the surfaces at the West Coast of Singapore from 27 February 2009 to 27 March 2009.5 All test samples were recorded by photography once a week. In conclusion, nanostructured surfaces formed by the self-assembly of amphiphilic block copolymers containing fluorinated and PEG segments were obtained and demonstrated to reduce adsorption of different types of protein. More importantly, the amphiphilic and dynamic surfaces proved to be resistant to the settlement of barnacles. These results should facilitate further development of anti-fouling and environmentally friendly coatings using heterogeneous surfaces.Acknowledgements
The authors gratefully acknowledge financial support from IMRE A*STAR and a Federation Fellowship from ARC.Notes and references
- S. Krishnan, C. J. Weinman and C. K. Ober, J. Mater. Chem., 2008, 18, 3405–3413 RSC; S. Krishnan, A. Ramakrishnan, A. Hexemer, J. A. Finlay, K. E. Sohn, R. Perry, C. K. Ober, E. J. Kramer, M. E. Callow, J. A. Callow and D. A. Fischer, Langmuir, 2006, 22, 5075–5086 CrossRef CAS.
- C. S. Gudipati, J. A. Finlay, J. A. Callow, M. E. Callow and K. L. Wooley, Langmuir, 2005, 21, 3044–3053 CrossRef CAS.
- E. Martinelli, S. Agostini, G. Galli, E. Chiellini, A. Glisenti, M. E. Pettitt, M. E. Callow, J. A. Callow, K. Graf and F. W. Bartels, Langmuir, 2008, 24, 13138–13148 CrossRef CAS.
- C. M. Grozea, N. Gunari, J. A. Finlay, D. Grozea, M. E. Callow, J. A. Callow, Z. H. Lu and G.C. Walker, Biomacromolecules, 2009, 10, 1004–1012 CrossRef CAS.
- N. Aldred and A. S. Clare, Biofouling, 2008, 24, 351–363 CrossRef CAS; I. Y. Phang, K. C. Chaw, S. S. H. Choo, R. K. C. Kang, S. S. C. Lee, W. Birch, S. L. M. Teo and G. J. Vancso, Biofouling, 2009, 25, 139–147 CrossRef CAS.
- B. H. Tan, H. Hussain, Y. Liu, C. B. He and T. P. Davis, Langmuir, 2009 DOI:10.1021/la902816b.
- G. D. Fu, Z. H. Shang, L. Hong, E. T. Kang and K. G. Neoh, Adv. Mater., 2005, 17, 2622–2626 CrossRef CAS; K. T. Powell, C. Cheng and K. L. Wooley, Macromolecules, 2007, 40, 4509–4515 CrossRef CAS.
- F. Zhang, E. T. Kang, K. G. Neoh, P. Wang and K. L. Tan, Biomaterials, 2001, 22, 1541–1548 CrossRef CAS.
- D. J. Gan, A. Mueller and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3531–3540 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- C. J. van Oss, R. J. Good and M. K. Chaudhury, J. Colloid Interface Sci., 1986, 111, 378–390 CAS; C. J. van Oss, R. J. Good and M. K. Chaudhury, Langmuir, 1988, 4, 884–891 CrossRef CAS.
- P. A. Dimilla, J. P. Folkers, H. A. Biebuyck, R. Harter, G. P. Lopez and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 2225–2226 CrossRef CAS.
- K. Sakai, G. B. Webber, C. D. Vo, E. J. Wanless, M. Vamvakaki, V. Butun, S. P. Armes and S. Biggs, Langmuir, 2008, 24, 116–123 CrossRef CAS.
- D. L. Elbert and J. A. Hubbell, Annu. Rev. Mater. Sci., 1996, 26, 365–394 CrossRef CAS.
- P. Landini and A. J. B. Zehnder, J. Bacteriol., 2002, 184, 1522–1529 CrossRef CAS.
- H. Z. Yu, J. W. Zhao, Y. Q. Wang, S. M. Cai and Z. F. Liu, J. Electroanal. Chem., 1997, 438, 221–224 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |