Towards poly(ester) nanoparticles: recent advances in the synthesis of functional poly(ester)s by ring-opening polymerization
Ryan J.
Pounder
and
Andrew P.
Dove
*
Department of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 4107
First published on 5th January 2010
Abstract
The recent trends in the synthesis of functional poly(ester)s by ring-opening polymerization (ROP) are reviewed. While the use of ROP processes for the synthesis of poly(lactide), poly(lactide-co-glycolide) and several poly(lactone)s has been well studied, the paucity of functional groups available for further reaction limits their application. Recent efforts to expand this available functionality are reviewed focusing on the application of renewable resources in the synthesis of new monomers and the utilization of click chemistry to provide common intermediate polymers in the manipulation of poly(ester) functionality. In turn these advances are leading to a new generation of precisely controlled nanoparticles comprised entirely of poly(ester)s.
 Ryan Pounder | Ryan Pounder completed his MChem studies at the University of Warwick in 2006. During this time he conducted his final year research project in the area of catalyst development for Dynamic Kinetic Resolution processes under the supervision of Dr Paul C. Taylor. Ryan began his PhD studies in October 2006 to investigate the synthesis of cyclic esters from renewable resources and their subsequent application as monomers in ring-opening polymerization. |
 Andrew Dove | Andrew Dove graduated from the University of York with an MChem (Hons) degree in Chemistry in 1999. He went on to study for his PhD under the supervision of Prof. Vernon C. Gibson at Imperial College London, graduating in 2003. Andrew then moved to Stanford University, California and subsequently IBM Almaden research centre to undertake periods of postdoctoral research. In 2005, Andrew returned to the UK to undertake an RCUK fellowship at Warwick, subsequently being appointed as an Assistant Professor in 2006 and then Associate Professor in 2009. Andrew's current research is focused on the synthesis of functional poly(ester)s using controlled polymerization methodology and their application in self-assembly. |
Introduction
The ring-opening polymerization (ROP) of lactones and lactides to produce aliphatic poly(ester)s provides versatile biocompatible and biodegradable polymers possessing good mechanical properties. These advantages have seen aliphatic poly(ester)s receive increasing attention over the last few years driven by their application as biodegradable substitutes for conventional commodity thermoplastics and applications in the biomedical field,1–6 where amongst the family of biodegradable polymers, aliphatic poly(ester)s possess the leading position as a consequence of the ready metabolization of the degradation products in most cases. This has led to poly(ester) homo- and copolymers being widely used as sutures, oral implants, and microspheres for drug encapsulation and delivery.1,2,4,7–11 Poly(ester)s can be prepared from a wide range of materials with judicious choice of monomer feedstock able to modulate the physio-chemical properties including glass transition temperatures, toughness, stiffness and degradability.Aliphatic poly(ester)s are prepared through one of two routes: the first is step-growth polycondensation of a hydroxy acid or between a diacid and a diol enabling access to a large range of monomer feedstocks. However, the molecular weights are generally limited and any minor deviations in the stoichiometry are detrimental to the chain length. Furthermore, long reaction times and high temperatures are often required resulting in unfavorable side reactions.12 The second route for the synthesis of aliphatic poly(ester)s is via ROP. By this methodology the preparation of high molecular weight aliphatic poly(ester)s is possible while maintaining high levels of control over their molecular characteristics under relatively mild conditions. There has been much research directed towards the controlled ROP of commercially available cyclic esters including glycolide, lactide, ε-caprolactone, δ-valerolactone and β-propiolactone resulting in aliphatic poly(ester)s with highly controlled molecular parameters. A variety of catalytic systems have been investigated to more efficiently mediate the ROP process including the development of well-defined metal complexes, organic catalysts and the study of enzymatic catalysis.8,12–14 High levels of control over polymer molecular weight, tacticity, polydispersity and end-group fidelity are vital if such polymers are to be applied in fields such as, amongst others, drug and gene delivery. Additionally, work has been directed towards the formation of architecturally diverse poly(ester)s, including stars, brushes, cycles, cross-linked materials and hyper branched poly(ester)s, to improve mechanical properties, hydrophilicity and degradability profiles.15–21 While the physical properties of these polymers can be further tailored via copolymerization,2,22–28 a major limitation towards application in new arenas results from the lack of readily accessible side-chain functionality and thus also their hydrophobicity. The introduction of functional groups throughout the polymer chain via ROP remains highly challenging but yields degradable polymers with tunable properties including increased hydrophilicity, post-polymerization modification as well as further fine tuning of the physical properties.
Many studies have focused on the synthesis of monomers and their subsequent ROP to produce functional poly(ester)s in a highly controlled manner and this area has been the subject of previous reviews.1,12,13,29–33 Herein we aim to focus on some current trends in the field, namely the synthesis of functional poly(ester)s from renewable resources, the application of click chemistries and the synthesis of functional nanoparticles comprised of only poly(ester)s produced by ROP.
Functional poly(ester)s from renewable resources
As a consequence of the structural similarity between amino acids and α-hydroxy acids and large range of terminal functional groups available, poly(ester)s have been commonly derived by conversion of suitable protected amino acids followed by cyclization and ROP. Additionally, given the commercial availability of malic acid both enantiomerically pure and as a racemic mixture (the α-hydroxy acid of aspartic acid) it has been the source of many studies into this area.Cyclic diesters
Two main strategies for the synthesis of functionalized cyclic diester monomers have been reported: (i) the self-condensation of α-hydroxy acids catalyzed by p-toluenesulfonic acid (pTsOH) or cracking of oligoesters by ZnO/Sn(Oct)2 (route 1) or (ii) the step-by-step condensation of an α-hydroxy acid and an α-haloacyl halide with subsequent base-mediated cyclization (route 2) (Scheme 1).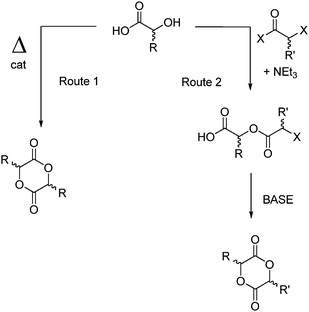 | ||
| Scheme 1 General synthetic routes for the synthesis of cyclic diesters from α-hydroxy acids. | ||
The synthesis of 3-(S)-[(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (BMD, 1) (Fig. 1) from L-aspartic acid was achieved by β-carboxylic acid protection with benzyl alcohol followed by diazotization with sodium nitrite (NaNO2) to afford the α-hydroxy acid that after coupling to bromoacetyl bromide with NEt3 was cyclized to BMD using NaHCO3 with a 26% yield over the final two steps (route 2—Scheme 1). Polymerization of 1 catalyzed by Sn(Oct)2, both in bulk at 160 °C and in toluene solution at 100 °C resulted in the isolation of polymers with Mn < 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 that displayed broad polydispersities (PDI > 1.4) attributed to a reversible depolymerization process.34 Copolymerization of 1 with L-lactide (95, 91 and 86 mol% LA) in bulk at 160 °C readily yielded copolymers. Deprotection via catalytic hydrogenolysis of the benzyl groups using both PtO2 and Pd/C catalysts was successful without any scission of the polymer chain as evidenced by size-exclusion chromatography. While the deprotected copolymers did not display any change in glass transition temperatures (Tg) they displayed enhanced in vitro hydrolysis rate compared to PLA as both bulk materials and within poly(ethylene glycol)-b-poly(lactide-co-BMD) nanoparticles.35,36 Films of deprotected random copolymers of BMD and analogously synthesized 3-(S)-[(dodecyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (DMD) (5 and 10 mol%) with L-lactide (PLBMD and PLDMD) enabled the attachment of cell-binding Arg-Gly-Asp tripeptide (RGD) via a dicyclohexylcarbodiimide (DCC) coupling reaction. These RGD-immobilized copolymers exhibited improved cell attachment with an increasing amount of immobilized RGD achieved from increasing the α-malate unit content in the copolymer.37 Dimerization of β-benzyl malate in the presence of a ZnO catalyst (route 1—Scheme 1) to yield 3,6-(S)-[di(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (malide, 2) (Fig. 1) was reported from both L-malic acid and L-aspartic acid (0.9–5.4% overall yield). Polymerization of malide in bulk at temperatures ranging from 100 to 220 °C with different organotin catalysts proved challenging with only low molecular weight poly(ester)s (Mn < 4000 g mol−1) being obtained, attributed to the steric hindrance present from the bulky pendant groups.38
000 g mol−1 that displayed broad polydispersities (PDI > 1.4) attributed to a reversible depolymerization process.34 Copolymerization of 1 with L-lactide (95, 91 and 86 mol% LA) in bulk at 160 °C readily yielded copolymers. Deprotection via catalytic hydrogenolysis of the benzyl groups using both PtO2 and Pd/C catalysts was successful without any scission of the polymer chain as evidenced by size-exclusion chromatography. While the deprotected copolymers did not display any change in glass transition temperatures (Tg) they displayed enhanced in vitro hydrolysis rate compared to PLA as both bulk materials and within poly(ethylene glycol)-b-poly(lactide-co-BMD) nanoparticles.35,36 Films of deprotected random copolymers of BMD and analogously synthesized 3-(S)-[(dodecyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (DMD) (5 and 10 mol%) with L-lactide (PLBMD and PLDMD) enabled the attachment of cell-binding Arg-Gly-Asp tripeptide (RGD) via a dicyclohexylcarbodiimide (DCC) coupling reaction. These RGD-immobilized copolymers exhibited improved cell attachment with an increasing amount of immobilized RGD achieved from increasing the α-malate unit content in the copolymer.37 Dimerization of β-benzyl malate in the presence of a ZnO catalyst (route 1—Scheme 1) to yield 3,6-(S)-[di(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (malide, 2) (Fig. 1) was reported from both L-malic acid and L-aspartic acid (0.9–5.4% overall yield). Polymerization of malide in bulk at temperatures ranging from 100 to 220 °C with different organotin catalysts proved challenging with only low molecular weight poly(ester)s (Mn < 4000 g mol−1) being obtained, attributed to the steric hindrance present from the bulky pendant groups.38
![3-(S)-[(Benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (BMD, 1) and 3,6-(S)-[di(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (malide, 2).](/image/article/2010/PY/b9py00327d/b9py00327d-f1.gif) | ||
| Fig. 1 3-(S)-[(Benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (BMD, 1) and 3,6-(S)-[di(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione (malide, 2). | ||
Benzyl protected L-serine, L-lysine and L-glutamic acid have also been applied in the synthesis of functional poly(ester)s via ROP. Following diazotization to the respective α-hydroxy acids a range of monomers have been synthesized including cyclic dimers and those derived from one functional α-hydroxy acid with either lactic or glycolic acid units via several synthetic methods in overall yields ranging from 20–48% (Scheme 2).
