Catalyzed chain growth (CCG) on a main group metal: an efficient tool to functionalize polyethylene
Jérôme
Mazzolini
,
Edgar
Espinosa
,
Franck
D'Agosto
* and
Christophe
Boisson
*
Université de Lyon, Univ. Lyon 1, CPE Lyon, CNRS UMR 5265, Laboratoire de Chimie Catalyse Polymères et Procédés (C2P2), Equipe LCPP Bat 308F, 43 Bd du 11 novembre 1918, F-69616, Villeurbanne, France. E-mail: dagosto@lcpp.cpe.fr; boisson@lcpp.cpe.fr; Fax: +33 4 72 43 17 68; Tel: +33 4 72 43 17 70
First published on 22nd January 2010
Abstract
The incorporation of functional groups at the end of polyolefin chains offers an opportunity to prepare polyolefin building blocks. The latter can be used to construct polymer architectures based on polyolefins with many desirable properties. For this purpose, the reactivity of the carbon-metal bond formed during a catalytic olefin polymerization process is particularly appealing. The possibility of taking advantage of this reactivity has indeed been enhanced by the discovery of systems in which fast and reversible chain transfer reactions between the active metal center and a main group metal centre are occurring. The recent developments of this catalyzed chain growth (CCG) concept are briefly reviewed. A specific system using a (C5Me5)2NdCl2Li(OEt2)2 complex in conjunction with n-butyloctylmagnesium is then employed to synthesize an array of end functional polyethylene chains. The potential of these building blocks to build up new macromolecular architectures is discussed.
 Jérôme Mazzolini | Jérôme Mazzolini was born in Nancy, France in 1984. He studied chemical engineering at the Ecole Supérieure de Chimie Physique Electronique de Lyon (CPE Lyon, France) from which he graduated in 2008. He completed his formation at the University of Lyon 1 with a Master of Science degree in polymer materials. He started his PhD in February 2009 under the supervision of Drs Christophe Boisson and Franck D'Agosto at C2P2 laboratory (Lyon, France). The focus of his research is to assess thiol-ene chemistry for functionalization of polyethylene. |
 Edgar Espinosa | Edgar Espinosa Rodriguez was born in México City in 1975 and studied chemical engineering at the Universidad Nacional Autonoma de México (1995–99). He completed his Master in Science at the Instituto de Investigaciones en Materiales (Dr Larissa Alexandrova 2001–03). He then worked as a researcher into the Centro de Investigaciones y Desarrollo Tecnologico in DESC Group in elastomers area (Alfonso Gonzalez and Javier Revilla 2002–08). Since October 2008, he has been completing a PhD (funded by CONACyT) under the supervision of Drs Christophe Boisson and Franck D'Agosto at C2P2 laboratory (Lyon, France). The focus of his research is to find original strategies for polyethylene functionalization through CCG. |
 Franck D'Agosto | Franck D'Agosto studied chemistry at the Ecole Nationale Supérieure de Chimie in Mulhouse (France). He completed a PhD in Polymer Chemistry at the joined unit between CNRS and bioMérieux (University of Lyon, France) before working at the University of Sydney (Australia) as a postdoctoral fellow in the Key Center for Polymer Colloids. Since 2002, he has been a researcher at the CNRS in the Chemistry and Process of Polymerization Team in C2P2 laboratory (Lyon, France). His research interests focus on the control of polymer architectures by the use of different polymerization chemistries, such as catalytic and controlled free radical polymerizations, either performed in solution or in dispersed media. |
 Christophe Boisson | Christophe Boisson studied chemistry at the Ecole Nationale Supérieure de Chimie in Montpellier (France). After completing a PhD in Organic Chemistry at the University of Paris-Sud 11 (Orsay, France), he joined the CNRS in the group of Dr Roger Spitz at the Ecole Supérieure de Chimie Physique Electronique de Lyon (CPE Lyon, France) where he is now “Directeur de Recherche” in the Chemistry and Process of Polymerization Team in C2P2 laboratory (Lyon, France). His research interests are the development of homogeneous and heterogeneous catalytic systems for the polymerization of olefins. |
1 Introduction
Polyolefins account for more than half of worldwide production of thermoplastics. Most of them are produced via coordination polymerization using catalytic systems. Among them, Ziegler/Natta catalysts are by definition formed by combination of a transition metal complex with a main group organometallic compound. During the growth of the chains, chain transfer reactions such as chain transfer to the organometallic compound can occur (Scheme 1).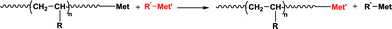 | ||
| Scheme 1 Chain transfer reaction to a main group organometallic compound (R′-Met') during olefin coordination polymerization in the presence of a transition metal catalyst (Met). | ||
