Polymer prodrug approaches applied to paclitaxel
Jeong Sun
Sohn
a,
Jung Il
Jin
b,
Michael
Hess
cd and
Byung Wook
Jo
*d
aDepartment of Polymer Science and Engineering, Chosun University, Gwangju, 501-759, South Korea. E-mail: jss4347@chosun.ac.kr; Fax: +82 62 232 2474; Tel: +82 62 230 7240
bDepartment of Chemistry, Korea University, Seoul, 136-701, South Korea. E-mail: jijin@korea.ac.kr; Fax: +82 2 928 3538; Tel: +82 2 928 3123
cDepartment of Macromolecular Chemistry, University Siegen, Siegen, D-57068, Germany. E-mail: emelel@hotmail.com; Tel: +49 271 740 2445
dDepartment of Chemical and Biochemical Engineering, Chosun University, Gwangju, 501-759, South Korea. E-mail: bwjo@chosun.ac.kr; Fax: +82 62 232 2474; Tel: +82 62 230 7214
First published on 3rd February 2010
Abstract
Paclitaxel possesses a unique mechanism of drug action as a new class of microtubule stabilizing agent which finally leads to disrupted mitosis and cell death. Recent findings have shown that paclitaxel initiates apoptosis through multiple mechanisms. However, the strong hydrophobicity of paclitaxel drastically limits its use in natural form. In addition, significant toxicity which is mainly ascribed to the non-specific, indiscriminate distribution among the tissues limited the application of paclitaxel in cancer therapy. Therefore, it remains of interest to improve their use by enhancing solubility, selectivity and in vivo delivery by preparing functional prodrugs selectively activable in tumour areas. This article summarizes results and information derived recently from paclitaxel prodrugs based mainly on macromolecular conjugates and their interactions with body fluids. We also explore the potential application of site-specific carriers in the development of tumor-targeting paclitaxel conjugates.
 Jeong Sun Sohn | Jeong-Sun Sohn obtained her PhD in 2005 from Chosun University in Gwangju, Korea. She was a researcher in Kumho Tire Technical Institute and in the University of Duisburg-Essen, Germany. She is now an Adjunct Professor of the Polymer Science and Engineering Department of the Chosun University, and her main research interests are biomaterial science, physicochemical properties of polymer drugs, and liquid crystal polymers. |
 Jung Il Jin | Prof. Jung-Il Jin obtained his PhD in 1969 from the City University of New York. Since 1974 he has been teaching at the Chemistry Department of the Korea University, Seoul. He was a visiting professor at the University of Massachusetts, Amherst, and a visiting scholar at the University of Cambridge, UK. He was president of the Korean Chemical Society and the Polymer Society of Korea. He is now the Professor Emeritus of the same department, and Past President of IUPAC. He has published ca. 400 papers on liquid crystalline compounds and polymers, conjugated polymers and materials properties of DNA. |
 Michael Hess | Prof. Michael Hess obtained his PhD in 1976 from the RWTH Aachen University, Germany. He is a senior scientist of the Macromolecular Chemistry Department of the University Siegen, and an Adjunct Professor at the University of North Texas. He is now a visiting professor at the Chosun University, and Secretary of IUPAC Polymer Division. |
 Byung Wook Jo | Prof. Byung-Wook Jo obtained his PhD in 1988 from Dankook University in Seoul, Korea. Since 1975 he has been teaching at the Chemical and Biochemical Engineering Department of the Chosun University, Gwangju. He was a visiting professor at the University of Massachusetts, Amherst, and an exchanging scientist at the E.S.P.C.I. CNRS, France. He was vice president of the Chosun University and the Polymer Society of Korea and a member of the National Science and Technology Council, Korea. He is now a Member National Consulting Committee for Science and Technology Korea and Director/Professor Marine Functional Materials Centre, BK21, Korea. |
Introduction
Paclitaxel (Taxol®), a natural diterpene extracted from the bark of the Pacific Yew tree (Taxus brevifolia),1,2 is one of the most effective anticancer drugs for treatment of various human solid tumors, such as metastatic breast cancer, head and neck carcinomas, advanced ovarian carcinoma, non-small cell lung cancer, melanoma, as well as AIDS-related Kaposi's sarcoma.2–11 It has a unique mechanism of drug action. In contrast to clinical antimitotic agents, such as the vinca alkaloids that inhibit the microtubule assembly process, paclitaxel promotes tubulin assembly into stable microtubules by preventing their depolymerization,12 thereby blocking cell replication in the late G2 mitotic phase of the cell cycle, resulting in apoptotic death.13–21Although paclitaxel has therapeutic potential as an anticancer drug, unfortunately, its drawbacks, such as poor solubility and lack of selectivity toward tumor cells, have limited its widespread use in cancer therapy.22
Paclitaxel is currently formulated in Taxol® and a mixture of polyoxyethyleneglycerol triricinoleate 35 (Cremophor EL®) and dehydrated ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). However, a large number of studies have reported various clinical side effects, such as severe anaphylactoid hypersensitivity, abnormal lipoprotein patterns, hyperlipidemia, aggregation of erythrocytes, neurotoxicity, and peripheral neuropathy, mainly due to the organic solvent used in drug formulation.23–27 Previously, a great number of efforts have concentrated on development of new delivery systems for paclitaxel, such as polymer conjugates, liposomes, polymeric micelles, parenteral emulsions, polymeric micro/nanoparticles, and water-soluble prodrugs, with the aim of overcoming the most serious problems.28–37
:1, v/v). However, a large number of studies have reported various clinical side effects, such as severe anaphylactoid hypersensitivity, abnormal lipoprotein patterns, hyperlipidemia, aggregation of erythrocytes, neurotoxicity, and peripheral neuropathy, mainly due to the organic solvent used in drug formulation.23–27 Previously, a great number of efforts have concentrated on development of new delivery systems for paclitaxel, such as polymer conjugates, liposomes, polymeric micelles, parenteral emulsions, polymeric micro/nanoparticles, and water-soluble prodrugs, with the aim of overcoming the most serious problems.28–37
Topical application of drugs is an important route of drug administration. Because the physicochemical properties of a drugs are not usually optimal for their delivery into or through membranes, the administration of drugs is not always effective. A prodrug strategy is usually used as a chemical/biochemical approach to overcome various barriers in their administration. A prodrug is a bioreversible derivative of an active drug. In the prodrug approach, the changes in the physiochemical properties and the pharmacological profiles of the drug are transient, so that once the prodrug has delivered the parent drug, one is left with a well-characterized and well-understood molecule with which to work. The necessary conversion or activation of the prodrug to parent drug molecules in the body takes place by a variety of reactions. Therefore, it is also important to understand the interaction between the prodrug and body fluids
In the past decade, there has been a large number of reports on paclitaxel-prodrugs concerning their synthesis, targeting, drug efficacy, water solubility, physicochemical properties and so on. There have also been excellent reports published recently on applications and clinical trials of the paclitaxel prodrugs.38,39 But they did not mention interactions of the prodrugs with body fluids. In this review, however some recent studies of the paclitaxel prodrugs based on macromolecular conjugates and their interactions with body fluids will be discussed.
Prodrugs of paclitaxel
Macromolecular conjugates
As mentioned above, the goal of most prodrug technologies is typically to increase solubility of hydrophobic drugs in an aqueous system for desirable administration.39 Additionally, it's also focused on low toxicity, improving specific cell targeting effect, and synergistic drug action due to the ability of the polymer to accumulate in tumor tissues as a result of an enhanced permeability and retention (EPR) effect. Micelles or lipophilic nanoparticle carriers based on polymers can also be used for suspension of these prodrugs in an aqueous environment.Micellar drug-polymer conjugates have distinctive advantages of high drug content and good solubility in water, adjusting drug half-life in the body, and enhancing antitumor effects.40,41
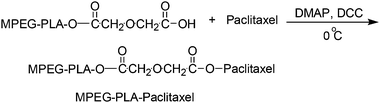 | ||
| Scheme 1 Synthesis scheme for MPEG-PLA-paclitaxel41 (reprinted with permission from X. Zhang, Y. Li, X. Chen, X. Wang, X. Xu, Q. Liang, J. Hu and X. Jing, Biomaterials, 2005, 26, 2121, Copyright (2004) Elsevier Ltd). | ||
Recently, Park and co-workers45 made hyaluronic acid (HA)-paclitaxel conjugates without using any blocking agents, ionic surfactants, and salts for functional groups in HA, by way of the HA solubilization method in a single organic phase. Hyaluronic acid (HA)-paclitaxel conjugates are known to form nanosized micellar aggregates self-assembly46 in an aqueous solution with poly(ethyleneglycol) (PEG), which can act as both hydrogen bonding donor and acceptor. Because of extreme hydrophilicity and poor solubility of HA with hydrophobic drug in most organic solvents, solubilization methods were used to induce a homogeneous mixture of HA and hydrophobic reactants. Hyaluronic acid (HA) is also related to angiogenesis of tumors. Since HA has a strong affinity with cell-specific surface markers i.e. CD44 and RHAMM,47 HA receptors are overexpressed on the surface. As a result, malignant cells which have high metastatic activities often show improved binding and uptake of HA.48–50 HA and its derivatives have been matters of concern in the field of target-specific drug delivery vehicles.51–53 Park et al. explained that HCT-116 cells, as studied by the confocal microscopy (Fig. 1A), were nonapoptotic and revealed strong red fluorescence homogeneously within the nucleus. In the case of Taxol (Fig. 1B) or HA-paclitaxel conjugate micelles (Fig. 1C), by contrast, HCT-116 cells displayed apparent evidence of the enhanced extent of apoptosis-inducing cell death. HA-paclitaxel conjugate micelles exhibited a greater cytotoxicity than the conventional paclitaxel formulation.45 They suggested that self-assembled and nanosized HA-paclitaxel conjugate micelles could be used as tumor-specific nanoparticulate therapeutic agents, or, in other words, efficient all-in-one carriers.