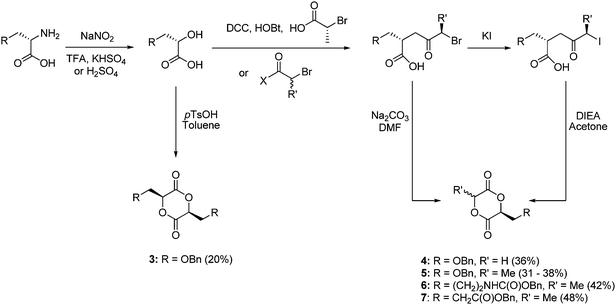 | ||
| Scheme 2 Synthesis of cyclic diester monomers 3–7 from amino acids (overall yields from amino acid in parentheses). | ||
High molecular weight polymers were realized for the copolymerization of 3–7 with rac-lactide by Sn(Oct)2 in bulk at 140 °C. The copolymers showed Tg values significantly below that of PLA (5 to 27 °C) and subsequent deprotection through either hydrogenation with Pd(OH)2 for the L-serine and L-glutamic acid polymers or via HBr(33%)–AcOH for the L-lysine polymers to reveal alcohol, carboxylic acid or amine functionality was demonstrated to occur without degradation of the polymer backbone.39
Of these monomers, the serine-derived cyclic diesters have received the most attention. Homo- and copolymerizations of 4 and 5 have been attempted with a range of catalysts including Sn(Oct)2 in bulk at 110 °C and 130 °C and an ethylzinc phenolate ((2-((dimethylamino)methyl)-4,6-dimethylphenoxy)(ethyl)zinc), 8, in solution at 35 °C. While homopolymerization under melt conditions with Sn(Oct)2 led to relatively low molecular weight polymers, ROP mediated by 8 resulted in high molecular weight poly(ester)s (Mn = 38000 g mol−1, PDI = 1.7). Notably, the Tg values of the polymers ranged from 15 to 30 °C depending on the polymer molecular weight. Both random and block copolymers have been prepared with L-lactide.40 Bulk copolymerization of 3 with L-lactide (95 mol%) using Sn(Oct)2 at 140 °C realized poly(ester)s with molecular weights up to 7.7 × 104 g mol−1 with a Tg of 56 °C. Deprotection of the homo- or copolymers containing 3 and 4via hydrogenation over either Pd/C (10%) or Pd(OH)2 gave the hydroxyl functionalized poly(ester)s with no significant change in Tg and without any chain scission.41 The deprotected poly((S)-3-(benzyloxymethyl)-1,4-dioxane-2,5-dione) (poly(4)) was semicrystalline with a Tg of −4 °C and a Tm of 135 °C while the deprotected poly((S)-6-methyl-3-(benzyloxymethyl)-1,4-dioxane-2,5-dione) (poly(5)) was an amorphous polymer with a Tg of 30 °C.40 Degradations of the deprotected poly(5) homo- and copolymers have also been studied. The deprotected poly(5-co-εCL) showed a decrease in degradation times with increasing L-serine monomer content, consistent with an increased hydrophilicity of the polymer. Poly(5) was observed to be completely degraded after 1 day; increasing the L-lactide content to 25, 50 and 75 mol% resulted in degradations requiring ∼1 week, ∼1 month and ∼2 months respectively.42 Additional modification of the deprotected poly(3) has been achieved through treatment with succinic anhydride to provide pendant carboxylic acid groups. Attachment of an amine-substituted biotin derivative and a RGD-containing peptide (GGRGDSPGGK) conjugated to a fluorescein derivative (FITC) via a dicyclohexylcarbodiimide (DCC) coupling led to poly(ester) films with increased epithelial cell adhesion performance compared to their respective unfunctionalized copolymer films.41
Random copolymers (50 : 50) of (S)-3-(benzyloxymethyl)-1,4-dioxane-2,5-dione, 4, with ε-caprolactone (εCL) were also prepared with Sn(Oct)2 at 110 °C, 130 °C and 150 °C resulting in amorphous copolymers with Tg values of −16, −29 and −28 °C respectively; εCL was chosen for its slow degradation rate, low Tg and crystallinity. Deprotection of the copolymers resulted in a subtle increase in the Tg values. In an attempt to prepare more crystalline materials, a triblock copolymer was synthesized through initiation of 4 from a telechelic poly(ε-caprolactone) (PCL) macroinitiator. The protected triblock copolymers were amorphous with Tg values ranging from −44 to −10 °C, however, upon deprotection the triblock copolymers showed phase separation and were semicrystalline with the PCL segments crystallizing with Tm values ranging from 39 to 46 °C.43 Such melting temperatures just above body temperature enabled their investigation as materials for stable scaffolds in tissue engineering. The wettability of these poly(ester)s was shown to be tunable by the percentage composition of 4 in the copolymer with increasing content resulting in an increase in hydrophilicity. The increased hydrophilicity resulted in an improved adherence of human mesenchymal stem cells (hMSCs) onto the polymeric surface with survival of the cells along with the ability to differentiate towards osteogenic lineage on the poly(4-b-PCL) surfaces.44
δ-Gluconolactone, a naturally occurring lactone derived from glucose, has also been applied in the synthesis of poly(ester)s via ROP. The synthesis of 3-(1,2:3,4-tetraoxobutyl-di-O-isopropylidene)- (DIPAGYL, 9) and 3-methyl-6-(1,2:3,4-tetraoxobutyl-di-O-isopropylidene)-1,4-dioxane-2,5-diones (DIPALYL, 10) has been achieved by ring-opening and protection of δ-gluconolactone with dimethoxypropane–methanol to yield an isopropylidene protected gluconic acid methyl ester. Hydrolysis of the methyl ester resulted in 3,4:5,6-di-O-isopropylidenegluconic acid that after addition to either bromoacetyl chloride or 2-bromopropanoyl chloride could be cyclized with NaHCO3 to yield DIPAGYL or DIPALYL in 30 and 49% yields over the final two steps respectively (Scheme 3).45
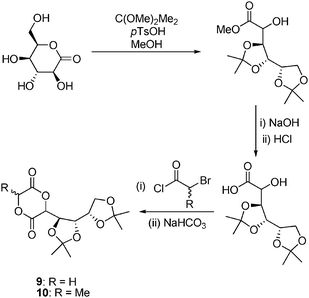 | ||
| Scheme 3 Synthesis of 3-(1,2:3,4-tetraoxobutyl-di-O-isopropylidene)- (DIPAGYL, 9) and 3-methyl-6-(1,2:3,4-tetraoxobutyl-di-O-isopropylidene)-1,4-dioxane-2,5-diones (DIPALYL, 10) from δ-gluconolactone. | ||
Bulk homopolymerization of DIPAGYL (9) with Sn(Oct)2 at 120 °C resulted in high molecular weight (Mwca. 20000 g mol−1) poly(DIPAGYL) as a brittle amorphous poly(ester) with a Tg ≈ 95 °C. Homopolymerization of DIPALYL (10), however, only resulted in oligomers with Mw ≈ 2000 g mol−1.45 Bulk copolymerization of DIPAGYL with both L- and rac-LA has been reported. Copolymerization of DIPAGYL with rac-LA (70 mol%) resulted in an amorphous copolymer with a Tg of 73 °C, incorporation of 17 mol% DIPALYL yielded a polymer with a Tg of 58 °C.46 The Tg values of the copolymers increased with increasing DIPAGYL content ranging from 61 to 77 °C while copolymers containing less than 90% lactide were amorphous. Deprotection of the isopropylidene groups to reveal hydroxyl groups proved difficult such that under optimized conditions using iodine in methanol or acetic acid, only ca. 60% of the protecting groups were cleaved, with more facile and complete cleavage being observed at the 5–6 position; only partial deprotection occurred at the 3–4 position along with partial degradation of the aliphatic poly(ester).45,47 Deprotection of poly(DIPAGYL-co-LLA) required treatment with trifluoroacetic acid (TFA) realizing copolymers with various degrees of hydroxylation with the Tg values of the deprotected copolymers increasing from 74 to 93 °C with rising number of hydroxyl groups.47 Further modification of the polymer backbones was demonstrated by coupling naphthoyl chloride to the exposed hydroxyl groups at the 6-position on a deprotected poly(DIPAGYL-co-DLLA) with conformation by SEC showing the binding of the fluorescent label.48