For a long time, ZnEt2 has been used with conventional Ziegler/Natta catalysts in order to control the molar mass and the chain end of polyolefins through chain transfer reactions to Zn.1 Beyond heterogeneous Ziegler/Natta systems, the last generation of Ziegler catalysts concerns molecular complexes with well-defined structures called single-site catalysts. Among them, the most popular ones are the metallocenes (group 4 and lanthanides) but other efficient catalysts have also been reported such as bis(phenoxy imine) complexes of group 4, phenoxy imine and α-diimine complexes of nickel or bis(imino)pyridine complexes of iron and cobalt which all display very high activity in olefin polymerization.2 This list is not exhaustive and one can easily understand that a wide variety of properties can result from a fine-tuning of the metal and/or electronic and steric effects of ligands. Considering this variety of structures, it is not surprising to also find catalysts displaying unusual behavior toward transfer reactions to organometallic compounds such as AlR3, MgR2 or ZnEt2. Indeed, such transfer reactions can, for example, become advantageously very rapid and/or reversible. When reversible and in absence of another transfer reaction, the polymerization is best described as pseudo living since polymers display a remarkably narrow molar mass distribution and molar masses increase linearly with the productivity. Organometallic compounds used in these systems indeed act as reversible chain transfer agent (CTA) according to a mechanism of degenerative chain transfer. Everything is happening as if the growth of the chains was occurring on these organometallic compounds while being catalyzed by the active metal center: this is the catalyzed chain growth (CCG) concept.3 The first important result in this field has been reported by Mortreux ans co-workers4 who described a system based on a lanthanide metallocene used in combination with a di-alkylmagnesium compound as CTA (Scheme 2).
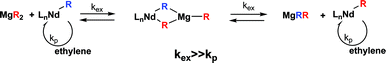 | ||
| Scheme 2 The catalyzed chain growth (CCG) concept. Nd Catalyst used in conjunction with di-alkylmagnesium organometallic species as CTA. | ||
The aim of the paper is to highlight our efforts in exploiting such a system as an efficient tool to end-functionalize polyethylene chains and to produce original reactive building blocks. After a short review on the development of CCG in polyolefin synthesis, the catalytic system we are employing to synthesize polyethylene chains will be discussed in more details. The various chemistries investigated to end functionalize polyethylene will then be depicted.
2. The catalyzed chain growth
The concept of catalyzed chain growth (CCG) was indeed defined and rationalized recently thanks to the work of Gibson3, who reported, in particular, the iron-catalyzed polyethylene chain growth on zinc. Related metal-alkyl systems were then investigated as potential CTA (AlR3, MgR2, BR3, GaR3, PbR4 and SnR4) and eventually screened a range of metallocenes and post-metallocenes catalysts.5 During the same period, CCG has been used to provide polyethylene oligomers using chromium catalysts in combination with CTA.6In the case of propylene, the first reported example satisfying a degenerative chain transfer based mechanism was reported by Busico and co-workers7 who described the preparation of stereoblock-isotactic polypropylene with a hafnium catalyst used in racemic form in conjunction with triethylaluminium as CTA. In all the aforementioned catalytic systems, the control of the polymerization via degenerative chain transfer is limited to rather low molar mass polyolefins. Although for many applications those molar masses are high enough (vide infra), the limitation in terms of molar masses can easily be explained. Actually, precipitation of polyolefins (polyethylene and isotactic polypropylene) impedes the control of the polymerization since the chain transfer reaction to the main group metal centre is no longer possible due to the dramatic decrease of the chain mobility. To get high molar mass polyolefins in a controlled way via CCG two solutions can be put forward. The first one consists of breaking the crystallinity of the polyolefins by performing either copolymerization of ethylene with other vinyl monomers or atactic homopolymerization of α-olefins. Interestingly, Zhang and Sita8 catalyzed atactic polypropylene chain growth on zinc using a hafnium catalyst and ZnEt2 as the CTA. High molar mass atactic polypropylene with an extremely narrow molar mass distribution was synthesized. Nevertheless, crystalline polyolefins are highly desirable due to their unique mechanical properties which make them the most relevant polymers for many applications. Consistent with this last statement, the only remaining valuable solution to target high molar mass polyolefins using CCG consists in keeping the polymer soluble by increasing the polymerization temperature. This however requires catalysts that are stable enough and that the chain transfer to the CTA is the only chain transfer reaction. This remains tricky since at high temperatures and in most cases β-H elimination reactions become predominant. This reaction occurs on the active metal center and produces vinyl terminated dead chains that cannot be involved in the degenerative transfer any more.