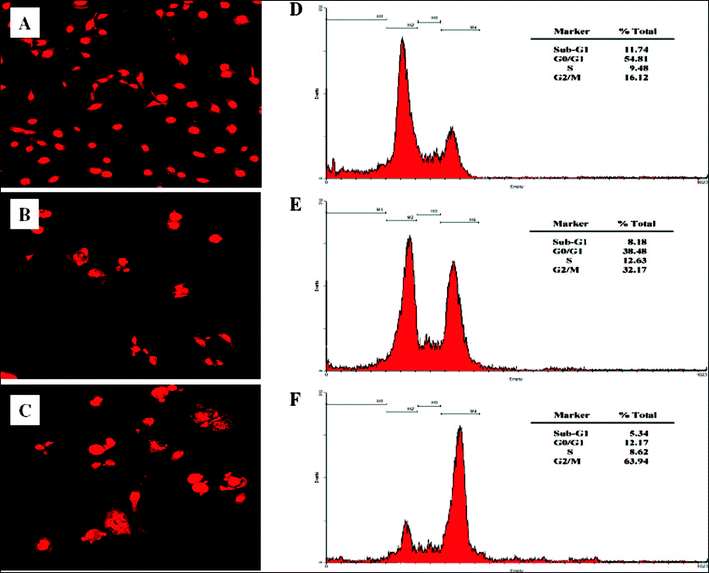 | ||
| Fig. 1 Confocal images of PI stained HCT-116 cells: (A) control, (B) Taxol, and (C) HA-paclitaxel. FACS analysis of apoptotic effects on HCT-116 cells: (D) control, (E) Taxol, and (F) HA-paclitaxel conjugate micelles. All paclitaxel formulations have 1 μg mL−1 of equivalent paclitaxel concentration45 (reprinted with permission from H. J. Lee, K. R. Lee and T. G. Park, Bioconjugate Chem., 2008, 19, 1319, Copyright (2008) American Chemical Society). | ||
In recent years, there has been increasing concern in chemotherapeutic treatment regimes involving multiple drugs. Many efforts have been made to produce functional lipophilic paclitaxel prodrugs.54–60 To address this issue, Ansell et al.61 have developed liposome-based delivery systems in lipophilic nanoparticles. In their study, the structure of conjugates was based on paclitaxel-linker-lipid using succinate or diglycolate cross-linkers (Scheme 2). They suggested that the releasing rate of prodrugs from nanoparticles could be controlled by modulating the hydrophobicity of the lipid anchor. Paclitaxel itself is nearly insoluble in water, while it has sufficient solubility that it is able to rapidly partition out of lipid-based delivery systems. The relative efficacies of nanoparticle formulations62–67 were presented in Fig. 2. The results from Ansell's group61 showed no evidence of efficacy for succinate prodrug nanoparticles, but the efficacy for diglycolate prodrug nanoparticles increased with increasing anchor hydrophobicity; that is, the efficacy of the prodrugs in vivo depends on the relative partitioning rate of the lipid anchor and the nature of the linkage.
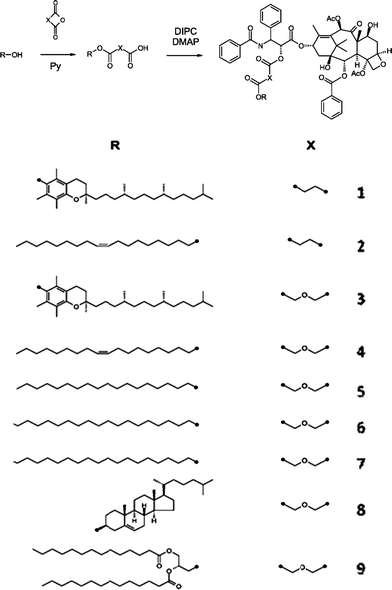 | ||
| Scheme 2 Synthesis of Lipophilic Paclitaxel Prodrugs61 (reprinted with permission from S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prud'homme and L. D. Mayer, J. Med. Chem., 2008, 51, 3288, Copyright (2008) American Chemical Society). | ||
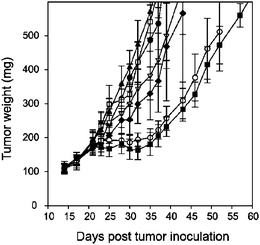 | ||
| Fig. 2 Relative efficacy of prodrugs formulated as prodrug/POPC/2kPS3k (1:1:2; w/w) nanoparticles when administered to athymic nude mice bearing HT29 human colon carcinoma xenographs. Drug was administered at a dose of 36 μmol kg−1 using a Q4D × 6 schedule beginning on day 14. The prodrugs and controls used were saline (●), 1 (□), 2 (▲), 4 (▽), 5 (◆), 6 (○), 7 (■). Error bars represent standard error of the mean (n = 6)61 (reprinted with permission from S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prud'homme and L. D. Mayer, J. Med. Chem., 2008, 51, 3288, Copyright (2008) American Chemical Society). | ||
Our group recently developed a new series of poly(ethyleneglycol)(PEG)-paclitaxel conjugates, namely PP7,68–77 which introduces a hydrophilic PEG, succinic acid mono-ester of poly(ethyleneglycol) monomethylether, with a self-immolating linker (L) at the carbon 7 position (Scheme 3).78 One possible way of overcoming poor water-solubility is the incorporation of a highly water-soluble nontoxic polymer, such as poly(ethyleneglycol) (PEG), at the C2′ or C7 position. Derivatives of paclitaxel with PEG, however, were too stable so that a high percentage of the polymer drug was excreted before the drug could be made bio-available by esterases at the site of their intended interaction. Therefore, we developed well designed self-immolating linkers (L) between a drug and a water soluble polymer moiety which undergo extremely rapid hydrolysis converting a prodrug into a parent drug without any reduction in drug efficacy.
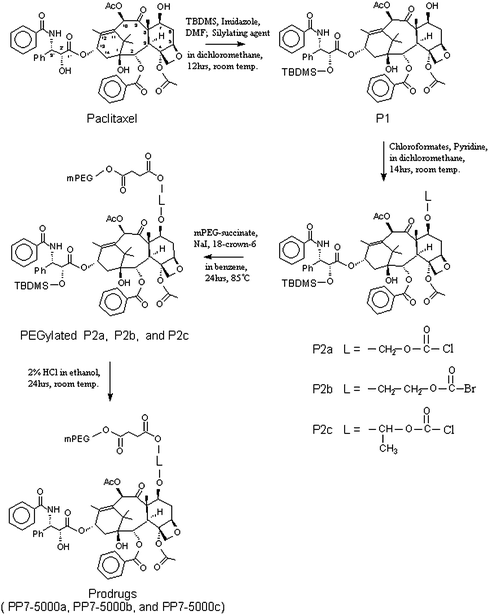 | ||
| Scheme 3 Synthesis route for the prodrugs (PP7-5000a: PP7 having a M.W. of 5000 PEG and P2a self-immolating group, L = –CH2–O–CO–).78 | ||
Self-immolating spacer groups were introduced between the solubilizing PEG and C7-hydroxyl group of paclitaxel in order to control the enzymatic hydrolysis rate. Fig. 3 shows the hydrolysis mechanism of prodrugs. It appeared that introduction of self-immolating spacer groups in conjugates which decreased steric hindrance in the enzymatic break down process of the prodrug could lead to rapid enzymatic hydrolysis. All these prodrugs had a water-solubility of 400 mg ml−1 or more (Table 1) and the rate of hydrolysis for prodrugs in rat plasma showed considerable variation with t½ ranging from 0.94 min to 42.7 min (Table 2). The prodrug was potent in inhibition of the growth of various tumor cell lines, and more efficacious than paclitaxel concerning development of melanoma lung colonies in C57B/6 mice following intravenous administration of metastatic murine B16/F10 melanoma cells.68–70 This kind of self-immolating groups can be advantageously utilized in controlling the rate of parent drug release from a prodrug in any other drug administrations.
| Compound | Amount dissolved in water/mol mL−1 |
|---|---|
| Paclitaxel | <1.17 × 10−8 |
| PP7-5000a | >6.77 × 10−5 |
| PP7-5000b | >6.70 × 10−5 |
| PP7-5000c | >6.75 × 10−5 |
| Compound | t1/2 Hydrolysis | ||
|---|---|---|---|
| Rat plasma | PBS | Distilled water | |
| a Greenwald, et al., U.S. Patent 5,614,549 (1997). | |||
| PP7-5000a | 0.94 min | 26.7 h | 201.5 h |
| PP7-5000b | 42.70 min | — | — |
| PP7-5000c | 1.12 min | — | — |
| Ester-based prodruga | ≥7 h | >96 h | >96 h |
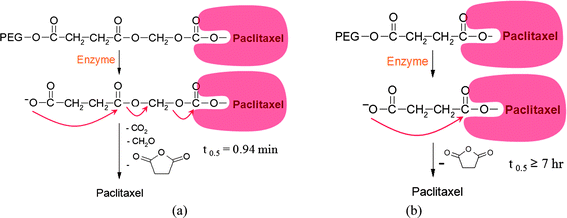 | ||
| Fig. 3 Rapid hydrolysis mechanism of prodrugs (a) with a self-immolating linker, succinyloxycarbonyl group (B.-W. Jo, U.S. Patent 6,703,417, 2004), (b) without a self-immolating linker (Neil P. Desai, U.S. Patent 5,648,506, 1997).78 | ||
They suggested that minor structural changes in the polyamine skeleton cause differences in their transport behavior. In the case of 2′-acyl derivatives, its cytotoxic activity could be lost by modification of 2′-hydroxyl functionality. Therefore, Battaglia et al.79 attached the spermine to the hydroxyl group at the carbon 7 of paclitaxel and at the carbon 10 of 10-deacetyl-paclitaxel via the carbamate bond (compounds 10 and 11, Fig. 4).