Recently Bourissou and co-workers have demonstrated that the synthesis and ROP of O-carboxyanhydrides (OCAs) (Scheme 4) provide facile access to functional poly(glycolide) derivatives.49,50 ROP mediated by 4-dimethylaminopyridine (DMAP) is entropically driven by loss of CO2 rather than enthalpically driven through release of ring strain and hence the thermodynamic effects of ring-substitution are lessened. Synthesis of an OCA derived from benzyl protected glutamic acid (L-gluOCA, 11) has been achieved by the diazotization of commercially available benzyl protected L-glutamic acid with NaNO2 in aqueous AcOH to afford the respective α-hydroxy acid. After conversion to the dicyclohexylamine salt, cyclization with diphosgene in the presence of polystyrene-supported diisopropylethylamine (PS-DIEA) resulted in the isolation of 11 in a 30% yield from O-benzyl-L-glutamic acid. Homopolymerization of L-gluOCA was achieved with DMAP at 25 °C within 5 min for a targeted [M]/[I] of 50 while maintaining excellent control over the polymerization (Mn = 6300 g mol−1, PDI = 1.18). Subsequent acetylation of the hydroxy end-group with acetic anhydride enabled the successful hydrogenolysis with Pd/C of the benzyl groups without any chain scission of the poly(ester) occurring. Block and statistical copolymerizations with L-lacOCA, an OCA derived from L-lithium lactate via a similar procedure, were also successful.
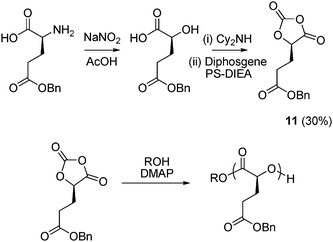 | ||
| Scheme 4 Synthesis and ring-opening polymerization of O-carboxyanhydride monomer derived from benzyl protected glutamic acid (L-gluOCA, 11), overall yield in parenthesis. | ||
ε- and δ-Lactones
Very few examples of the derivation of ε- or δ-lactones from renewable resources have been reported. (−)-Menthide, 12, prepared from the simple mCPBA oxidation of the natural product (−)-menthone (74% yield) is one of the few examples of functional εCLs derived from renewable resources.51 Homopolymerization of (−)-menthide was carried out with a discrete zinc alkoxide catalyst at 25 °C under an inert atmosphere yielding an amorphous high molecular weight polymer with a Tg of −25 °C. Further work has focused on the synthesis of telechelic ABA triblock copolymers with lactide to alter the mechanical properties of PLA.51–53 Hydrolytic degradation of poly((−)-menthide) at 37 °C revealed a slower degradation than PLA with little mass loss even after 48 weeks.54 (+)-Dihydrocarvone has also been applied in the synthesis of 7-methyl-4-(2-methyloxiran-2-yl)oxepan-2-one, 13 (Scheme 5).55 Oxidation of the natural product with mCPBA resulted in both ring-expansion of the 6-membered ring and epoxidation of the pendant propenyl group resulting in an epoxide-functional εCL, 13, in 25% yield. Homopolymerization of 13 was carried out with ZnEt2 and Sn(Oct)2 at temperatures ranging from 20 to 120 °C resulting in both lactone and epoxide ring-opening to yield low molecular weight branched oligomers. Copolymerization with εCL and δVL by ZnEt2 and Sn(Oct)2 at 60 and 120 °C also resulted in branched polymers with increasing Tg and decreasing Tm with increasing 13 content. Copolymers containing less than 10% incorporation of 13 exhibited excellent shape memory behavior even after repeated bending.55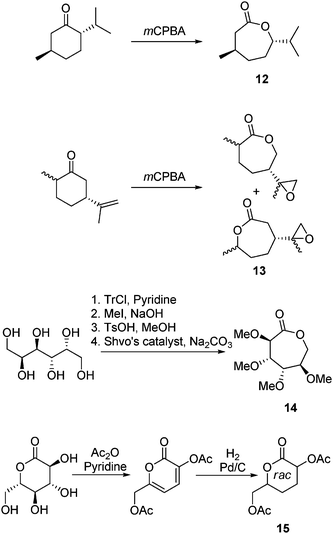 | ||
| Scheme 5 Synthesis of (−)-menthide, 12, from (−)-menthone, 7-methyl-4-(2-methyloxiran-2-yl)oxepan-2-one, 13, from (+)-dihydrocarvone, 2,3,4,5-tetra-O-methyl-D-glucono-1,6-lactone, 14, from D-dulcitol and carbohydrate δ-lactone, 15, from D-gluconolactone. | ||
The ε-lactone, 2,3,4,5-tetra-O-methyl-D-glucono-1,6-lactone, 14, has also been synthesized and studied as a monomer in ROP. Synthesis of 14 has been reported by three routes. In the first reported synthesis, protection of the primary alcohol group of methyl α-D-glucopyranoside with trityl chloride was followed by the subsequent protection of the secondary alcohol groups with methyl iodide. Removal of the 6-O-triphenyl group by acidic hydrolysis was then followed by protection with benzyl bromide. Further acid hydrolysis with acetic anhydride and subsequent oxidation using dimethyl sulfoxide gave 6-O-benzyl-2,3,4-tri-O-methyl-D-glucono-1,5-lactone. Ring-opening of this δ-lactone with methyl iodide and potassium hydroxide, removal of the benzyl group and lactonization with dicyclohexyldiimide (DCC) and 4-dimethylaminopyridine (DMAP) yielded 14 in a 21% overall yield.56 Alternatively this monomer has been prepared from D-glucose diethylmercaptal by first protecting the primary and secondary alcohols with trityl chloride and methyl iodide respectively before removal of the diethylmercaptal protecting group and oxidation of the resulting aldehyde with Hg(ClO4)2 and pyridinium dichromate (PDC) respectively. The triphenylmethyl protected alcohols of the resultant 6-O-triphenylmethyl-2,3,4,5-tetra-O-methyl-D-gluconic acid were liberated in acidic conditions before lactonization with DCC and DMAP yielded 14 (37% overall yield).56 The final reported synthetic route for this monomer applies the commercially available reduced sugar D-dulcitol as its starting material. Initial protection of the primary and secondary alcohols with trityl chloride and methyl iodide respectively is followed by removal of the trityl groups under acidic conditions with subsequent cyclization by oxidation using Shvo's catalyst (60% final step).57 The monomer was initially copolymerized with L-lactide (∼16 mol%) in bulk using Sn(Oct)2 at 110 °C. The resultant copolymers contained differing amounts of carbohydrate monomer with the highest incorporation being 2.2% 14 (Mn = 14900 g mol−1 and PDI = 1.2).56 Further screening of catalysts for the ROP of 14 resulted in the discovery that Y(OiPr)3 was able to efficiently catalyze its ROP at 25 °C in a living manner to obtain amorphous poly(14) displaying a Tg of 52 °C. Block copolymerizations with εCL were also successful. Surface plasmon resonance (SPR) sensograms demonstrated that both the homo- and block copolymers exhibit excellent resistance to fibrinogen and lysozyme and therefore could be extended to use in biomaterials applications.57
Recently, Williams and co-workers have reported the synthesis and ROP of a δ-lactone derived directly from D-gluconolactone.58 The carbohydrate δ-lactone, 15, bearing acetyl protected hydroxyl groups at the α- and δ-positions was prepared in good yield by treatment of D-gluconolactone with acetic anhydride and pyridine resulting in isolation of 3-acetoxy-6-acetoxymethyl-pyran-2-one that upon hydrogenolysis with Pd/C yielded 15 as a racemic mixture in 90% overall yield (Scheme 5). Despite the steric hindrance around the ester, ROP of 15 was achieved with Sn(OBu)2 at 80 °C in toluene realizing amorphous poly(ester)s with modest molecular weights (Mn ranging from 1800–7300 g mol−1) and a Tg of 18 °C; higher activities were realized using a well-defined zinc-ethyl initiator in toluene at 25 °C. The absence of end-group resonances in NMR spectra and MALDI-ToF analysis led to the determination of the major product as cyclic polymers that was attributed to be a result of the rate of propagation and transesterification being comparable as a consequence of the low ring strain of the monomer.