Nevertheless, Kempe et al.9 recently described ethylene polymerization systems successfully operating by degenerative chain transfer in presence of yttrium cations and tetraisobutylaluminoxane at 100 °C with nearly no β-H elimination. Narrowly distributed polyethylene chains of 15600 g mol−1 (PDI = 1.4) were obtained. Indeed, high throughput technology is probably the best tool for probing the potential candidates for such conditions. Researchers at The Dow Chemical Company screened and discovered two interesting catalysts that were both able to operate at high temperature, without β-H elimination and in the presence of ZnEt2 as the CTA: one catalyst for ethylene polymerization and another one able to copolymerize α-olefins. By combining these catalysts in the presence of ZnEt2, polymer chains were reversibly transferred from one metal center to the other via the alkylzinc transfer agent. This “double” catalyzed chain growth was denoted Chain Shuttling.10 Olefin block copolymers (OBCs) bearing several “hard” (polyethylene) and “soft” (poly(ethylene-co-α-olefins) segments were prepared. It is certainly the first industrial application of degenerative chain transfer mechanism in catalytic polymerization.
3. Functionalization strategy
3.1 The system employed
It has been shown that a pseudo-living ethylene polymerization via a chain transfer mechanism between di-alkylmagnesium derivatives and the lanthanidocene complex (C5Me5)2LnCl2Li(OEt2)2 could be achieved (Ln = Nd, Sm).4 As mentioned above, the polymerization system that we decided to investigate is based on a neodymium catalyst used in conjunction with n-butyloctylmagnesium. As chains are generated on the catalyst, a constant and reversible transfer is operating on the CTA where the chains are stored during the growth of other chains at the Nd center. This is an example of Nd-catalyzed polyethylene chain growth on Mg. Note that this system does not require the use of a cocatalyst. The interesting features of this system is that it produces di-alkylmagnesium compounds, Mg(PE)2 (PE = polyethylene chain, Scheme 3) having narrowly distributed molar masses that increase with the ethylene consumption.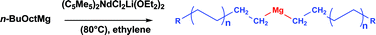 | ||
| Scheme 3 Production of Mg(PE)2 with a neodymium catalyst. | ||
As mentioned earlier, the pseudo-living behaviour is, however, stopped by the crystallization of Mg(PE)2 that occurs at the optimum polymerization temperature (80 °C) when the molar mass of the corresponding PE chains has reached a limit value. The precipitation of Mg(PE)2 deprives the polymerization system of reversible CTA and the control is rapidly lost. Still, by tuning the polymerization conditions such as the pressure of ethylene and/or the concentrations of catalyst and MgR2, we managed to produce a range of PE chains exhibiting controlled molar masses between 500 and 5000 g mol−1 with polydispersity indices lower than 1.3. In addition, an accurate 1H NMR analysis of the chain ends of these polymers showed that only 2% to 7% of the chains were unsaturated. This vinyl end group is indeed formed by the unavoidable β-H elimination side reaction that occurs on the Nd center during the polymerization. This last result leaves us with more than 93% of the chains saturated, thus originally bound to a magnesium center in the polymerization medium. They are excellent candidates for the further chemistry that is now discussed.