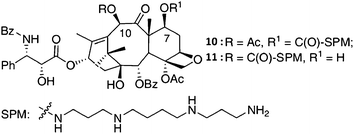 | ||
| Fig. 4 Synthesis of 7- and 10-spermine conjugates of paclitaxel79 (reprinted with permission from A. Battaglia, A. Guerrini, E. Baldelli, G. Fontana, G. Varchi, C. Samorı and E. Bombardelli, Tetrahedron Lett., 2006, 47, 2667, Copyright (2006) Elsevier Ltd). | ||
Several paclitaxel-peptide conjugates were successfully synthesized combining solid-phase peptide synthesis (SPPS) and chemoselective ligation methods by Tsikaris and co-workers. The 2′-bromoacetyl paclitaxel was the most effective for the formation of the thioether bond in the sequential oligopeptide carriers. They presented 2′-paclitaxel-substituted derivatives by the covalent attachment of paclitaxel to peptides or as multiple copies to synthetic carriers using 2′-halogeno-acetylated paclitaxel derivatives.
These paclitaxel conjugates showed greater a solubility in water than paclitaxel itself and inhibited proliferation of human cancer cells. They synthesized Ac-[Lys-Aib-Cys(CH2CO-2′-Pac)]4-NH2 using 2′-halogenoacetylated paclitaxel derivatives, which showed increased biological activity as compared with paclitaxel in prostate and cervical human cancer cells because it permits covalent attachment of a modified paclitaxel in multiple copies with a thioether bond (Fig. 5). Li reported a paclitaxel prodrug (Paclitaxel poliglumex, Opaxio®) which consists of paclitaxel conjugated to a long chain of poly-L-glutamic acid.90 This prodrug is one of the most successful prodrugs that demonstrates stability in plasma, nonimmunogenicity and water-solubility.91,92 Preclinical and clinical studies supported that OPAXIO® metabolism by lung cancer cells may be influenced by estrogen, which could lead to enhanced release of paclitaxel and efficacy in women's lung cancer compared to standard therapies, and to superior safety according to existing phase III clinical trials data (http://ww.cticseattle.com/).
![A schematic representation of Ac-[Lys-Aib-Cys (CH2CO-2′-Pac)]4-NH284 (reprinted with permission from S. Papas, T. Akoumianaki, C. Kalogiros, L. Hadjiarapoglou, P. A. Theodoropoulos and V. Tsikaris, J. Pept. Sci., 2007, 13, 662, Copyright (2007) European Peptide Society and John Wiley & Sons, Ltd).](/image/article/2010/PY/b9py00351g/b9py00351g-f5.gif) | ||
| Fig. 5 A schematic representation of Ac-[Lys-Aib-Cys (CH2CO-2′-Pac)]4-NH284 (reprinted with permission from S. Papas, T. Akoumianaki, C. Kalogiros, L. Hadjiarapoglou, P. A. Theodoropoulos and V. Tsikaris, J. Pept. Sci., 2007, 13, 662, Copyright (2007) European Peptide Society and John Wiley & Sons, Ltd). | ||
Tsikaris et al.84 examined nuclear morphology and nuclear envelope integrity of HeLa and DU cancer cells treated first with either paclitaxel or As–PA and then cultured in a drug-free medium. Many of the treated HeLa and DU (Fig. 6) cells had abnormally shaped nuclei that often presented with nuclear envelope discontinuity. This phenotype was shown to cause, at least partially, by defects in cell division, such as the incorrect cytokinesis and sorting of nuclear envelope components (Fig. 6). They found that the treated one was more effective than paclitaxel itself in producing cells with multinucleated phenotype in both cell lines. Serum proteins offer promises of selective delivery of anticancer agents because of their accumulation in tumor tissues. Albumin, a human protein, demonstrates preferential tumor uptake in various animal models due to the EPR effect of macromolecules.93,94 An albumin-bound nanoparticle of paclitaxel (Abraxane® or ABI-007) which has been approved for the treatment of breast cancer, showed significant greater efficacy in a phase III clinic trial than free paclitaxel.95 In a clinical trial, the tumor response rate was nearly double for patients who received Abraxane® compared to those who received free paclitaxel. But, this prodrug revealed undesirable side effects such as low blood cell count, numbness, tingling or burning in the hands or feet, fatigue and weakness.
![Representative images of DU cells treated with paclitaxel (A) and Ac-[Lys-Aib-Cys(CH2CO-2′-Pac)]4-NH2 (tetra) (B)84 (reprinted with permission from S. Papas, T. Akoumianaki, C. Kalogiros, L. Hadjiarapoglou, P. A. Theodoropoulos and V. Tsikaris, J. Pept. Sci., 2007, 13, 662, Copyright (2007) European Peptide Society and John Wiley & Sons, Ltd).](/image/article/2010/PY/b9py00351g/b9py00351g-f6.gif) | ||
| Fig. 6 Representative images of DU cells treated with paclitaxel (A) and Ac-[Lys-Aib-Cys(CH2CO-2′-Pac)]4-NH2 (tetra) (B)84 (reprinted with permission from S. Papas, T. Akoumianaki, C. Kalogiros, L. Hadjiarapoglou, P. A. Theodoropoulos and V. Tsikaris, J. Pept. Sci., 2007, 13, 662, Copyright (2007) European Peptide Society and John Wiley & Sons, Ltd). | ||
In order to develop novel prodrugs for synergistic drug efficacy, it would be desirable to bind two different drugs on enzymatically cleavable substrates. The Kratz group96 recently reported synthesis of a so-called dual-acting prodrug containing paclitaxel and doxorubicin as drugs and two arms consisting of a carboxyl group and an activated 1,6-self-immolative para-aminobenzyloxy carbonyl spacer together with a cathepsin B cleavable dipeptide Phe-Lys as linkers (Fig. 7). Although they did not show any anticancer treatment potential of the prodrugs, the prodrug binds rapidly and selectively to the cysteine-34 position of endogenous serum albumin and is cleaved specifically by cathepsin B releasing the free drug.
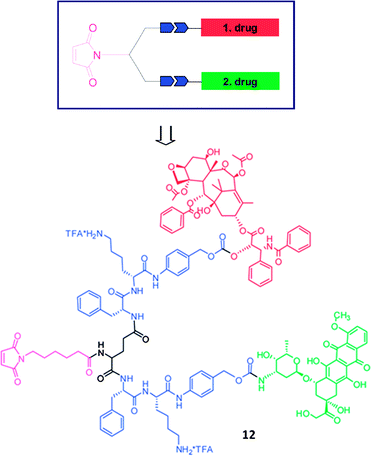 | ||
| Fig. 7 General structure of a dual-acting prodrug and the structure of the target compound 1296 (reprinted with permission from K. Abu Ajaj, M. L. Biniossek and F. Kratz, Bioconjugate Chem., 2009, 20, 390, Copyright (2009) American Chemical Society). | ||
Paclitaxel has been considered to induce cumulative neurotoxicity which may be attributed to its capability to block the polymerization of microtubules that are involved in axoplasmic transport.97 It appears to be related to dose level and cumulative dose98 that may be controlled by polymer-conjugated prodrugs well designed like this dual-acting prodrugs. However, problems associated with neurotoxicity against paclitaxel may not be so easily overcome by the prodrug strategy.
Tumor-targeting photoresponsive conjugates
As described in the Introduction section, one of paclitaxel's drawbacks is that it has little or no specificity for target tumor cells. In order to solve this problem, prodrug strategy99–103 focused on tumor-targeting prodrugs conjugates which can achieve a high local concentration of antitumor drugs. Maeda et al. reported that prodrugs conjugated to macromolecules showed interesting properties accumulating in tumor tissues because of an EPR effect which is applicable tumor tissue at a much higher concentration than that only to polymers.104,105 They elucidated that tumor-selective macromolecules and monoclonal antibodies (mAbs) can be used as carriers to tumor tissues which are ideal for delivery of macromolecular anticancer agents. Some of them showed good prospects, but none of them are yet available for clinical use.106,107Kiso and co-workers,108–111 by using the concept of photodynamic therapy (PDT) and so-called ‘caged’ compounds,112–116 prepared the first photoresponsive prodrug of paclitaxel. Caged compounds found various applications in life sciences to monitor biological processes.117–121 Especially, to study Alzheimer's disease122,123 the photo-triggered amyloid β peptide 1–42 (Aβ 1–42), isopeptide containing a photocleavable group, was used.124 They designed a tumor-targeting photoresponsive prodrug of paclitaxel, namely phototaxel, that has a coumarin derivative conjugated to an amino group moiety of isotaxel (O-acyl isoform of paclitaxel).
The prodrug, phototaxel, was expected to be selectively activated to isotaxel through photoirradiation by 430 nm light with cleavage of a coumarin derivative which has been reported to be thermally stable, water-soluble, and rapidly photolyzed by visible light.114,119,120 Conclusively, paclitaxel was released with a reasonable conversion time by O–N intramolecular acyl migration (Fig. 8).124–126
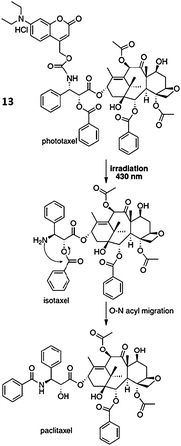 | ||
| Fig. 8 Releasing mechanism of paclitaxel from its photoresponsive prodrug 13108 (reprinted with permission from M. Skwarczynski, M. Noguchi, S. Hirota, Y. Sohma, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2006, 16, 4492, Copyright (2006) Elsevier Ltd.). | ||
This photolytic cleavage and release of coumarin derivative was investigated by HPLC analysis, as shown in Fig. 9 and Fig. 10, and the results indicate the time dependency of photolysis of prodrug 13 and release of coumarin derivative and paclitaxel. A new photoresponsive prodrug is practically applicable to the current photodynamic therapy.