β-Lactones
The conceptually facile ring-closure of β-hydroxyl acids such as malic acid (or thereby derived by the diazotization of aspartic acid derivatives) has led to a great deal of work being reported in the synthesis and ROP of β-lactones from renewable resources. Several methods exist for the derivatization of the α-carboxylic acid group and generally involve the formation of a 5-membered cyclic anhydride that can be selectively ring-opened to form an ester at the α-carboxylate thus enabling cyclization to the propiolactone via the β-hydroxy acid ranging in yields from 5–44% (Scheme 6). Some initial syntheses reported the protection of both carboxylic acid groups with benzyl alcohol by mesylation of the free hydroxyl group using mesyl chloride. The subsequent removal of the benzyl groups, cyclization with trifluoroacetic anhydride (TFAA) and reaction of the pendant β-carboxylic acid with an alcohol affords the desired product. An improvement on this route involves the formation of a cyclic anhydride species from L-malic acid with TFAA and subsequent ring-opening with an alcohol resulting in the respective β-hydroxy acid that can be ring closed by treatment with diisopropylazodicarboxylate (DIAD) and triphenylphosphine. Monomers synthesized from aspartic acid have also been realized via a β-bromo acid synthesized by the diazotization of the amino acid with NaBr and NaNO2 that can be cyclized with sodium hydroxide. In this manner a range of β-malolactonate monomers have been prepared including benzyl (MLABz, 16),59–64 benzyloxyethanol (MABE, 17),65 allyl (MLAAllyl, 18),66 benzyloxypropyl,67 dodecyl,68 butyl69 and hexyl66 amongst others.70,71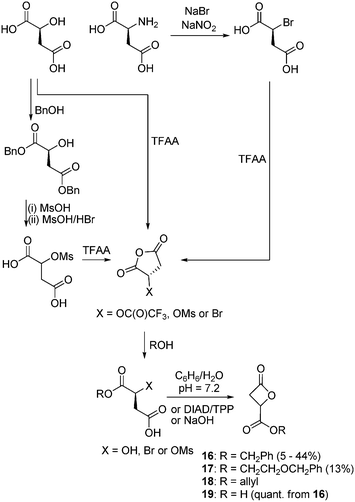 | ||
| Scheme 6 General routes for the synthesis of β-malolactones from L-aspartic acid or L-malic acid. | ||
Benzyl β-malolactone (MLABz, 16) has seen much attention providing a controlled route into highly desirable poly(β-malic acid) (PMA). Early work in which the ROP of MLABz, 16, was preformed with NEt3 at 70 °C yielded only low molecular weight poly(benzyl β-malolactone) (PMLABz) with Mn ≈ 6000 g mol−1.62 Preparation of high molecular weight PMLABz with reproducible results was achieved by extensive purification of MLABz before polymerization with tetraethylbenzoate at 37 °C realizing PMLABz's (Mn > 170000 g mol−1) close to theoretical molecular weights.60 Conversion of PMLABz to PMA was carried out by catalytic hydrogenolysis with Pd/C at 25 °C yielding water-soluble poly(ester)s.60,62 Additionally, partial hydrogenolysis of PMLABz was achieved by controlling the reaction time and conditions to yield copolymers of MLABz and malic acid (MA) with non-random distribution throughout the polymer forming “blocky” copolymers.61 Oligomer formation during the degradation of PMA was monitored and quantified by aqueous size-exclusion chromatography (SEC) and high performance capillary electrophoresis (HPCE) respectively. The degradation rate was found to increase with increasing acid group content in the poly(ester).59 Poly((R,S)-benzyloxyethyl-β-malolactonate) has been prepared by the ROP of MABE (17) using Sn(Oct)2 at 130 °C. Upon hydrogenolysis of the polymer amorphous copolymers with a pendant hydroxyethyl group are realized. These groups increase the hydrophilicity while maintaining relatively long-term degradation stability compared to carboxylic acid functionalized poly(ester)s.65
Statistical and block copolymers of MLABz with L-lactide and εCL have also been prepared.63,64,72 Copolymerization with L-lactide by Sn(Oct)2 at 110 °C and subsequent removal of the benzyl ester through hydrogenolysis resulted in PLA copolymers bearing pendant carboxylic acid groups; the composition was easily controlled by adjusting the MLABz : LLA ratio. The morphology of poly(MLABz-co-LLA) changed from crystalline to amorphous with increasing MLABz content (8 to 41 mol%) with decreases in Tg values from 59 to 45 °C. Tg values of poly(MLA-co-LLA) were higher than those of the parent protected copolymers that increased with increasing MA content.63 These correlations between MLABz/MLA content and Tg values were further observed in both analogously prepared poly(MLABz-co-DLLA) and poly(MLA-co-DLLA).64
The pendant carboxylic acid group of malolactonic acid, MLA, 19 (Scheme 6), has also been applied to produce further functional polymers. Hydroxy-terminated PLA,66 low molecular weight PCL–OH chain (5 repeat units)73 and cholesterol66 have been grafted to MLA by coupling in the presence of DCC. Polymerization of these monomers was achieved with either tetraethylammonium benzoate or potassium 11-hydroxydodecanoate at 0 °C.
L-Serine has also been applied in the synthesis of functional β-lactones. In an initial report, an N-tritylated-L-serine-βPL (20) was synthesized by tritylation of the amine with triphenylchloromethane followed by cyclization with DCC and DMAP resulting in the N-tritylated L-serine βPL (Scheme 7).74 Homopolymerization of 20 was studied using tetrabutylammonium acetate at 80 °C, 110 °C and 130 °C and resulted in the isolation of poly(N-tritylated L-serine) with narrow polydispersities between 1.2 and 1.5. Deprotection of the trityl group was possible with trifluoroacetic acid, however, some degradation of the polymer backbone was observed. In a further study a functional βPL was derived from L-serine via cyclization of the commercially available N-(benzyloxycarbonyl)-D-serine (N-Z-L-serine) with N-phosphonium adduct of Ph3P and dimethyl azodicarboxylate (DMAD) to yield N-(benzyloxycarbonyl)-L-serine-βPL (N-Z-L-serine-βPL, 21) (Scheme 7). The thermal initiated melt polymerization of 21 at 135 °C resulted in the formation of low molecular weight oligomers along with thermal degradation of the monomer. Solution polymerization at 30 °C with tetraethylammonium benzoate and potassium acetate–dicyclohexyl-18-crown-6 ether resulted in the synthesis of reproducible high molecular weight poly(N-Z-L-serine-βPL)s.75 Homopolymerization of N-Z-L-serine-βPL has also been achieved with mesyl chloride at 25 °C and Na2CO3 at 20 °C, however, no relationship between experimental conditions and molecular weight was observed and polymers with Mnca. 20000 g mol−1 were isolated.76 Deprotection of poly(N-Z-L-serine-βPL) via catalytic hydrogenolysis with formic acid yielded the corresponding poly(L-serine ester).75 Improved deprotection was achieved with HBr–AcOH mixtures with combination of deprotected poly(N-Z-L-serine-βPL) bromide salt with poly(β-malic acid) and/or its respective sodium salt formed polyelectrolyte complexes.76 Copolymerization of N-Z-L-serine-βPL with N-(tert-butyloxycarbonyl)-L-serine-β-lactone (N-boc-L-serine-βPL, 22) at 20 °C realized copolymers with molecular weights of 40000 g mol−1.75
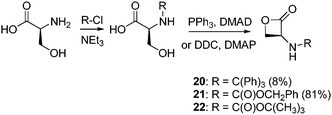 | ||
| Scheme 7 Synthesis of β-malolactones from L-serine (reported overall yields in parentheses). | ||
Click-functional polyesters
A great deal of research has focused on the introduction of side-chain functionality into poly(ester)s. Recently, the application of click chemistry within polymer chemistry has proved to be an effective tool for the preparation of libraries of polymers from universal prepolymers.77 The click concept relies on the application of highly efficient reactions that occur stoichiometrically and do not require laborious purification techniques such as column chromatography. The most commonly known example of this type of chemistry is the Cu(I) catalyzed Huisgen-1,3-dipolar cycloaddition between an azide and a terminal alkyne.78 The family of reactions are wide ranging, however, and include Diels–Alder reactions, ring-opening of epoxides by amines, reaction of aminooxy compounds with aldehydes or ketones and a range of so-called ‘thiol–ene’ reactions whereby the reaction of a sulfhydryl compound with an activated alkene occurs via either an anionic chain, Michael addition or radical mechanism.79,80 Given the tailor-made nature of many cyclic ester monomers, click chemistry approaches are especially attractive in poly(ester) synthesis.Cyclic diesters