In the following sections, functionalization reactions performed on Mg(PE)2 compounds will be shown. The results commented on only concern PE chains with molar masses ranging from 500 up to 2000 g mol−1. We have very often been asked by the reviewers of our works if the molar mass could be increased. We constantly paid attention to work on polymers that can behave as such and we would like to take the opportunity in this paper to mention that the critical entanglement of PE has been determined to be 1100 g mol−1.11 In addition, thermal analyses performed on our polymers (2000 g mol−1) revealed 85% crystallinity with melting points as high as 135 °C. The molar mass really depends on the targeted applications and high molar masses are required if the polymer is used for its structural properties. The situation is not the same if the product is meant to be used as an additive, which is expected for such modified PE.
3.2 End functionalized PE
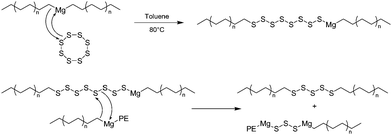 | ||
| Scheme 4 The proposed mechanism of PE-Sk-PE species formation after addition of elemental sulfur into the polymerization medium. | ||
A polysulfur based product (PE-Sk-PE) was recovered which, after reduction under appropriate conditions, gave the desired thiol end functionalized polyethylene chains in high yield (>80%) and according to a versatile and cheap route. It is worth noting here that this chemistry also resulted in the formation of unavoidable and non-reductable PE-S-PE species, the amount of which has been reduced to 14% by optimizing the reaction conditions. Alternative strategies that do not produce such PE-S-PE species were then evaluated by taking advantage of the synthesis of end thiocarbonyl thio PE and are mentioned in section 3.3.3.
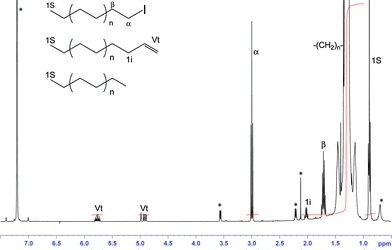 | ||
| Fig. 1 1H NMR (C6D6/TCE ½ v/v, 400 MHz, number of scans = 512, 363 K) spectrum of PE-I. Signals marked by an asterisk are solvent impurities. | ||
The obtained PE-I chains in which the chain end carbon is now electrophilic, open the way to a much broader range of possible chemistries including those involving nucleophilic attack.
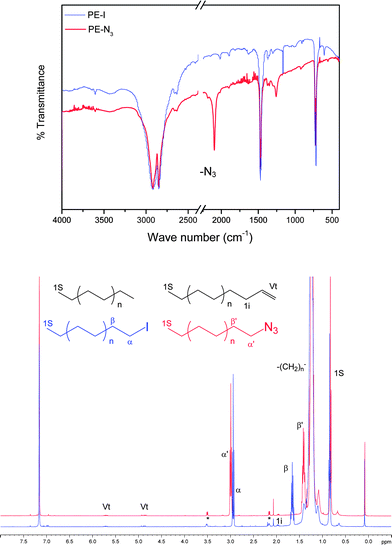 | ||
| Fig. 2 FT-IR analyses and 1H NMR (C6D6/TCE ½ v/v, 400 MHz, number of scans = 512, 363 K) spectra of PE-I (blue) and PE-N3 (red). | ||
This versatile introduction of an azido group onto polyethylene was of paramount interest to us, particularly in the context of click chemistry.19 Among the different chemistries fitting this concept, the copper catalyzed 1,3-dipolar Huisgen type cycloaddition between azides and alkynes has been shown to be a powerful tool to efficiently and quantitatively bind two organic moieties. Indeed, the polymer science field has witnessed fantastic academic efforts in taking advantage of these click chemistries in order to build up macromolecular architectures in these past years.20 PE-N3 is thus considered to be a very promising building block for generating architectures incorporating PE segments. Parts of the developments performed in our group in this area will be described in the next paragraph and in section 3.3.
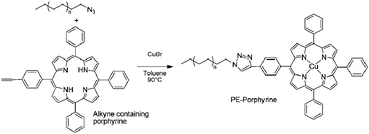 | ||
| Scheme 5 1,3-Dipolar Huisgen cycloaddition of PE-N3 with an alkyne containing porphyrine. | ||
The product of the reaction performed in the presence of copper(I) was characterized by different techniques such as 1H, 13C and 1H/1H COSY NMR, FT-IR, UV near infra red analyses and MALDI-ToF mass spectrometry. PE-Porphyrine was quantitatively obtained with a copper atom complexed into the porphyrine moiety and coming from the copper source used to catalyze the cycloaddition.