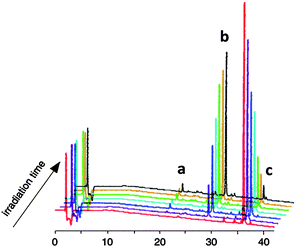 | ||
| Fig. 9 HPLC charts for prodrug 13 (c) subjected to irradiation followed by incubation at RT to induce migration, detected at 230 nm (a: 7-N,N-diethylamino-4-hydroxymethyl coumarin (DECM), b: paclitaxel) (line colors correspond to the irradiation time in the same manner as for absorption spectra)108 (reprinted with permission from M. Skwarczynski, M. Noguchi, S. Hirota, Y. Sohma, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2006, 16, 4492, Copyright (2006) Elsevier Ltd). | ||
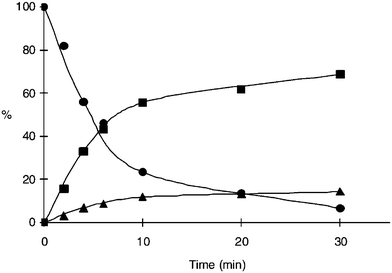 | ||
| Fig. 10 Time course of photolysis of prodrug 13 (●) and release of 7-N,N-diethylamino-4-hydroxymethyl coumarin (DECM) (▲) and paclitaxel (■) in 0.1 M PB/MeOH (1:1) at 430.6 nm. The percentage was determined by HPLC108 (reprinted with permission from M. Skwarczynski, M. Noguchi, S. Hirota, Y. Sohma, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2006, 16, 4492, Copyright (2006) Elsevier Ltd). | ||
Phototaxel, which was developed for photoresponsible and deliverable prodrug application, is completely insoluble in water. Such a prodrug can cause serious problems in the practical application. Thus, more recently, Kiso's group expanded their approach to prepare a water-soluble photoresponsive prodrug of paclitaxel.127 They designed new phototaxels (carbonate-type prodrug 14 and carbamate-type prodrug 15, Scheme 4), with a new coumarin-based highly watersoluble photocleavable protecting group, one of which indicated high water solubility, high stability in the dark and appropriate kinetics for parent drug release by UV light irradiation (prodrug 15, Scheme 4). These novel coumarin-based photoresponsive conjugates had a water-solubility of 100 mg ml−1 or more. These paclitaxel conjugates released the parent drug, paclitaxel, rapidly and efficiently when exposed to 365 nm UV light irradiation, while laser activation at 355 nm revealed to lead extensive decomposition of the conjugates. Tumor-tissue targeting after administration could be achieved for such prodrugs by selective light delivery, like to that used in photodynamic therapy.
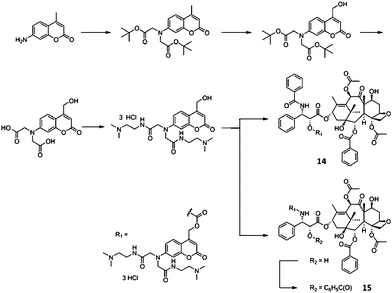 | ||
| Scheme 4 Synthesis of phototaxels 14 and 15127 (reprinted with permission from M. Noguchi, M. Skwarczynski, H. Prakash, S. Hirota, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem., 2008, 16, 5389, Copyright (2008) Elsevier Ltd). | ||
Saccharide conjugates
Tabrizian and co-workers128 reported the first prodrug, where the drug is linked to a biocompatible polymer through a hydrolyzable bond, and the layer-by-layer (LbL) technique, which had been used in polyelectrolyte multilayers (PEMs) fabrications129–132 (Fig. 11). In other words, they studied the possibility of self-assembling paclitaxel-loaded PEMs with a hyaluronan ester prodrug of paclitaxel.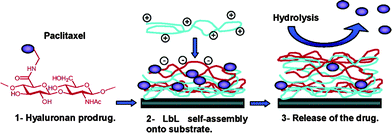 | ||
| Fig. 11 Delivery platform for hydrophobic drugs: prodrug approach combined with self-assembled multilayers128 (reprinted with permission from B. Thierry, P. Kujawa, C. Tkaczyk, F. M. Winnik, L. Bilodeau, and M. Tabrizian, J. Am. Chem. Soc., 2005, 127, 1626, Copyright (2005) American Chemical Society). | ||
Fig. 12 shows the structure of the HA prodrug of paclitaxel (HA-Pac), which is formed by linking paclitaxel to hyaluronan (HA) containing a labile succinate ester linkage.128,133 Multilayers made by HA and chitosan (CH) have previously been shown to recover blood compatibility of injured arteries, reducing the risks of post-angioplasty restenosis in blood vessels.134 Combining the LbL technique with complex geometries, living tissues, nanoparticles or microparticles, this approach could be of a matter of grave concern in the design of new therapeutic strategies for drug delivery.
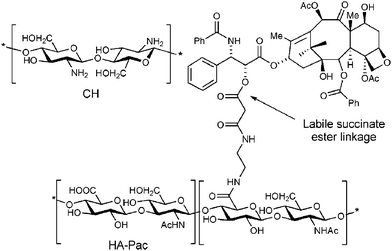 | ||
| Fig. 12 The structure of the HA prodrug of paclitaxel (HA-Pac) and CH128 (reprinted with permission from B. Thierry, P. Kujawa, C. Tkaczyk, F. M. Winnik, L. Bilodeau, and M. Tabrizian, J. Am. Chem. Soc., 2005, 127, 1626, Copyright (2005) American Chemical Society). | ||
Also the Schmidt group135 recently synthesized new glucuronide conjugates with allyl carbonates and allyl ester, which were used as protecting groups of the glucuronic moiety. The use of conventional protection of the glucuronyl unit could not be applied in the presence of acid- or alkali-sensitive groups in the drug framework. In contrast, they reported the use of glucuronyl derivatives with a fully allyl protection group and its final deprotection step with palladium leading to the planned, tripartite prodrug, whose structure is presented in Fig. 13.
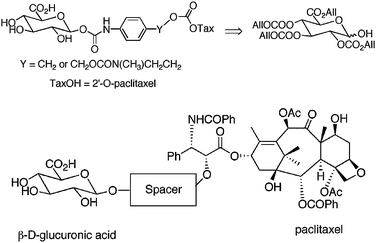 | ||
| Fig. 13 The structure of paclitaxel prodrugs135 (reprinted with permission from A. E. Alaoui, F. Schmidt, C. Monneret, and J. C. Florent, J. Org. Chem., 2006, 71, 9628, Copyright (2006) American Chemical Society). | ||
Glucose uptake into cells is performed mainly by GLUTs, that is a family of membrane proteins. Cancer cells generally have higher expression levels of GLUTs than normal cells.106,136–138 Most recently, the paclitaxel based prodrugs with glycan conjugation, 16–19 in Fig. 14, using either ether or ester bonds as linkers between the 2′-position of paclitaxel and glucose or glucuronic acid, were introduced by the Chen group.139Fig. 14 shows that the structures of two glucose and two glucuronic acid-derived paclitaxel prodrugs: 16, 17, 18, and 19. These prodrugs are more water soluble than paclitaxel, and they also have improved specific cell targeting effect. Prodrug 16 showed the most cytotoxicity against cancer cells and was able to cause morphological changes in tubulin and chromosomal aggregation in cells (NPC-TW01), which could induce cell cycle arrest or cell death through apoptosis. Fluorescence microscopy images show the relative position of fluorescence-labeled prodrug 16 and tubulin. Panel a and b represent the distributions of tubulin and the prodrug, respectively. Panel c shows the merged images. Fig. 15 shows colocalization of the prodrug and tubulin. According to their claim these prodrugs may be potent drug candidates for therapeutic applications and the glycan conjugation approach is a useful method for improvement of the targeted delivery of other drugs to cancer cells that express higher levels of GLUTs.
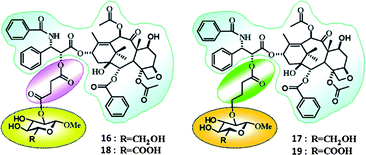 | ||
| Fig. 14 Structures of glucose-based (16 and 17) and glucuronic acid-based (18 and 19) paclitaxel prodrugs139 (reprinted with permission from Y. S. Lin, R. Tungpradit, S. Sinchaikul, F. M. An, D. Z. Liu, S. Phutrakul and S. T. Chen, J. Med. Chem., 2008, 51, 7428, Copyright (2008) American Chemical Society). | ||
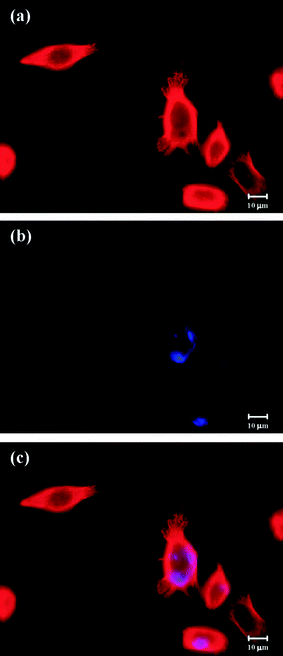 | ||
| Fig. 15 Colocalization of prodrug 16 and tubulin in NPC-TW01 cells under fluorescence microcopy139 (reprinted with permission from Y. S. Lin, R. Tungpradit, S. Sinchaikul, F. M. An, D. Z. Liu, S. Phutrakul and S. T. Chen, J. Med. Chem., 2008, 51, 7428, Copyright (2008) American Chemical Society). | ||
One of the most recent reports describes a new platform for oral delivery of paclitaxel, which was developed through treating the drug with a chemosensitizer which interacts with the cytosolic domains of P-gp and its ATP binding site140–142 or chemical conjugation of paclitaxel to a low molecular weight chitosan (LMWC).143 Chitosan has been widely used in the pharmaceutical field due to its biodegradable, biocompatible, mucoadhesive, and nontoxic properties. Moreover, LMWC has lower toxicity, higher water solubility, and a narrower molecular weight distribution in comparison with the conventional high molecular weight chitosan (HMWC).144–146 The LMWC-paclitaxel conjugate (Scheme 5) showed higher water solubility (>1 mg mL−1) than the native paclitaxel and had much higher IC50 values against human cancer cell lines.143 Also, the conjugate-treated group indicated suppression of tumor growth, that was comparable to the current clinical formulation of paclitaxel after oral administration to mice bearing xenograft or allograft.