The synthesis of an allyl functionlized aliphatic poly(ester) has been realized from the synthesis and ROP of 3-allyl-1,4-dioxane-2,5-dione (allylglycolide, 23). A simple procedure from glyoxylic acid monohydrate involving a Barbier-type addition of allyl bromide yielded allylglycolic acid that was converted to allylglycolide via coupling with bromoacetyl bromide in the presence of NEt3 and subsequent cyclization with Na2CO3 in 43% overall yield (Scheme 8). Bulk homo- and copolymerization with L-lactide (25, 50 and 75%) by Sn(Oct)2 at 110 °C resulted in amorphous poly(ester)s with Tg values of 14 °C for poly(allylglycolide) and ranging from 19 to 42 °C for poly(allylglycolide-co-LLA) with increasing L-LA content. Modification of the allyl groups was investigated by oxidation with m-chloroperoxy benzoic acid (mCPBA) to reveal synthetically versatile epoxide groups; these amorphous polymers displayed Tg values higher than the parent allyl polymers.81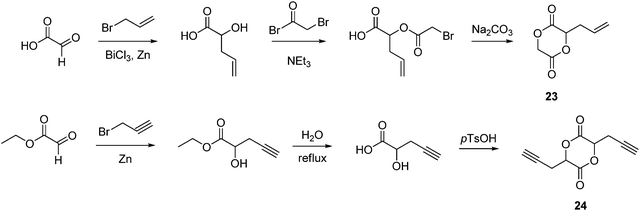
In a comparable synthetic procedure, 3,6-di-2-propynyl-1,4-dioxane-2,5-dione (dipropargyl glycolide, 24) has been synthesized from ethyl glyoxylate.82 Coupling of propargyl bromide via a Reformatsky-type reaction with subsequent hydrolysis in water provided propargylglycolic acid that was cyclized with p-toluenesulfonic acid in refluxing toluene (Scheme 8) to afford dipropargyl glycolide, 24, in 15% overall yield. Bulk homopolymerizations and copolymerizations with rac-lactide were performed with Sn(Oct)2 at 130 °C yielding poly(propargyl glycolide) (PPGL). Pendant alkyne groups enabled the quantitative attachment of both a polyethyleneglycol-550 monomethyl ether azide (mPEG-550 azide) and 1-azidodecane via Cu(I)-catalyzed Huisgen-1,3-dipolar cycloaddition click reactions (CuSO4–sodium ascorbate). Importantly no polymer degradation was observed. Furthermore, grafting mixtures of 10-azido-2,5,8-trioxadecane (mDEG) and 1-azidodecane provided water-soluble polymers that demonstrated lower critical solution temperature (LCST) behavior. Simple and precise adjustment of the cloud point temperature between 25 and 65 °C was possible by varying the mole fraction of mDEG and 1-azidodecane.82
ε-Lactones
Azide-functional poly(ε-caprolactone) copolymers have been prepared by both the direct polymerization of an azide containing monomer and the post-polymerization modification of chloro- and bromo-containing PCLs (Fig. 2).83–85 The synthesis of α-azide-εCL, 25, was realized in a quantitative yield from the reaction of NaN3 with α-chloro-εCL, 26, that in turn was prepared by oxidation of 2-chlorohexanone with mCPBA in 85% yield.83 ROP of both of these monomers was achieved using 2,2-dibutyl-2-stanna-1,3-dioxepane (DSDOP). The Tg values of the copolymers were dependent on the α-azide-εCL content with values between −60 °C for PCL and −43 °C for poly(α-azide-εCL). Treatment of these azide-functional polymers with a range of terminal alkyne-containing compounds in the presence of DBU and CuI led to functionalization of the polymers without degradation being observed. In this manner a range of alkynes have also been attached to poly(α-azide-εCL-co-εCL) to moderate the polymer properties and uses including propargyl benzoate, 3-amino-dimethyl-1-propyne and N,N,N-triethylpropargyl ammonium, but-3-yn-1-ol, and propargyl acrylate to enable UV crosslinking, propargyl bromoisobutyrate to enable grafting with poly(styrene) via ATRP and α-methoxy-ω-alkyne-PEG synthesized from PEG and 4-pentynoic acid. The one-pot synthesis of functional PCLs has also been realized by reaction of poly(αClεCL-co-εCL) and NaN3 with subsequent addition of 3-(dimethylamino)-1-propyne in the presence of CuI.83,84The synthesis of 4-(acryloyloxy)-εCL (ACL, 27) has been reported in yields ranging from 24–36% by coupling of cyclohexane-1,4-diol with acryloyl chloride and subsequent treatment with pyridinium chlorochromate (PCC) and mCPBA.86 ACL has also been prepared directly from 4-hydroxy-cyclohexanone, avoiding the need for the oxidation of the hydroxyl group.87 The methacrylic derivative, γ-methacryloyloxy-εCL, 28, has also been synthesized using methacryloyl chloride in an analogous fashion.87 ACL has been selectively polymerized through both atom transfer radical polymerization (ATRP) using NiBr2(PPh)3 at 90 °C to realize a poly(acrylate) with pendant εCL units and also via ROP with Al(OiPr)3 at 25 °C realizing amorphous polymers with Tg values of 95 and −60 °C respectively. Notably, the bulk copolymerization with εCL by Sn(Oct)2 at 110 °C proceeded with no cross-linking of the pendant acrylate groups despite the high polymerization temperatures.86 The pendant acrylate functionalities in poly(ACL-co-εCL) have been utilized in further pyridine-catalyzed Michael additions. Mercaptoacetic acid and an oligomeric thiol, α-methoxy-ω-mercapto-poly(ethylene glycol) (PEG-SH) have been conjugated to the poly(ACL-co-εCL) at ambient temperature to yield functional PCLs without any polymer degradation.88
Wooley and co-workers have studied aminoxy/ketone reaction for the synthesis and intramolecular crosslinking of εCL copolymers from polymers prepared by the copolymerization of γ-ketone-εCL (KCL, 29, Fig. 2) and εCL using Al(OiPr) at 25 °C.89–91 In early attempts, reductive amination of a poly(γ-ketone-εCL-co-εCL) (poly(KCL-co-CL)) with primary diamines to afford covalently cross-linked polyester materials was unsuccessful due to side reactions that resulted in chain scission and rearrangement products through intramolecular lactamization.91 The application of 1,6-bis(aminooxy)hexane afforded an improved methodology for these transformations such that cross-linking of the poly(KCL-co-εCL) catalyzed with pTsOH was successful resulting in an insoluble cross-linked PCL gel observing melting temperatures lower than their polymer precursors.90 Applying these conditions, a mixture of O-dodecylhydroxylamine, O-benzylhydroxylamine and dansyl hydrazine, a sulfonylhydrazine-terminated fluorophore, have been grafted to poly(KCL-co-εCL) via both sequential and single-step processes without degradation of the polymer backbone. Dansyl hydrazine resulted in considerable deviations in the product composition relative to the feed stoichiometries resulting from the reduced nucleophilicity along with the differences in relative stabilities of the sulfonyl hydrazone and ketoxime ether linkages under the acidic conditions. Application of a single-step, one-pot strategy for this grafting was also successful with varying mixtures of the three compounds leading to the simple preparation of multifunctional polyesters.89
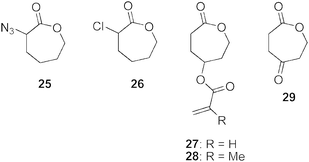 | ||
| Fig. 2 Click-functional ε-caprolactone monomers. | ||
δ- and β-Lactones
A general synthetic method for the functionalization of δVLs relies on the increased acidity of the α-methylene group on δVL such that lithiation with lithium diisopropylamide (LDA) and subsequent quenching with an alkyl halide in hexamethylphosphoramide (HMPA) yields the desired functional δVL in 40–75% yields (Scheme 9). An early example of this was the synthesis of α-methyl-δVL in which the excipient enolate was quenched with methyl iodide at −40 °C.92 Homopolymerization of this monomer has only been achieved in the bulk under argon with a lipase catalyst derived from Candida antarctica at 35 °C obtaining a Mn of 8500 g mol−1 and PDI of 2.1.93 More recently α-allyl-δVL, 30, was synthesized in an analogous manner from δVL and allyl bromide.94 Polymerizations performed with Sn(OTf)2 at 25 °C resulted in an amorphous poly(α-allyl-δVL) whereas the respective poly(α-allyl-δVL-co-εCL) and poly(α-allyl-δVL-co-δVL) copolymers showed decreasing Tm values correlating with increased α-allyl-δVL incorporation; no melting transitions were observed at >25% and >15% incorporation in εCL and δVL copolymers respectively. Dihydroxylation of poly(α-allyl-δVL-co-εCL) with OsO4 resulted in complete conversion of the allyl groups to hydroxyalkyl groups without polymer degradation.94 The allyl groups have been applied in radical thiol–ene couplings within nanoparticles and have been converted to epoxides by treatment of the α-allyl-δVL with mCPBA that have been subsequently applied in the synthesis of poly(ester) nanoparticles by reaction with diamines under dilute conditions.95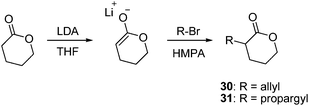 | ||