3.3 Macromonomers and macrocontrol agents
Our aim was then twofold: (i) the synthesis of macromonomers based on polyethylene that would easily be copolymerizable in a free radical process and (ii) the synthesis of macrocontrol agents based on PE.
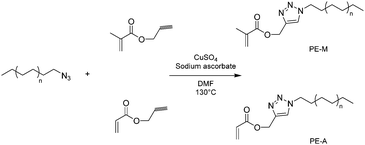 | ||
| Scheme 6 Macromonomers based on polyethylene obtained by 1,3-dipolar Huisgen cycloaddition. | ||
The extraordinarily short reaction times required (less than 15 min) to perform the Huisgen type 1,3-dipolar cycloaddition make this polyethylene functionalization pathway a very attractive one. The proof of concept that such compounds can genuinely be used as building blocks can be provided by showing that these macromonomers are readily (co)polymerized for example by free radical polymerization. Thus, PE-M was copolymerized with methyl methacrylate (MMA) in toluene using 2,2′-azobis(isobutyronitrile) (AIBN) as the initiator (Scheme 7). After three hours of reaction at 85 °C, MMA was completely consumed. 76% of the starting PE-M was converted. High temperature SEC analyses of the polymerization medium revealed the presence of two populations: unreacted PE-M (Mn = 1040, PDI = 1.19, PE calibration) and an additional population with Mn of 20000 g mol−1 (PDI = 2.0, universal calibration). Additional characterizations by 1H NMR confirmed that this population corresponded to a statistical copolymer of PE-M and MMA with 2.4% of PE unit molar content.
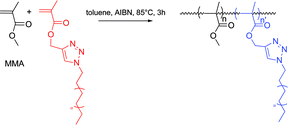 | ||
| Scheme 7 Copolymerization of MMA with PE-M and production of a poly(methyl methacrylate) bearing long branch of PE chains. | ||
In that respect, we decided to check whether simple and efficient strategies could be employed to introduce alkoxyamine or thiocarbonyl thio groups at the end of polyethylene chains in a one step reaction with Mg(PE)2.
Macroalcoxyamines based on PE. Alkoxyamines used in NMP can be synthesized using Grignard chemistry. For example, C6H14MgBr can be reacted with two equivalents of a stable nitroxyl radical (O–N(R1R2)) and form an C6-alkyl based alkoxyamine.25 This organic chemistry result was transposed to our Mg(PE)2 compounds. 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) and N-(2-methyl-2-propyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxy (DEPN or SG1) stable radicals were first investigated in this strategy. PE chains terminated with an alkoxyamine based on TEMPO26 or DEPN were obtained with a yield of 70% and 45%, respectively.26 However, the homolysis of the C–ON bonds in these compounds required very high temperatures (180 °C and 160 °C respectively) and do not allow the performance of the free radical polymerization step under mild conditions. Taking advantage of this high bond dissociation energy, two nitroxides (DD1 and DD2 in Fig. 3) based on TEMPO and carrying an alkoxyamine based on DEPN were designed.27 The TEMPO part is used to ensure a strong link between the PE and the alkoxyamine, the latter being homolyzed between 70 and 100 °C. Although the functionalization yield remained to be optimized (17% for DD1 and 45% for DD2), block copolymerization using n-butyl acrylate (n-BuA) was undertaken. The controlled behaviour of the polymerization was confirmed by SEC analyses on samples withdrawn from the polymerization medium at different times (SEC using trichlorobenzene as eluent at 150 °C) and on the block copolymer extracted from these samples with THF (SEC using THF as eluent at 40 °C).