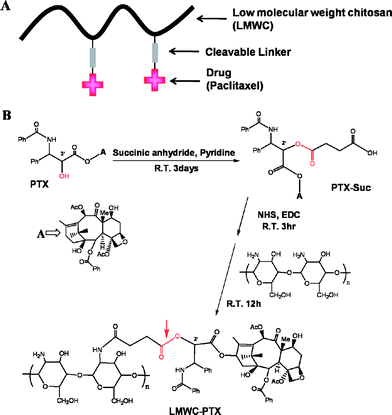 | ||
| Scheme 5 A schematic diagram (A) and synthesis scheme (B) of LMWC-PTX; the arrow indicates the cleavable bond between LMWC and PTX143 (reprinted with permission from E. H. Lee, J. J. Lee, I. H. Lee, M. K. Yu, H. J. Kim, S. Y. Chae, and S. Y. Jon, J. Med. Chem., 2008, 51, 6442, Copyright (2008) American Chemical Society). | ||
The synthesis of paclitaxel prodrug with ester-linked oligosaccharide by chemo-enzymatic methods, which include enzymatic transglycosylations with α-glucosidase and cyclodextrin glucanotransferase (CGTase), is shown in Scheme 6.147 The water-solubility of paclitaxel–sugar conjugates was higher than that of paclitaxel. The modification of paclitaxel with a longer oligosaccharide chain at the carbon 7 position decreased the in vitro cytotoxicity of paclitaxel against KF human ovarian cancer cells (Table 3).
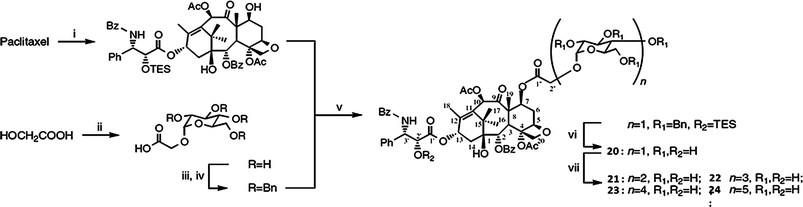 | ||
| Scheme 6 A synthesis scheme for paclitaxel-sugar conjugates: (i) TESCl, imidazole, DMAP; (ii) a-glucosidase, maltose, DMSO–H2O (1:4, v/v); (iii) BnBr, NaH, DMF; (iv) KOH; (v) EDCI, DMAP, CH2Cl2; (vi) H2, Pd black, HOAc–H2O (9:1, v/v); (vii) CGTase, starch, Na–Pi buffer (25 mM)147 (reprinted with permission from K. Shimoda, H. Hamada and H. Hamada, Tetrahedron Lett., 2008, 49, 601, Copyright (2007) Elsevier Ltd). | ||
Interaction between protein and prodrugs
The interaction between drugs and constituents of body fluids such as serum proteins is of particular importance because it strongly affects the in vivo activity of drugs and their transport. Drug binding to proteins such as human serum albumin (HSA) can be an important determinant of pharmacokinetics, restricting the unbound concentration, affecting distribution and excretion as well as competition in transport processes. Bovine serum albumin (BSA) is often used as model protein.Paclitaxel is known for strong interactions, for example, with mammalian tubulin preventing the cell division during mitosis. Nogales et al.148 showed that tubulin is a heterodimer protein consisting of α- and β-tubulin containing three domains, one being a binding site for other proteins, the second one that interacts with nucleotides and the third one to bind paclitaxel. While Nogales et al. found that the protein loses its flexibility through the interaction with paclitaxel,148,149 they concluded from molecular dynamics studies that paclitaxel affects the interaction between the M and the H1-S2 loops of adjacent tubulin dimers (in the vicinity of the amino acids Lys-19, Val-23, Asp-26, His-227, and Phe 270150), which leads to a more stable inter-protofilament interaction.
At the same time this causes, however, a significant increase in mobility and flexibility of the β-tubulin that surrounds the nucleotide and makes tight contact with the α-tubulin of the tubulin dimer in the protofilament.
As a result any conformational changes that might be caused by nucleotide hydrolysis are hampered, and the protofilament remains in a straight and stable conformation unable to dissociate.71–76,151–155
The interaction of paclitaxel and mPEG can change when both are combined to a prodrug where steric effects might prevent mutual interaction of the terpenoid taxane structure element or interaction with binding sites in albumin or other proteins. For interaction with tubulin the polymer chain has to be removed from the drug. The PEG-albumin interaction, as suggested by Azegami et al.,156 however, were not studied in detail. One study157 found predominantly repulsive interaction between albumin and PEG by high-resolution force spectroscopy.
From our studies of the behaviour of PP7 in aqueous solution71–75 and in particular from the phase diagram76 it was concluded that the PEG-chain coils around the paclitaxel moiety of the prodrug, thus preventing the drug from interacting with other paclitaxel molecules since neither a crystalline nor a glassy phase of paclitaxel (Tm = 220 °C, Tg = 160 °C)152 is observed in the prodrug where paclitaxel is bound to PEG with a degree of polymerization around 114 (equivalent to an average molar mass of 5,000 g mol−1, polydispersity index Đ = 1.05).
From the examples given above it is clear that paclitaxel interacts with proteins of different type. It was also shown by Trynda-Lemiesz158 that paclitaxel changes the conformation of albumin such that the CD-curve with the typical double-peak signature at 209 nm and 220 nm (n → π*-transition in an α-helical structure) becomes smaller with increasing paclitaxel content. It was recently shown by Hess et al.71–76,151–155 that this effect occurs even more strongly with PP7 and that pure mPEG 5000 does not cause this effect. It leaves the albumin conformation unchanged. Competition with other substances that can interact with the docking sites of albumin is possible. That can be of relevance in a multi-drug therapy as well as blocking agent for the mitosis as in transport processes, since albumin is – among other proteins – a transport protein for lipophilic substances in the polar serum. Correspondingly, an anticancer formulation with paclitaxel and albumin is commercially available as ABRAXANE™ (American BioScience, Inc.).159,160
The interaction of paclitaxel and the prodrug PP7 with albumin (HSA and BSA) has been investigated in aqueous buffer solution using the intrinsic and the extrinsic fluorescence of the amino acid Trp.151–155 Enzymatic activity of albumin and degradation of PP7 by the protein was not observed. Sudlow site I houses one paclitaxel molecule in Subdomain IIA near tryptophan Trp 214. Subdomain IIA ranges from the amino acids 199 (Lys) to 291 (Ala) and forms a hydrophobic pocket inside the water-soluble protein.161 Because of its chemical structure and morphology, albumin is known as one of the most important transport proteins. The amino acid Trp in the core of the structure is an important fluorescence probe in the centre of BSA and HSA. BSA has a second Trp that, in contrast, is located at number 134, close to the surface of the protein and it is believed that it is in a much more polar environment when compared with Trp 214. A clear spectroscopic distinction between the two sites by fluorescence or UV-spectroscopy is not described. Excitation at 295 nm discriminates from the fluorescence of the aminoacids of Phe and Tyr and yields an intrinsic fluorescence around 340 nm, bathochromic shifted in a more polar environment. The strong extrinsic fluorescence that is produced by a warfarin-Trp 214 complex is excited at 330 nm with a broad emission maximum at 380 nm and is very useful for the analysis of drug-albumin interaction in Sudlow site I.
Both, the intrinsic and the extrinsic fluorescence are influenced (quenched) by the presence of paclitaxel and PP7 and it was shown71–76,151–155 that even the paclitaxel of the prodrug can penetrate albumin and come closer to warfarin than 5 Å.154,155 It was shown that the protons on carbon C3, C4, C5, C7 and C20 (see Fig. 16) are close to the CH3-protons near the keto/semiketal group of warfarin.
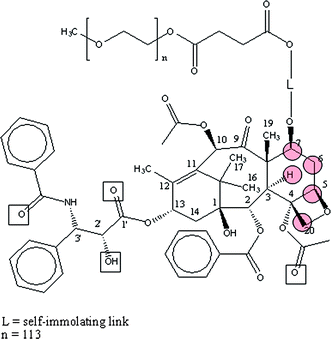 | ||
| Fig. 16 Prodrug PP7, a water-soluble PEG-paclitaxel conjugate (squares: hydrogen bonds to tubulin Arg 218, Arg 222, Lys 199, Hs 242; circles: interaction with hydrogen in warfarin (in albumin)).154 | ||
Paal and co-workers162 have identified a secondary paclitaxel docking site in the cleft between Sudlow domains I and II at the entrance to the hydrophobic core of albumin. They locate the primary docking site at the interface of subdomain IIA and IIIA, near Trp 214, with the side group emanating from C13 (see Fig. 16) buried in the drug binding site I in Subdomain IIA, so that the core of the molecule is more or less pointing upwards in the channel.
With warfarin occupying site I and considering the interactions of paclitaxel-warfarin concentrated in the taxane core part of paxlitaxel the orientation of the drug appears to be inverted, with the taxane core buried in the hydrophobic albumin channel and close to the interacting groups of the warfarin. In particular, this seems to be sterically preferred when paclitaxel is PEGylated and the polymer chain requires additional space.155 At least warfarin as a competitor for docking sites of albumin is not replaced by paclitaxel or the prodrug PP7. It can be sufficient to dock PP7 onto a different site with the result of deformation of the main drug binding cavities in the hydrophobic interior of albumin to cause changes in the fluorescence of the complex.