| Scheme 9 Synthesis of allyl (30) and propargyl (31) functional δ-valerolactone monomers. | ||
In a further extension of this chemistry, an alkyne-functional δVL has also been synthesized in analogous fashion using propargyl bromide to yield α-propargyl-δVL, 31.96 Homopolymerization with Sn(OTf)2 at 25 °C led to the synthesis of well controlled polymers and copolymerization with εCL enabled control over pendant alkyne density. Application of these alkyne-functional polymers in click reactions with α-monomethyl ether-ω-azidoPEG1100 in the presence of CuSO4 and sodium ascorbate at 80 °C in water resulted in grafting to the alkyne functionalized poly(ester) without degradation of the poly(ester). The grafted amphiphilic copolymers were crystalline with a Tm of 32 °C and were further shown to be biocompatible. An azide-terminated oligopeptide was also grafted onto the alkyne-functional copolymers under similar conditions with slightly elevated temperatures (100 °C) without poly(ester) degradation.96 In a further study, an azide-functionalized camptothecin was grafted to poly(α-propargyl-δVL-co-εCL). As a consequence of the poor aqueous solubility of the functionalized camptothecin, this was performed using bromotris(triphenylphosphine)copper(I)N,N-diisopropylethylamine in dichloromethane. The hydrophobic polymer–drug conjugates can be used to prepare microparticles, further grafting of the α-monomethyl ether-ω-azidoPEG1100 resulted in a highly water-soluble poly(ester)–camptothecin conjugate.97 Finally, phosphorylcholine (PC) moieties have been grafted to poly(α-propargyl-δVL) by further click reactions using CuSO4 and sodium ascorbate at 70 °C under constant microwave radiation. PC-azide reaction with the terpolymers of poly(α-propargyl-δVL) with εCL and L-lactide required the application of CuBr–PMDETA at 35 °C to achieve complete grafting of the PC-azide. The resulting poly(ester)s demonstrated good cell viability suggesting their usefulness for integration into medical devices, biomaterials, and drug delivery vehicles.98
While no specific reports of the application of click chemistries with poly(ester)s prepared from β-lactones have been detailed, poly(MLAAllyl-co-MLABz) has been prepared and the pendant allyl groups converted to hydroxyl groups by a radical reaction with mercaptoethanol in the presence of AIBN at 70 °C. This modification enabled grafting of εCL from the backbone mediated by AlEt3 at 25 °C. Subsequent hydrogenolysis of the MLABz components realized the amphiphilic poly(MLA-g-PCL).73 An allyl-functional βPL, 32, has also been prepared by the carbonylation of the 1,2-epoxy-5-hexene catalyzed by [(C6H5)3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ]2NCo(CO)4 and BF3·Et2O.99 Homo- and copolymerization of 32 with βBL by a discrete amino-alkoxybis(phenolate)yttrium amido complex at 20 °C yielded highly syndiotactic polymers. Poly(32) is an amorphous polyester with a Tg of −44 °C while increasing the content of 32 in the copolymer resulted in a decrease in both the Tm and Tg. Quantitative hydroxylation, dihydroxylation and epoxidation of the pendant allyl groups were performed with either pinacolborane in the presence of Wilkinson's catalyst (RhCl(PPh3)3), OsO4 and N-methylmorpholine-N-oxide (NMO) or mCPBA respectively without any polyester degradation.100
]2NCo(CO)4 and BF3·Et2O.99 Homo- and copolymerization of 32 with βBL by a discrete amino-alkoxybis(phenolate)yttrium amido complex at 20 °C yielded highly syndiotactic polymers. Poly(32) is an amorphous polyester with a Tg of −44 °C while increasing the content of 32 in the copolymer resulted in a decrease in both the Tm and Tg. Quantitative hydroxylation, dihydroxylation and epoxidation of the pendant allyl groups were performed with either pinacolborane in the presence of Wilkinson's catalyst (RhCl(PPh3)3), OsO4 and N-methylmorpholine-N-oxide (NMO) or mCPBA respectively without any polyester degradation.100
Poly(ester) nanoparticles
Given the attraction of using biocompatible, biodegradable materials in the biomedical field, poly(ester)s are attractive materials. As a consequence of the increased versatility and more facile syntheses of functional poly(ester)s, the realization of nanoparticles synthesized by controlled approaches to yield well-defined, narrowly polydisperse poly(ester) nanoparticles has been possible. Furthermore, the application of functional poly(ester)s has enabled the post-particle formation tuning of the particle properties and functionalities including controlling hydrophilicity and biotargeting/adhesion. Three main approaches for the synthesis of functional poly(ester) particles have been reported: intramolecular chain crosslinking, dilute solution self-assembly and nanoprecipitation particle formation. All three methods do not require additional surfactants or salts to enable particle formation and hence provide clean poly(ester) nanoparticles. Given the focus of this article on the application of functional poly(ester)s this aspect of the review will focus on the first two techniques. The nanoprecipitation method is a powerful technique for poly(ester) nanoparticle formation but has so far been limited to the formation of PLA,101,102 PLGA,24,103 PCL104 and poly(β-MA)105 nanoparticles but has great potential for the synthesis of functional poly(ester) nanoparticles.Dilute solution self-assembly
Self-assembly of block copolymers in solution relies on the use of non-covalent interactions to drive the process. In the case of amphiphilic block copolymers, this assembly is most commonly driven by the hydrophobic effect whereby the selective solvation of one polymer block in water drives the polymers to arrange in a manner that most efficiently reduces their free energy, i.e. whereby the water-soluble blocks are solvated in the aqueous phase and the hydrophobic blocks are protected in the core of the particle. The dimensions of the resultant self-assembled particles are largely controlled by the molecular weight of the polymer blocks whereas the mole fraction of the blocks has a significant effect on the morphology of the resultant particles.106Given the ready accessibility of water-soluble poly(ester)s by the ROP and deprotection of β-benzylmalolactonate (MLABz, 16) several studies have reported its application in block copolymers with hydrophobic poly(ester)s in self-assembly to form nanoparticles (Fig. 3). An ABA triblock copolymer of εCL and MLABz was prepared by initial homopolymerization of MLABz with potassium 11-hydroxydodecanoate at 0 °C with initiation from 18-crown-6 ether (HDD) affording PMLABz with α-hydroxyl and ω-carboxylic acid end-group. Reduction of the carboxylic acid end-group with a borane–tetrahydrofuran complex (BH3·THF) at 0 °C resulted in a macroinitiator used in the telechelic polymerization of εCL with AlEt3 at 25 °C realizing the triblock copolymer poly(εCL-b-MLABz-b-εCL). Hydrogenolysis of the MLABz groups realized the amphiphilic triblock copolymer poly(εCL-b-MLA-b-εCL) without poly(ester) degradation. UV spectroscopy with pyrene demonstrated that poly(εCL-b-MLA-b-εCL) formed ‘flower’ micelles in pure water.72 A range of amphiphilic block copolymers have been prepared from a hydrophobic malolactone derivative and MLABz that upon deprotection reveals a carboxylic acid functionality. The characteristics of the macromolecular micelles formed are dependent on block chain length and chemical structure of the hydrophobic block demonstrated.66,69
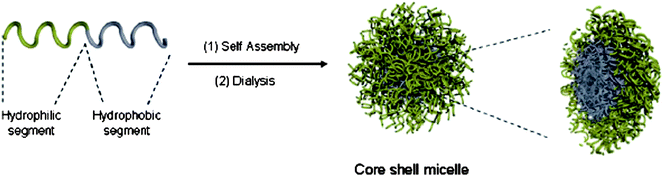 | ||
| Fig. 3 Dilute solution self-assembly of block copolymers into spherical micelles (reproduced by permission of The Royal Society of Chemistry).106 | ||