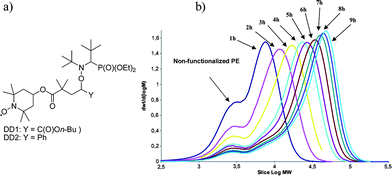 | ||
| Fig. 3 (a) Alkoxyamine based on DEPN. (b) High temperature SEC (PE Standards) chromatograms of samples withdrawn during n-buytl acrylate polymerization mediated by PE-DD2. | ||
End thiocarbonyl thio PE. Armed with our experience in the field of polyethylene end-functionalization with nitroxides, and with the knowledge that thiocarbonyl thio compounds can also be obtained using Grignard chemistry,28 our group also investigated the functionalization of polyethylene carrying thiocarbonyl thio end groups. The general strategy adopted was to react a thiocarbonyl thio disulfide with Mg(PE)2 (Scheme 8). Dithiocarbonate (PEa), dithiocarbamate (PEb), dithioester (PEc) and trithiocarbonate (PEd) were introduced at the end of polyethylene chains.29 Functionalization yields ranged from 40% (PEc) up to 95% (PEb). However, PEd (60% functionalization yield) is the only structure that can mediate a RAFT polymerization. Polyethylene-b-poly(n-BuA) block copolymers were indeed synthesized and the control of the polymerization was validated through SEC analyses using both trichlorobenzene (150 °C) or THF as eluents.
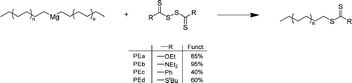 | ||
| Scheme 8 Functionalization of polyethylene with thiocarbonyl thio compounds by reacting thiocarbonyl thio disulfide with Mg(PE)2 species. | ||
As mentioned in the PE-SH section (3.2), these thiocarbonyl thio PE were very valuable compounds for the synthesis of thiol end functionalized PE. Indeed, there are a number of examples in the literature for the removal of the thiocarbonyl thio end group, reported first by Rizzardo et al. in 1999.30 Among them, reduction via metal hydride compounds such as LiAlH4 or NaBH4 is a classical methodology that provides one with a thiol group. Since PEa carrying a dithiocarbonate chain end was obtained in high yields (higher than 80%), we decided to evaluate its reduction with LiAlH4. A quantitative transformation of the dithiocarbonate chain end into a thiol end group was indeed observed. The efficiency of the overall transformation was recently pushed ahead. Since the synthesis of PE-I gave functionality higher than 95%, the further substitution of the iodine by the commercial and cheap xanthic acid was performed and also occurred quantitatively. The quantitative reduction into PE-SH of the corresponding dithiocarbonate end functionalized PE gave a unique, simple and cheap route for producing such end functionalized PE and enlarged the possibilities of conveniently producing PE-SH (see section 3.2).16
The original goal of being able to synthesize in a direct manner polyethylene chains that would carry a control agent for RAFT as an end group was thus achieved. The control ability of the corresponding structure (PE-SC(=S)S-t-Bu) was established by the syntheses of polyethylene-b-poly(n-butyl acrylate) copolymers. However, due to the relatively low functionalization rates (PE-SC(=S)S-t-Bu: 60%) unreacted PE had to be removed – sometimes with difficulty – from the formed block copolymers. In addition, when examining the chemical structures of the final block copolymers (Scheme 9), a trithiocarbonate group coming PE-SC(=S)S-t-Bu, links the polyethylene block to the poly(n-butyl acrylate) block. This fragile bond could be a considerable drawback, particularly in the case of macromolecules that would have to be processed at high temperatures, which is the case for polyolefin based materials.
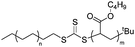 | ||
| Scheme 9 Polyethylene-b-poly(n-butyl acrylate) via RAFT polymerization mediated by PE-SC(=S)S-t-Bu. | ||
With this in mind, we decided to further focus on the design of macrocontrol agents that would be highly functionalized and/or exhibit such a chemical structure that the resulting link between the polyethylene and the block grown from it would be strong enough to handle high temperature processing.
Chemistry involving PE-NH2 – obtained by reduction of PE-N3 – and activated ester containing RAFT agents according to an already established procedure31 was indeed successful. The resulting macroRAFT based on PE was employed to grow a poly(n-butyl acrylate) segment according to a RAFT mechanism (Scheme 10).18 The resulting block copolymers exhibited a strong amide link between PE and poly(n-butyl acrylate) segment.