The efficacy of pharmaceutical drugs strongly depends on their solubility in aqueous media, stability, and distribution in the body. Once administered to the body, any substance will interact with many other substances, experiencing competing reactions and defence mechanisms present in a living biological system. These properties are not only influenced by their intrinsic solution properties but also by their specific or unspecific interaction with constituents of the body fluid such as blood or serum, and their transport properties through membranes and interaction with tissue. The information about binding of paclitaxel to serum albumin and the affinity binding site of the drug on the protein molecule is of special importance as the multi-drug therapy, for example, in combinations of paclitaxel with cisplatin or carboplatin, two other powerful cytostatica.
What can presently be stated is that there is some interaction between albumin and PP7 that causes a major change of the close environment of the Trp 214-warfarin complex (Fig. 17). The influence of the presence of a ligand on the conformation of a protein was, for example, shown for warfarin-albumin using CD spectroscopy,162,163 and also our own CD experiments have shown a concentration-depending influence of the presence of PP7 on the Cotton-effect of BSA.164 Warfarin is not necessarily replaced by the paclitaxel coming with PP7. It can be sufficient to dock PP7 onto a different site with the result of a deformation of the main drug binding cavities in the hydrophobic interior of albumin to cause changes of the fluorescence of the complex.
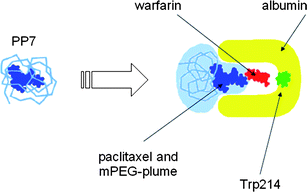 | ||
| Fig. 17 Dethreading of the paclitaxel core from the mPEG shell and filing of paclitaxel into albumin retracting warfarin from its docking site near TRP 214, hence decreasing the extrinsic fluorescence of the Trp-warfarin complex.153 | ||
Conclusions and perspectives
In this review we have summarized and discussed the various advantages and achievements of paclitaxel prodrug strategies including macromolecular conjugates carrier linked prodrugs, bioprecursors, and tumor-targeting chemical delivery systems. Use of polymer conjugates and their implications in delivering paclitaxel seem to be crucial in drug administration and therapeutics. The bioactive macromolecules can be delivered more efficiently by being converted into a prodrug conjugated form. But improved efficacy of the prodrugs over paclitaxel has not been confirmed in clinical trials. Only a few prodrugs of paclitaxel have reached clinical trials. The polymer prodrug approaches applied to paclitaxel, however, have shown a promising future for the development of anticancer therapy for several reasons such as targeting, drug efficacy, water solubility and physicochemical properties.The water soluble paclitaxel conjugated to polymers would allow us to utilize higher equivalent doses of paclitaxel in cancer treatment and the prodrugs can be effective in paclitaxel-resistant cancer cells because of their high hydrophilicity.
Even though many problems in the design and development of paclitaxel prodrugs were successfully overcome as mentioned in this review, more study is still needed to develop highly effective and clinically acceptable prodrugs of paclitaxel. However, we expect that prodrugs with more advanced properties, such as those with micellar conjugates, self-immolating linkers for adjusting the hydrolysis rate, albumin-bound nanoparticles, protein conjugated or tumor-targeting photoresponsive conjugates, those that are activated by cancer cell overexpressed enzymes, and those with drug conjugated to polymers, should be able to make critical contributions to anticancer chemotherapy in the foreseeable future.
The interaction between drugs and body fluids is of particular importance in prodrug application because it strongly affects the in vivo activity of drugs and their transport. Improved efficacy of the prodrugs over paclitaxel has not been yet confirmed in clinical trials. We strongly feel that much more extensive structural and clinical studies of paclitaxel prodrugs are worthwhile to pursue.
Acknowledgements
This study was supported by research funds from Chosun University, 2008.References
- J. Wang, L. S. Li, Y. L. Feng, H. M. Yao and X. H. Wang, Chin. Med. J., 1993, 35, 441 Search PubMed.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325 CrossRef.
- W. P. McGuire, E. K. Rowinsky, N. B. Rosenheim, F. C. Grumbine, D. S. Ettinger, D. K. Armstrong and R. C. Donehower, Ann. Int. Med., 1989, 111, 273 Search PubMed.
- E. K. Rowinsky, L. A. Cazenave and R. C. Donehower, J. Natl. Cancer Inst., 1990, 82, 1247 CrossRef CAS.
- F. A. Holmes, R. S. Walters, R. L. Theriault, A. D. Forman, L. K. Newton, M. N. Raber, A. U. Buzdar, D. K. Frye and G. N. Hortobagyi, J. Natl. Cancer Inst., 1991, 83, 1797 CrossRef CAS.
- G. Sarosy, E. Kohn, D. A. Stone, M. Rothenberg, J. Jacob, D. O. Adamo, F. P. Ognibene, R. E. Cunnion and E. Reed, J. Clin. Oncol., 1992, 10, 1165 CAS.
- E. K. Rowinsky and R. C. Donehower, N. Engl. J. Med., 1995, 332, 1004 CrossRef CAS.
- C. M. Spencer and D. Faulds, Drugs, 1994, 48, 794 CrossRef CAS.
- J. T. Thigpen, Semin. Oncol., 2000, 27(3 Suppl. 7), 1 CAS.
- A. Y. Chang, J. Rubins, R. Asbury, L. Boros and L. F. Hui, Semin. Oncol., 2001, 28(4 Suppl. 14), 10 CrossRef CAS.
- M. Ishitobi, E. Shin and N. Kikkawa, Int. J. Clin. Oncol., 2001, 6, 55 CrossRef CAS.
- R. Panchagnula, Int. J. Pharm., 1998, 172, 1 CrossRef CAS.
- N. Kumar, J. Biol. Chem., 1981, 256, 10435 CAS.
- J. J. Manfredi and S. B. Horwitz, Pharmacol. Ther., 1984, 25, 83 CrossRef CAS.
- P. Schiff, J. Fant and S. B. Horwitz, Nature, 1979, 277, 665 CAS.
- E. K. Rowinsky, R. C. Donehower, R. J. Jones and R. W. Tucker, Cancer Res., 1988, 48, 4093 CAS.
- M. V. Blagosklonny and T. Fojo, Int. J. Cancer, 1999, 83, 151 CrossRef CAS.
- P. A. Theodoropoulos, H. Polioudaki, O. Kostaki, S. P. Derdas, V. Georgoulias, C. Dargemont and S. D. Georgatos, Cancer Res., 1999, 59, 4625 CAS.
- C. M. Spencer and D. Faulds, Drugs, 1994, 48, 794 CrossRef CAS.
- E. K. Rowinsky, N. Onetto, R. M. Canetta and S. G. Arbuck, Semin. Oncol., 1992, 19, 646 CAS.
- J. T. Thigpen, J. A. Blessing, H. Ball, S. J. Hummel and R. J. Barrett, J. Clin. Oncol., 1994, 12, 1748 CAS.
- R. V. Chari, Adv. Drug Delivery Rev., 1998, 31, 89 CrossRef CAS.
- H. Gelderblom, J. Verweij, K. Nooter and V. Sparreboom, Eur. J. Cancer, 2001, 37, 1590 CrossRef CAS.
- N. Onetto, R. Canetta, B. Winograd, R. Catane, J. Grechko, J. Burroughs and M. Rozencweig, J. Natl. Cancer Inst. Monogr., 1993, 15, 131.
- D. Mazzo, J. J. Nguyen-Huu, S. Pagniez and P. Denis, Am. J. Health Syst. Pharm., 1997, 54, 566 CAS.
- R. Gregory and A. F. DeLisa, Clin. Pharm., 1993, 12, 401 Search PubMed.
- E. K. Rowinsky, E. A. Eisenhauer, V. Chaudhry, S. G. Arbuck and R. C. Donehower, Semin. Oncol., 1993, 20, 1 CAS.
- P. Crosasso, M. Ceruti, P. Brusa, S. Arpicco, F. Dosio and L. Cattel, J. Controlled Release, 2000, 63, 19 CrossRef CAS.
- K. H. Bae, Y. Lee and T. G. Park, Biomacromolecules, 2007, 8, 650 CrossRef CAS.
- B. D. Tarr, T. G. Sambandan and S. H. Yalkowsky, Pharm. Res., 1987, 4, 162 CrossRef CAS.
- S. K. Dordunoo, J. K. Jackson, L. A. Arsenault, M. C. Oktaba, W. L. Hunter and H. M. Burt, Cancer Chemother. Pharmacol., 1995, 36, 279 CAS.
- P. Kan, Z. B. Chen, C. J. Lee and I. M. Chu, J. Controlled Release, 1999, 58, 271 CrossRef CAS.
- A. Sharma and R. M. Straubinger, Pharm. Res., 1994, 11, 889 CrossRef CAS.
- R. T. Liggins and H. M. Burt, Adv. Drug Delivery Rev., 2002, 54, 191 CrossRef CAS.
- S. S. Feng, L. Mu, K. Y. Win and et al, Curr. Med. Chem., 2004, 11, 413 CrossRef CAS.
- Y. C. Dong and S. S. Feng, Biomaterials, 2004, 25, 2843 CrossRef CAS.
- H. M. Deutsch, J. A. Glinski, M. Hermandez, R. D. Haugwitz, L. V. Narayanan, M. Suffness and L. H. Zalkow, J. Med. Chem., 1989, 32, 788 CrossRef CAS.
- Y. S. Lin, R. Tungpradit, S. Sinchaikul, F. M. An, D. Z. Liu, S. Phutrakul and S. T. Chen, J. Med. Chem., 2008, 51, 7428 CrossRef CAS.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253 CrossRef CAS.
- H. Maeda, L. W. Seymour and Y. Miyamoto, Bioconjugate Chem., 1992, 3, 351 CrossRef CAS.
- X. Zhang, Y. Li, X. Chen, X. Wang, X. Xu, Q. Liang, J. Hu and X. Jing, Biomaterials, 2005, 26, 2121 CrossRef CAS.
- R. B. Greenwald, C. W. Gibert, A. Pendri, C. D. Conover, J. Xia and A. Martinez, J. Med. Chem., 1996, 39, 424 CrossRef CAS.
- C. Li, D. F. Yu, R. A. Newman, F. Cabral, L. C. Stephen, N. Hunter, L. Milas and S. Wallace, Cancer Res., 1998, 58, 2404 CAS.
- N. Mongell, E. Pesenti, A. Suarato and G. Biasoli, U.S. Pat., 5,473,055, 1994, p. 831 Search PubMed.
- H. J. Lee, K. R. Lee and T. G. Park, Bioconjugate Chem., 2008, 19, 1319 CrossRef CAS.
- H. Mok and T. G. Park, Bioconjugate Chem., 2006, 17, 1369 CrossRef CAS.
- T. C. Laurent, Acta Oto-Laryngol., 1987, 104, 7 CrossRef.
- P. Herrlich, J. Sleeman, D. Wainwright, H. Konig, L. Sherman and H. Ponta, Cell Commun. Adhes., 1998, 6, 141 Search PubMed.
- C. L. Hall, B. Yang, X. Yang, S. Zhang, M. Turley, S. Samuel, L. A. Lange, C. Wang, G. D. Curpen, R. C. Savani, A. H. Greenburg and E. A. Turley, Cell, 1995, 82, 19 CrossRef CAS.
- Q. Hua, C. B. Knudson and W. J. Knudson, J. Cell Sci., 1993, 106, 365 CAS.
- H. Lee, H. Mok, S. Lee, Y. K. Oh and T. G. Park, J. Controlled Release, 2007, 119, 245 CrossRef CAS.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, J. Shu and H. Uyama, Chem. Commun., 2005, 4312 RSC.
- N. E. Larsen and E. A. Balazs, Adv. Drug Delivery Rev., 1991, 7, 279 CrossRef CAS.
- S. M. Ansell, U.S. Pat. 5, 534, 499, 1996 Search PubMed.
- K. Y. Hostetler and N. C. Sridhar, U.S. Pat. 5,484,809, 1996 Search PubMed.
- P. J. Stevens, M. Sekido and R. J. Lee, Pharm. Res., 2004, 21, 2153 CrossRef CAS.
- W. R. Perkins, I. Ahmad, X. Li, D. J. Hirsh, G. R. Masters, C. J. Fecko, J. K. Lee, S. Ali, J. Nguyen, J. Schupsky, C. Herbert, A. S. Janoff and E. Mayhew, Int. J. Pharm., 2000, 200, 27 CrossRef CAS.
- D. C. Rodrigues, D. A. Maria, D. C. Fernandes, C. J. Valduga, R. D. Couto, O. C. M. Ibanez and R. C. Maranhao, Cancer Chemother. Pharmacol., 2005, 55, 565 CrossRef CAS.
- T. Y. Zakharian, A. Seryshev, B. Sitharaman, B. E. Gilbert, V. Knight and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 12508 CrossRef CAS.
- M. O. Bradley, N. L. Webb, F. H. Anthony, P. Devanesan, P. A. Witman, S. Hemamalini, M. C. Chander, S. D. Baker, L. He, S. B. Horwits and C. S. Swindell, Clin. Cancer Res., 2001, 7, 3229 CAS.
- S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prud'homme and L. D. Mayer, J. Med. Chem., 2008, 51, 3288 CrossRef CAS.
- T. O. Harasym, P. G. Tardi, N. L. Harasym, P. Harvie, S. A. Johnstone and L. D. Mayer, Oncol. Res., 2007, 16, 361.
- L. D. Mayer and A. S. Janoff, Mol. Interventions, 2007, 7, 216 CrossRef CAS.
- T. O. Harasym, P. G. Tardi, S. A. Johnstone, M. B. Bally, A. S. Janoff and L. D. Mayer, Fixed Drug Ratio Liposome Formulations of Combination Cancer Therapeutics, in Liposome Technology, New York, USA, 2007 Search PubMed.
- A. Fahr, P. Hoogevest, S. May, N. Bergstand and M. L. S. Leigh, Eur. J. Pharm. Sci., 2005, 26, 251 CrossRef CAS.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253 CrossRef CAS.
- K. L. Hennenfent and R. Govindan, Ann. Oncol., 2006, 17, 735 CrossRef CAS.
- B. W. Jo, Kolon, U.S. Pat. 6,703,417, 2004 Search PubMed.
- B. W. Jo, Kolon, EP 01926177.5, 2002.
- B. W. Jo, Kolon, JP 3538606, 2004.
- M. Hess, B. W. Jo and M. Zähres, Mater. Res. Innovations, 2003, 7, 178 Search PubMed.
- J. S. Sohn, S. K. Choi, B. W. Jo, M. Hess and M. Zhäres, Macromol. Symp., 2003, 201, 163 CrossRef CAS.
- M. Hess, M. Zhäres and B. W. Jo, Macromol. Symp., 2004, 214, 351 CrossRef CAS.
- J. S. Sohn, S. K. Choi, B. W. Jo and M. Hess, Mat. Res. Innovations, 2005, 9, 85 Search PubMed.
- J. S. Sohn, B. W. Jo, M. Hess, K. Schwark, S. Dehne and M. Zhäres, Macromol. Symp., 2005, 225, 31 CrossRef CAS.
- J. S. Sohn, S. K. Choi, K. Schwark and M. Hess, e-Polymers, 2005, 007, 1 Search PubMed.
- J. S. Choi and B. W. Jo, Int. J. Pharm., 2004, 280, 221 CrossRef CAS.
- B. Y. Ryu, J. S. Sohn, M. Hess, S. K. Choi, J. K. Choi and B. W. Jo, J. Biomater. Sci., Polym. Ed., 2008, 19, 311 CrossRef CAS.
- A. Battaglia, A. Guerrini, E. Baldelli, G. Fontana, G. Varchi, C. Samorı and E. Bombardelli, Tetrahedron Lett., 2006, 47, 2667 CrossRef CAS.
- V. Guillemard and H. U. Saragovi, Cancer Res., 2001, 61, 694 CAS.
- S. S. Cohen, A Guide to the Polyamines, Oxford University Press, Oxford, 1998 Search PubMed.
- D. G. Vassylyev, H. Tomitori, K. Kashiwagi, K. Morikawa and K. Igarashi, J. Biol. Chem., 1998, 273, 17604 CrossRef CAS.
- R. A. Gardner, J. G. Delcros, F. Konate, F. Breitbeil, B. Martin, M. Sigman, M. Huang and O. Phanstiel, J. Med. Chem., 2004, 47, 6055 CrossRef CAS.
- S. Papas, T. Akoumianaki, C. Kalogiros, L. Hadjiarapoglou, P. A. Theodoropoulos and V. Tsikaris, J. Pept. Sci., 2007, 13, 662 CrossRef CAS.
- H. M. Deutsch, J. A. Glinski, M. Hernandez, R. D. Haugwitz, V. L. Narayanan, M. Suffness and L. H. Zalkow, J. Med. Chem., 1989, 32, 788 CrossRef CAS.
- T. A. Kirschberg, C. L. VanDeusen, J. B. Rothbard, M. Yang and P. A. Wender, Org. Lett., 2003, 5, 3459 CrossRef CAS.
- J. W. Lee, J. Y. Lu, P. S. Low and P. L. Fuchs, Bioorg. Med. Chem., 2002, 10, 2397 CrossRef CAS.
- X. Chen, C. Plasencia, Y. Hou and N. Neamati, J. Med. Chem., 2005, 48, 1098 CrossRef CAS.
- S. Wang, N. Z. Zhelev, S. Duff and P. M. Fischer, Bioorg. Med. Chem. Lett., 2006, 16, 2628 CrossRef CAS.
- C. Li, Adv. Drug Delivery Rev., 2002, 54, 695 CrossRef CAS.
- J. W. Singer, B. Baker, P. De Vries, A. Kumar, S. Shaffer, E. Vawter, M. Bolton and P. Garzone, Adv. Exp. Med. Biol., 2003, 519, 81 CAS.
- Y. Zou, H. Fu, S. Ghosh, D. Farquhar and J. Klostergaard, Clin. Cancer Res., 2004, 10, 7382 CrossRef CAS.
- F. Kratz and U. Beyer, Drug Delivery, 1998, 5, 281 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23, 7794 CrossRef CAS.
- K. Abu Ajaj, M. L. Biniossek and F. Kratz, Bioconjugate Chem., 2009, 20, 390 CrossRef CAS.
- E. K. Rowinsky, V. Chaudhry, D. R. Cornblath and R. C. Donehower, J. Natl. Cancer Inst. Monographs, 1993, 15, 107 Search PubMed.
- K. Kuroi and K. Shimozuma, Breast Cancer, 2004, 11, 92 CrossRef.
- P. S. Huang and A. Oliff, Curr. Opin. Genet. Dev., 2001, 11, 104 CrossRef CAS.
- M. J. Ferguson, F. Y. Ahmed and J. Cassidy, Drug Resist. Updates, 2001, 4, 225 CrossRef CAS.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253 CrossRef CAS.
- D. G. I. Kingston and D. J. Newman, Curr. Opin. Drug Discov. Dev., 2007, 10, 130 Search PubMed.
- S. Jaracz, J. Chen, L. V. Kuznetsova and I. Ojima, Bioorg. Med. Chem., 2005, 13, 5043 CrossRef CAS.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189 CrossRef CAS.
- H. Maeda, T. Sawa and T. Konno, J. Controlled Release, 2001, 74, 47 CrossRef CAS.
- C. Ferlini, I. Ojima, M. Distefano, D. Gallo, A. Riva, P. Morazzoni, E. Bombardelli, S. Mancuso and G. Scambia, Curr. Top. Med. Chem., 2003, 3, 133 CrossRef CAS.