The copolymerization of (R,S)-4-benzyloxycarbonyl-3,3-dimethyl-2-oxetanone (dMMLABz) with β-butyrolactone (βBL) via metal-free catalysis with 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (triazole carbene) at 50 °C in the presence of t-BuOH enabled the synthesis of polymers with narrow polydispersities between 1.09 and 1.27 and excellent end-group control. The resulting α,ω-dihydroxy poly(dMMLABz-co-βBL) was used as a macroinitiator for the telechelic polymerization of L-lactide at 90 °C yielding α,ω-dihydroxy PLLA-b-poly(dMMLABz-co-βBL)-b-PLLA that was subjected to catalytic hydrogenolysis resulting in the respective amphiphilic triblock PLLA-b-poly(dMMLA-co-βBL)-b-PLLA. Self-assembly of the amphiphilic triblock poly(ester) at low temperatures (4 °C) resulted in ‘flower’ type micelles with microgelation occurring at 25 °C and dissolution of the microgel at 40 °C.107
Intramolecular chain crosslinking
This approach to nanoparticle synthesis relies on the intramolecular crosslinking of individual polymer chains and is achieved by the dropwise addition of a dilute polymer solution to a solution containing a crosslinking agent (if required). In this way, copolymers of 4-(acryloyloxy)-εCL (ACL, 27) and εCL prepared with Al(OiPr)3 at 25 °C were transformed into unimolecular particles in ultradilute conditions by self-crosslinking using the radical initiator 2,2-azo-bis-isobutyronitrile (AIBN) at 65 °C. A reduction in molecular weight (determined by size-exclusion chromatography) from 9900 to 9000 g mol−1 indicated a reduction in hydrodynamic volume of the polymers consistent with a contraction resulting from nanoparticle formation. Additionally the nanoparticles showed an increase in Tg from −62 °C in the copolymer to −45 °C.108Recently Harth and co-workers have described the copolymerization of δVL, α-allyl-δVL, α-propargyl-δVL and 2-oxepane-1,5-dione (ODP) using Sn(Oct)2 at 105 °C to realize multifunctional poly(ester) particles with a diverse range of pendant functionalities (Fig. 4). The pendant allyl groups were converted to epoxides with mCPBA and after subsequent addition to a refluxing DCM solution of 2,2′-(ethylenedioxy)bis(ethylamine) cross-linked amorphous nanoparticles were isolated. The nanoscopic size dimensions of the particles were dependent on the amount of diamine present during the cross-linking process and could be further tailored with higher incorporation of epoxides leading to larger particles.95 Utilization of the integrated functionalities in the nanoparticle was demonstrated by reductive amination of the keto groups introduced from ODP units with N-boc-ethylenediamine. Partial oxidation of the allyl groups before nanoparticle formation enabled the preparation of allyl functionalized polyester nanoparticles via a novel one-pot reaction with no significant change in dimensions. Conjugation of dye-labeled NHS ester Alexa Fluor® 594 to the free amine groups present from the cross-linking diamine enabled the monitoring of uptake and transport of these nanoparticles. Applying thiol–ene chemistry, a thiol functionalized dendritic molecular transporter and targeting peptides for radiated and non-radiated tumor vasculature, HVGGSSV and a novel CRGD, with incorporated cysteine residues were successfully attached to the allyl functionalized nanoparticles. Combination of this with reductive amination of the keto functionality enabled the preparation of conjugate materials including nanoparticle–peptide–dye (NP–P–dye/NP–P), nanoparticle–dendritic-molecular transporter–dye (NP–MT–dye) and nanoparticle–peptide–molecular transporter–dye (NP–P–MT–dye).109
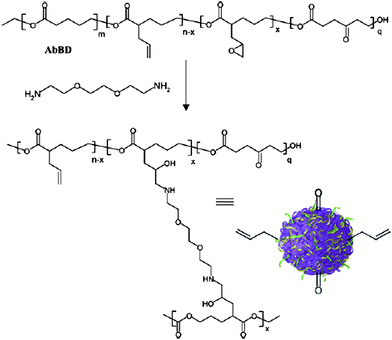 | ||
| Fig. 4 Nanoparticle formation from functional poly(ester) with residual alkene and alcohol groups for further functionalization (reproduced by permission of The Royal Society of Chemistry).109 | ||
Conclusions
The synthesis and ROP of functional cyclic esters provide a controlled route to the synthesis of biodegradable and biocompatible polymers with controllable physical and biological properties. The diversity of starting materials available from renewable resources and facile polymer modification strategies available via the application of click chemistries have enabled the realization of a multitude of poly(ester)s with controlled molecular weights, narrow PDIs and high end-group fidelities as well as access to block and statistical copoly(ester)s. While scope remains for further functionalization of poly(ester) backbones, the application of these polymers as biomaterials or in nanoscale self-assembly is opening up exciting new channels for investigation. To date the advances in functional poly(ester) synthesis have not been truly harnessed to influence nanoparticle properties. With the increasingly accessible range of functional poly(ester)s now reported, more advanced poly(ester) nanoparticles can be accessed and studied.References
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- A. U. Daniels, M. K. O. Chang, K. P. Andriano and J. Heller, J. Appl. Biomater., 1990, 1, 57–78 Search PubMed.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- M. I. Sabir, X. X. Xu and L. Li, J. Mater. Sci., 2009, 44, 5713–5724 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS.
- D. Bendix, Polym. Degrad. Stab., 1998, 59, 129–135 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Adv. Polym. Sci., 2002, 157, 1–40 CAS.
- C. Jerome and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef CAS.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- Y. R. Choi, Y. H. Bae and S. W. Kim, Macromolecules, 1998, 31, 8766–8774 CrossRef CAS.
- H. R. Kricheldorf and S. Eggerstedt, Macromol. Chem. Phys., 1999, 200, 587–593 CrossRef CAS.
- H. R. Kricheldorf and B. Fechner, Macromolecules, 2001, 34, 3517–3521 CrossRef CAS.
- H. R. Kricheldorf and B. Fechner, Biomacromolecules, 2002, 3, 691–695 CrossRef CAS.
- H. R. Kricheldorf and D. Langanke, Macromol. Chem. Phys., 1999, 200, 1174–1182 CrossRef CAS.
- M. Ryner, A. Finne, A. C. Albertsson and H. R. Kricheldorf, Macromolecules, 2001, 34, 7281–7287 CrossRef CAS.
- M. Ryner, A. Valdre and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2049–2054 CrossRef CAS.
- C. C. Chen, J. Y. Chueh, H. Tseng, H. M. Huang and S. Y. Lee, Biomaterials, 2003, 24, 1167–1173 CrossRef CAS.
- D. K. Gilding and A. M. Reed, Polymer, 1979, 20, 1459–1464 CrossRef CAS.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS.
- R. A. Miller, J. M. Brady and D. E. Cutright, J. Biomed. Mater. Res., 1977, 11, 711–719 CrossRef CAS.
- Y. Q. Shen, W. L. Sun, K. J. Zhu and Z. Q. Shen, J. Biomed. Mater. Res., 2000, 50, 528–535 CrossRef CAS.
- J. M. Vion, R. Jerome, P. Teyssie, M. Aubin and R. E. Prudhomme, Macromolecules, 1986, 19, 1828–1838 CrossRef CAS.
- M. Malin, M. Hiljanen, T. Karjalainen and J. Seppala, J. Appl. Polym. Sci., 1994, 52, 1327–1332 CrossRef.
- G. L. Baker, E. B. Vogel and M. R. Smith, Polym. Rev., 2008, 48, 64–84 Search PubMed.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 Search PubMed.
- D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775–794 CrossRef CAS.
- X. D. Lou, C. Detrembleur and R. Jerome, Macromol. Rapid Commun., 2003, 24, 161–172 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- Y. Kimura, K. Shirotani, H. Yamane and T. Kitao, Macromolecules, 1988, 21, 3338–3340 CrossRef CAS.
- Y. Kimura, K. Shirotani, H. Yamane and T. Kitao, Polymer, 1993, 34, 1741–1748 CrossRef CAS.
- J. Y. Lee, E. C. Cho and K. Cho, J. Controlled Release, 2004, 94, 323–335 CrossRef CAS.
- T. Yamaoka, Y. Hotta, K. Kobayashi and Y. Kimura, Int. J. Biol. Macromol., 1999, 25, 265–271 CrossRef CAS.
- T. Ouchi and A. Fujino, Makromol. Chem. Macromol. Chem. Phys., 1989, 190, 1523–1530 Search PubMed.
- W. W. Gerhardt, D. E. Noga, K. I. Hardcastle, A. J. Garcia, D. M. Collard and M. Weck, Biomacromolecules, 2006, 7, 1735–1742 CrossRef CAS.
- M. Leemhuis, C. F. van Nostrum, J. A. W. Kruijtzer, Z. Y. Zhong, M. R. ten Breteler, P. J. Dijkstra, J. Feijen and W. E. Hennink, Macromolecules, 2006, 39, 3500–3508 CrossRef CAS.
- D. E. Noga, T. A. Petrie, A. Kumar, M. Weck, A. J. Garcia and D. M. Collard, Biomacromolecules, 2008, 9, 2056–2062 CrossRef CAS.