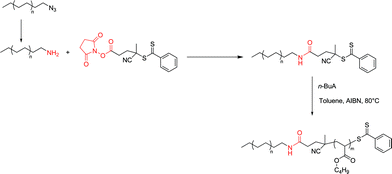 | ||
| Scheme 10 The synthesis of a macroRAFT based on PE using PE-NH2 and an activated ester containing RAFT agent. Application to the synthesis of a block copolymer with n-BuA. | ||
Our efforts are currently based on the fact that the literature strongly suggests that a radical based on a methyl triazole group is (i) stable enough to be involved in a free radical polymerization since vinyl triazole monomers are efficiently (co)polymerized by this technique32 (ii) an efficient reinitiating R group for the RAFT process if incorporated in the chemical structure of a reversible chain transfer agent (ZC(=S)-SR).33 As a result our efforts are now focused toward the direct coupling on PE-N3 of alkyne containing thiocarbonyl thio molecules of structures depicted in Scheme 11. Our first results show that highly functionalized PE are obtained.16 In addition, the triazole ring is a strong link between the PE and the envisioned segments that can be produced by RAFT polymerizations.
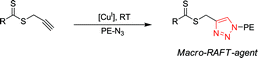 | ||
| Scheme 11 The coupling reaction between PE-N3 and alkyne containing thiocarbonyl thio molecules. | ||
4. Conclusion
This paper has highlighted the potential of polyethylene catalyzed chain growth (CCG) on magnesium to functionalize polyethylene chains. CCG makes chemistry that is selective and quantitative and that allows the introduction of versatile functional groups possible. These functionalities include reactive or polymerizable groups and initiators or controlling agents for other polymerization techniques. The resulting polyethylene building blocks can further be incorporated into more complex architectures (Scheme 12).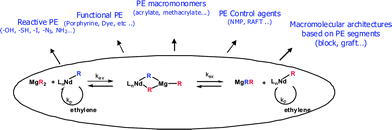 | ||
| Scheme 12 A general overview of the potential of polyethylene CCG on magnesium. | ||
The transposition of these results to catalytic systems that are operating according to a CCG at higher temperatures10 would permit the functionalization of higher molar mass polyolefins. This work is under progress in our laboratories.
Acknowledgments
This paper is dedicated to Dr Roger Spitz for his exceptional scientific input at the head of the LCPP Team.The authors would like to thank D. Bertin and D. Gigmes (UMR 6517, CROPS team, Marseille, France) for collaboration on NMP, Drs R. Charvet and J. Hill (National Institute for Material Science, Japan) for porphyrine related works, F. Boisson (UMR 5223 IMP, Service de RMN, Villeurbanne, France) for NMR characterization, F. Delolme (UMR 5086, IBCP, Lyon, France) for MALDI-ToF analyses, C. Novat and O. Boyron (LCPP Team) for SEC analyses, J.P. Broyer (LCPP Team) for reactor engineering. The financial supports from CONACYT programs, the French Ministry for Research, the French National Agency for Research (ANR 08-JCJC-0115-01), the competitiveness clusters Plastipolis and Axelera are acknowledged.
References
- E. Agouri, C. Parlant, P. Mornet, J. Rideau and J. F. Teitgen, Makromol. Chem., 1970, 137, 229–243 CrossRef; D.R. Burfield, Polymer, 1984, 25, 1817–1822 CrossRef CAS; T. Shiono, H. Kurosawa and K. Soga, Makromol. Chem., 1992, 193, 2751–2761 CrossRef CAS; S. G. M. Perin, J. R. Severn, C. E. Koning and J. C. Chadwick, Macromol. Chem. Phys., 2006, 207, 50–56 CrossRef CAS.
- For an exhaustive review on post-metallocene catalysts see: V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 Search PubMed and references therein.
- V. C. Gibson, Science, 2006, 312, 703–704 CrossRef CAS.
- S. Bogaert, T. Chenal, A. Mortreux and J. F. Carpentier, J. Mol. Catal. A: Chem., 2002, 190, 207–214 CrossRef CAS; J. F. Pelletier, A. Mortreux, X. Olonde and K. Bujadoux, Angew. Chem., Int. Ed. Engl., 1996, 35, 1854–1856 CrossRef CAS.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson, P. J. Maddox and M. van Meurs, Angew. Chem., Int. Ed., 2002, 41, 489–491 CrossRef CAS; G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef CAS; M. Van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS.