- J. M. Nabholtz, J. M. Vannetzel, J. F. Llory and P. Bouffette, Clinical Breast Cancer, 2003, 4, 187 Search PubMed.
- M. Skwarczynski, M. Noguchi, S. Hirota, Y. Sohma, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem. Lett., 2006, 16, 4492 CrossRef CAS.
- M. Skwarczynski, Y. Sohma, M. Noguchi, T. Kimura, Y. Hayashi, Y. Hamada and Y. Kiso, J. Med. Chem., 2005, 48, 2655 CrossRef CAS.
- Y. Sohma, Y. Hayashi, M. Skwarczynski, Y. Hamada, M. Sasaki, T. Kimura and Y. Kiso, Biopolymers, 2004, 76, 344 CrossRef CAS.
- Y. Hayashi, M. Skwarczynski, Y. Hamada, Y. Sohma, T. Kimura and Y. Kiso, J. Med. Chem., 2003, 46, 3782 CrossRef CAS.
- K. Hayashi, K. Hashimoto, N. Kusaka, A. Yamazoe, H. Fukaki, M. Tasaka and H. Nozaki, Bioorg. Med. Chem. Lett., 2006, 16, 2470 CrossRef CAS.
- Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267 CrossRef CAS.
- V. R. Shembekar, Y. Chen, B. K. Carpenter and G. P. Hess, Biochemistry, 2005, 44, 7107 CrossRef CAS.
- S. Hirota, Y. Fujimoto, J. Choi, N. Baden, N. Katagiri, M. Akiyama, R. Hulsker, M. Ubbink, T. Okajima, T. Takabe, N. Funasaki, Y. Watanabe and M. Terazima, J. Am. Chem. Soc., 2006, 128, 7551 CrossRef CAS.
- T. Furuta, S. S. H. Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193 CrossRef CAS.
- Y. Sohma, Y. Hayashi, M. Skwarczynski, Y. Hamada, M. Sasaki, T. Kimura and Y. Kiso, Biopolymers, 2004, 76, 344 CrossRef CAS.
- Y. Hayashi, M. Skwarczynski, Y. Hamada, Y. Sohma, T. Kimura and Y. Kiso, J. Med. Chem., 2003, 46, 3782 CrossRef CAS.
- D. Geissler, Y. N. Antonenko, R. Schmidt, S. Keller, O. O. Krylova, B. Wiesner, J. Bendig, P. Pohl and V. Hagen, Angew. Chem., Int. Ed., 2005, 44, 1195 CrossRef CAS.
- R. O. Schonleber, J. Bendig, V. Hagen and B. Giese, Bioorg. Med. Chem., 2002, 10, 97 CrossRef CAS.
- M. Skwarczynski, Y. Sohma, M. Kimura, Y. Hayashi, T. Kimura and Y. Kiso, Bioorg. Med. Chem. Lett., 2003, 13, 4441 CrossRef CAS.
- W. M. Kazmierski, P. Bevans, E. Furfine, A. Spaltenstein and H. Yang, Bioorg. Med. Chem. Lett., 2003, 13, 2523 CrossRef CAS.
- K. B. Buck and J. Q. Zheng, J. Neurosci., 2002, 22, 9358 CAS.
- A. Taniguchi, Y. Sohma, M. Kimura, T. Okada, K. Ikeda, Y. Hayashi, T. Kimura, S. Hirota, K. Matsuzaki and Y. Kiso, J. Am. Chem. Soc., 2006, 128, 696 CrossRef CAS.
- Y. Sohma, A. Taniguchi, T. Yoshiya, Y. Chiyomori, F. Fukao, S. Nakamura, M. Skwarczynski, T. Okada, K. Ikeda, Y. Hayashi, T. Kimura, S. Hirota, K. Matsuzaki and Y. Kiso, J. Pept. Sci., 2006, 12, 823 CrossRef CAS.
- M. Skwarczynski and Y. Kiso, Curr. Med. Chem., 2007, 14, 2813 CrossRef CAS.
- M. Noguchi, M. Skwarczynski, H. Prakash, S. Hirota, T. Kimura, Y. Hayashi and Y. Kiso, Bioorg. Med. Chem., 2008, 16, 5389 CrossRef CAS.
- B. Thierry, P. Kujawa, C. Tkaczyk, F. M. Winnik, L. Bilodeau and M. Tabrizian, J. Am. Chem. Soc., 2005, 127, 1626 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- T. Serizawa, M. Yamaguchi and M. Akashi, Biomacromolecules, 2002, 3, 724 CrossRef CAS.
- J. F. Quinn and F. Caruso, Langmuir, 2004, 20, 20 CrossRef CAS.
- C. A. Constantine, S. V. Mello, A. Dupont, X. Cao, D. Santos Jr., O. N. Oliveira Jr., F. T. Strixino, E. C. Pereira, T. C. Cheng, J. J. Defrank and R. M. Leblanc, J. Am. Chem. Soc., 2003, 125, 1805 CrossRef CAS.
- K. C. Nicolaou, C. Riemer, M. A. Kerr, D. Rideout and W. Wrasidlo, Nature, 1993, 364, 464 CrossRef CAS.
- B. J. Thierry, F. M. Winnik, Y. Mehri and M. Tabrizian, J. Am. Chem. Soc., 2003, 125, 7494 CrossRef CAS.
- A. E. Alaoui, F. Schmidt, C. Monneret and J. C. Florent, J. Org. Chem., 2006, 71, 9628 CrossRef.
- M. M. Abouzied, E. S. Crawford and H. A. Nabi, J. Nucl. Med. Technol., 2005, 33, 145 Search PubMed.
- X. C. Nguyen, W. W. Lee, J. H. Chung, S. Y. Park, S. W. Sung, Y. K. Kim, Y. So, D. S. Lee, J. K. Chung, M. C. Lee and S. E. Kim, Eur. J. Radiol., 2007, 62, 214 CrossRef.
- J. M. Nabholtz, J. M. Vannetzel, J. F. Llory and P. Bouffette, Clin. Breast Cancer, 2003, 4, 187 Search PubMed.
- Y. S. Lin, R. Tungpradit, S. Sinchaikul, F. M. An, D. Z. Liu, S. Phutrakul and S. T. Chen, J. Med. Chem., 2008, 51, 7428 CrossRef CAS.
- J. S. Choi, B. W. Jo and Y. C. Kim, Eur. J. Pharm. Biopharm., 2004, 57, 313 CrossRef CAS.
- J. S. Choi and B. W. Jo, Int. J. Pharm., 2004, 280, 221 CrossRef CAS.
- J. S. Choi and S. C. Shin, Int. J. Pharm., 2005, 292, 149 CrossRef CAS.
- E. H. Lee, J. J. Lee, I. H. Lee, M. K. Yu, H. J. Kim, S. Y. Chae and S. Y. Jon, J. Med. Chem., 2008, 51, 6442 CrossRef CAS.
- M. Thanou, J. C. Verhoef and H. E. Junginger, Adv. Drug Delivery Rev., 2001, 52, 117 CrossRef CAS.
- M. N. Kumar, R. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. Domb, Chem. Rev., 2004, 104, 6017 CrossRef.
- S. Y. Chae, M. K. Jang and J. W. Nah, J. Controlled Release, 2005, 102, 383 CrossRef CAS.
- K. Shimoda, H. Hamada and H. Hamada, Tetrahedron Lett., 2008, 49, 601 CrossRef CAS.
- E. Nogales, S. Wolf and K. Downing, Nature, 1998, 391, 199 CrossRef CAS.
- A. Mitra and D. Sept, Biophys. J., 2008, 95, 3252 CrossRef CAS.
- M. Gupta, C. J. Bode, G. I. Georg and R. H. Himes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6394 CrossRef CAS.
- B. Wermeckes, M. Hess, S. Dehne, B. W. Jo and J. S. Sohn, Mater. Res. Innov., 2006, 10, 88.
- M. Hess, B. W. Jo, B. Wermeckes, S. Dehne, J. S. Sohn, S. Wunderlich and M. Zähres, Macromol. Symp., 2005, 231, 28 CrossRef.
- S. Wunderlich, M. Zähres, B. W. Jo and M. Hess, Macromol. Symp., 2006, 242, 71 CrossRef CAS.
- S. Wunderlich, M. Zähres, B. W. Jo and M. Hess, Macromol. Symp., 2007, 258, 101 CrossRef CAS.
- M. Hess, B. W. Jo and S. Wunderlich, Pure Appl. Chem., 2009, 81, 439 CrossRef CAS.
- S. Azegami, A. Tsuboi, T. Izumi, M. Hirata, P. L. Dublin, B. Wang and E. Kokufuta, Langmuir, 1999, 15, 940 CrossRef CAS.
- M. A. Rixman, D. Dean and C. Ortiz, Langmuir, 2003, 19, 9357 CrossRef CAS.
- L. Trynda-Lemiesz, Bioorg. Med. Chem., 2004, 12, 3269 CrossRef CAS.
- N. K. Ibrahim, B. Samuels, R. Page, D. Doval, K. M. Patel, M. K. Nair, S. C. Rao, P. Bhar, N. Desai and G. N. Hortobagyi, J. Clin. Oncol., 2005, 23, 6019 CrossRef CAS.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O'Shaughnessy, J. Clin. Oncol., 2005, 23, 7794 CrossRef CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052 CAS.
- K. Paal, A. Shkarupin and L. Beckford, Bioorg. Med. Chem., 2007, 15, 1323 CrossRef CAS.
- N. A. Brown and W. E. Muller, Pharmacology, 1978, 17, 233 CrossRef CAS.
- M. Hess, T. Paululat, J. Lauterbach, J. S. Sohn, B. W. Jo, to be published.
| This journal is © The Royal Society of Chemistry 2010 |