- M. Leemhuis, J. A. W. Kruijtzer, C. F. van Nostrurn and W. E. Hennink, Biomacromolecules, 2007, 8, 2943–2949 CrossRef CAS.
- C. A. M. Loontjens, T. Vermonden, M. Leemhuis, M. J. van Steenbergen, C. F. van Nostrum and W. E. Hennink, Macromolecules, 2007, 40, 7208–7216 CrossRef CAS.
- H. Seyednejad, T. Vermonden, N. E. Fedorovich, R. van Eijk, M. J. van Steenbergen, W. J. A. Dhert, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3048–3054 CrossRef CAS.
- K. M. Benabdillah, J. Coudane, M. Boustta, R. Engel and M. Vert, Macromolecules, 1999, 32, 8774–8780 CrossRef CAS.
- K. M. Benabdillah, M. Boustta, J. Coudane and M. Vert, Biomacromolecules, 2001, 2, 1279–1284 CrossRef CAS.
- B. Saulnier, J. Coudane, H. Garreau and M. Vert, Polymer, 2006, 47, 1921–1929 CrossRef CAS.
- B. Saulnier, S. Ponsart, J. Coudane, H. Garreau and M. Vert, Macromol. Biosci., 2004, 4, 232–237 CrossRef CAS.
- O. T. du Boullay, C. Bonduelle, B. Martin-Vaca and D. Bourissou, Chem. Commun., 2008, 1786–1788 RSC.
- O. T. du Boullay, E. Marchal, B. Martin-Vaca, F. P. Cossio and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 16442–16443 CrossRef CAS.
- D. H. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS.
- C. L. Wanamaker, M. J. Bluemle, L. M. Pitet, L. E. O'Leary, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2904–2911 CrossRef CAS.
- C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS.
- C. L. Wanamaker, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 443–448 CrossRef CAS.
- J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef CAS.
- I. M. Pinilla, M. B. Martinez and J. A. Galbis, Carbohydr. Res., 2003, 338, 549–555 CrossRef CAS.
- H. Urakami and Z. Guan, Biomacromolecules, 2008, 9, 592–597 CrossRef CAS.
- M. Tang, A. J. P. White, M. M. Stevens and C. K. Williams, Chem. Commun., 2009, 941–943 RSC.
- C. Braud and M. Vert, Polym. Bull., 1992, 29, 177–183 CrossRef CAS.
- S. Cammas, I. Renard, V. Langlois and P. Guerin, Polymer, 1996, 37, 4215–4220 CrossRef CAS.
- A. Caron, C. Braud, C. Bunel and M. Vert, Polymer, 1990, 31, 1797–1802 CrossRef CAS.
- P. Guerin, M. Vert, C. Braud and R. W. Lenz, Polym. Bull., 1985, 14, 187–192 CrossRef CAS.
- B. He, J. Z. Bei and S. G. Wang, Polymer, 2003, 44, 989–994 CrossRef CAS.
- B. He, Y. F. Poon, J. Feng and M. B. Chan-Park, J. Biomed. Mater. Res., Part A, 2008, 87, 254–263 CrossRef.
- L. Wang, X. H. Jia, Y. S. Chen, Y. Z. Che and Z. Yuan, J. Biomed. Mater. Res., Part A, 2008, 87, 459–469 CrossRef.
- S. Cammas-Marion and P. Guerin, Macromol. Symp., 2000, 153, 167–186 CrossRef CAS.
- C. Barbaud, S. Cammas-Marion and P. Guerin, Polym. Bull., 1999, 43, 297–304 CrossRef CAS.
- C. Bouclier, L. Moine, H. Hillaireau, V. Marsaud, E. Connault, P. Opolon, P. Couvreur, E. Fattal and J. M. Renoir, Biomacromolecules, 2008, 9, 2881–2890 CrossRef CAS.
- S. Cammas-Marion, M. M. Bear, A. Harada, P. Guerin and K. Kataoka, Macromol. Chem. Phys., 2000, 201, 355–364 CrossRef CAS.
- M. A. LeboucherDurand, V. Langlois and P. Guerin, Polym. Bull., 1996, 36, 35–41 CrossRef CAS.
- R. Bizzarri, F. Chiellini, R. Solaro, E. Chiellini, S. Cammas-Marion and P. Guerin, Macromolecules, 2002, 35, 1215–1223 CrossRef CAS.
- O. Coulembier, P. Degee and P. Dubois, Macromol. Chem. Phys., 2006, 207, 484–491 CrossRef CAS.
- O. Coulembier, P. Degee, P. Gerbaux, P. Wantier, C. Barbaud, R. Flammang, P. Guerin and P. Dubois, Macromolecules, 2005, 38, 3141–3150 CrossRef CAS.
- I. Fietier, A. Leborgne and N. Spassky, Polym. Bull., 1990, 24, 349–353 CrossRef CAS.
- Q. X. Zhou and J. Kohn, Macromolecules, 1990, 23, 3399–3406 CrossRef CAS.
- H. Rossignol, M. Boustta and M. Vert, Int. J. Biol. Macromol., 1999, 25, 255–264 CrossRef CAS.
- M. G. Finn, H. C. Kolb, V. V. Fokin and K. B. Sharpless, Prog. Chem. (Beijing, China), 2008, 20, 1–4 Search PubMed.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- M. Leemhuis, N. Akeroyd, J. A. W. Kruijtzer, C. F. van Nostrum and W. E. Hennink, Eur. Polym. J., 2008, 44, 308–317 CrossRef CAS.
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS.
- R. Riva, P. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome and P. Lecomte, Chem. Commun., 2005, 5334–5336 RSC.
- R. Riva, S. Schmeits, C. Jerome, R. Jerome and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef CAS.
- N. Xu, R. Wang, F. S. Du and Z. C. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3583–3594 CrossRef CAS.
- D. Mecerreyes, J. Humes, R. D. Miller, J. L. Hedrick, C. Detrembleur, P. Lecomte, R. Jerome and J. San Roman, Macromol. Rapid Commun., 2000, 21, 779–784 CrossRef CAS.
- C. Vaida, P. Mela, H. Keul and M. Moller, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6789–6800 CrossRef CAS.
- J. Rieger, K. Van Butsele, P. Lecomte, C. Detrembleur, R. Jerome and C. Jerome, Chem. Commun., 2005, 274–276 RSC.
- B. A. Van Horn, R. K. Iha and K. L. Wooley, Macromolecules, 2008, 41, 1618–1626 CrossRef CAS.
- B. A. Van Horn and K. L. Wooley, Soft Matter, 2007, 3, 1032–1040 RSC.
- B. A. Van Horn and K. L. Wooley, Macromolecules, 2007, 40, 1480–1488 CrossRef.
- J. L. Herrmann and R. H. Schlessinger, J. Chem. Soc., Chem. Commun., 1973, 711–712 RSC.
- K. Kullmer, H. Kikuchi, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1998, 19, 127–130 CrossRef.
- B. Parrish, J. K. Quansah and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1983–1990 CrossRef CAS.
- A. E. van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS.
- B. Parrish and T. Emrick, Bioconjugate Chem., 2007, 18, 263–267 CrossRef CAS.
- B. M. Cooper, D. Chan-Seng, D. Samanta, X. F. Zhang, S. Parelkar and T. Emrick, Chem. Commun., 2009, 815–817 RSC.
- J. T. Lee, P. J. Thomas and H. Alper, J. Org. Chem., 2001, 66, 5424–5426 CrossRef CAS.
- N. Ajellal, C. M. Thomas and J. F. Carpentier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3177–3189 CrossRef CAS.
- S. S. Guterres, H. Fessi, G. Barratt, J. P. Devissaguet and F. Puisieux, Int. J. Pharm., 1995, 113, 57–63 CrossRef CAS.
- M. Chorny, I. Fishbein, H. D. Danenberg and G. Golomb, J. Controlled Release, 2002, 83, 389–400 CrossRef CAS.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS.
- J. Molpeceres, M. Guzman, M. R. Aberturas, M. Chacon and L. Berges, J. Pharm. Sci., 1996, 85, 206–213 CrossRef CAS.
- J. A. Portilla-Arias, M. Garcia-Alvarez, J. A. Galbis and S. Munoz-Guerra, Macromol. Biosci., 2008, 8, 551–559 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- O. Coulembier, L. Mespouille, J. L. Hedrick, R. M. Waymouth and P. Dubois, Macromolecules, 2006, 39, 4001–4008 CrossRef CAS.
- D. Mecerreyes, V. Lee, C. J. Hawker, J. L. Hedrick, A. Wursch, W. Volksen, T. Magbitang, E. Huang and R. D. Miller, Adv. Mater., 2001, 13, 204–208 CrossRef CAS.
- A. van der Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
| This journal is © The Royal Society of Chemistry 2010 |