- J. S. Rogers and G. C. Bazan, Chem. Commun., 2000, 1209–1210 RSC; G. C. Bazan, J. S. Rogers and C. C. Fang, Organometallics, 2001, 20, 2059–2064 CrossRef CAS; G. Mani and F. P. Gabbai, Angew. Chem., Int. Ed., 2004, 43, 2263–2266 CrossRef CAS; M. Ganesan and F. P. Gabbaï, Organometallics, 2004, 23, 4608–4613 CrossRef CAS.
- F. Alfano, H. W. Boone, V. Busico, R. Cipullo and J. C. Stevens, Macromolecules, 2007, 40, 7736–7738 CrossRef CAS.
- W. Zhang and L. R. Sita, J. Am. Chem. Soc., 2008, 130, 442–443 CrossRef CAS.
- W. P. Kretschmer, A. Meetsma, B. Hessen, T. Schmalz, S. Qayyum and R. Kempe, Chem.–Eur. J., 2006, 12, 8969–8978 CrossRef CAS; R. Kempe, Chem.–Eur. J., 2007, 13, 2764–2773 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS; M. Zintl and B. Rieger, Angew. Chem., Int. Ed., 2007, 46, 333–335 CrossRef CAS; R. L. Kuhlman and T. T. Wenzel, Macromolecules, 2008, 41, 4090–4094 CrossRef CAS; T. T. Wenzel, D. J. Arriola, E. M. Carnahan, P. D. Hustad and R. L. Kuhlman, Top. Organomet. Chem., 2009, 26, 65–104 CAS.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef CAS.
- C. J. Han, M. S. Lee, D. J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923–8925 CrossRef CAS.
- H. Kaneyoshi, Y. Inoue and K. Matyjaszewski, Macromolecules, 2005, 38, 5425–5435 CrossRef CAS.
- M. S. Kharasch, A. T. Read and F. R. Mayo, Chem. Ind., 1938, 57, 752 Search PubMed; C. S. Marvel and R. R. Chambers, J. Am. Chem. Soc., 1948, 70, 993–998 CrossRef CAS; C. S. Marvel and P. D. Caesar, J. Am. Chem. Soc., 1951, 73, 1097–1099 CrossRef CAS; The Chemistry of the Thiol Group, ed. S. Patai, Wiley, New York, 1974; A. F. Jacobine, in Radiation Curing in Polymer Science and Technology III ed. J. P. Fouassier and J. F. Rabek, Elsevier, London, 1993, ch. 7, pp. 219–268 Search PubMed.
- J. F. Lutz and H. Schlaad, Polymer, 2008, 49, 817–824 CrossRef CAS; A. Lowe, Polym. Chem., 2010 10.1039/b9py00216b; A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- Manuscripts in preparation.
- Y. Doi, F. Nozawa, M. Murata, S. Suzuki and K. Soga, Makromol. Chem., 1985, 186, 1825–1834 CrossRef CAS.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311–9313 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2020 CrossRef CAS; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- C. Barner Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS; M. Meldal, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS; J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS; W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- R. Godoy Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419–454 CrossRef.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS; W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS; S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS; G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- T. Nagashima and D. P. Curran, Synlett, 1996, 330–332 CrossRef CAS.
- R. Godoy, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson, D. Bertin and P. Tordo, Macromolecules, 2004, 37, 6540–6542.
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson, D. Gigmes and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2705–2718 CrossRef.
- J. R. Grunwell, J. Org. Chem., 1970, 35, 1500–1501 CrossRef CAS.
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spitz and F. Boisson, Macromol. Rapid Commun., 2006, 27, 173–181 CrossRef.
- E. C. J. Rizzardo, B. Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad and S. H. Thang, Macromol. Symp., 1999, 143, 291–307 CAS.
- M. Bathfield, F. D'Agosto, R. Spitz, M. T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546–2547 CrossRef CAS.
- R. J. Thibault, K. Takizawa, P. Lowenheilm, B. Helms, J. L. Mnyar, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2006, 128, 12084–12085 CrossRef CAS.
- N. Akeroyd, R. Pfukwa and B. Klumperman, Macromolecules, 2009, 42, 3014–3018 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |