Polymer brushes: Applications in biomaterials and nanotechnology
Neil
Ayres
*
The University of Cincinnati, Department of Chemistry, 301 Clifton Court, PO Box 210172, Cincinnati, OH 45221. E-mail: ayresni@ucmail.uc.edu; Fax: +1 (513) 556-9239; Tel: +1 (513) 556-9280
First published on 11th December 2009
Abstract
Surface-confined macromolecules known as polymer brushes are being increasing applied to a variety of areas. As more information is gained on the molecular structure of polymer brushes and how they respond to environmental stimuli, these applications are becoming wider ranging and better defined. This review seeks to highlight recent contributions in two broad areas: biotechnology and nanotechnology. These are positions in which polymer brushes are well-suited to offer performance gains. Examples are given that describe the benefits of using a well defined, covalently bound, and densely grafted polymer including in areas such as prevention of bacterial adherence, cell attachment, electrochemistry, and formation of colloidal crystals.
Neil Ayres received his PhD in chemistry from The University of Warwick in 2003 where he worked with Prof. David Haddleton. After a post-doc with Charles McCormick at The University of Southern Mississippi he worked with William Brittain at The University of Akron studying stimuli-responsive polymer brushes. From there he spent two years as a post-doc at The University of Utah with Prof. David Grainger before becoming an assistant professor at The University of Cincinnati in 2008. His research interests include using polymer brushes to prevent bacterial and cell adherence to surfaces. |
1. Introduction
Research interest in surface-confined polymers has lead to many novel approaches to their synthesis, characterization, and application.1–8 These surface-confined macromolecular architectures are referred to as polymer brushes. Attachment of the chains by one end in close proximity forces the chains to stretch out into an extended conformation to minimize segment–segment overlaps.9Polymer brushes are typically prepared through physisorption or covalent attachment. Physisorption, unlike covalent attachment, suffers through thermal and solvent instability. Covalent attachment can be achieved using “grafting to” and “grafting from” techniques. “Grafting to” requires the tethering of preformed polymer chains to a surface. However, this technique often suffers from low grafting density (surface coverage) and film thickness due to the requirement of polymer molecules to diffuse through an existing attached polymer layer to reach the reactive sites decorating the surface. Therefore, steric hindrance for surface attachment increases as the thickness of the polymer film increases. As a result of these limitations, the “grafting from” approach has become the preferred option for the synthesis of polymer brushes. This approach uses a surface-immobilized initiator layer and subsequent in situpolymerization to generate the polymer brush. This is referred to as surface-initiated polymerization (SIP) (Fig. 1). | ||
| Fig. 1 “Grafting from” SIP consists of attaching a covalently bound initiator functionalized adlayer to a surface and performing an in situpolymerization. | ||
This technique has been used to prepare thick polymer films that are covalently anchored to the substrate with a high grafting density. This review will consider only surface-initiated “grafted from” prepared polymer brushes. The topic has been further constrained to focus on recent papers dealing with the case of non-patterned surfaces. However, many excellent papers have still been omitted and the interested reader is directed to pertinent reviews where possible (for example; refs. 1–8). The review is organized into three sections. First an overview is given of the macromolecular structure and surface characterization of polymer brushes; the ongoing convergence of polymer brushes and biomaterials is summarized; and finally a survey of the use of polymer brushes in selected areas of nanotechnology is presented.
2. Structure and surface characterization of polymer brushes
A description of the conformation of polymer brushes in good solvents was presented by Alexander10 and later refined by de Gennes.11 As grafting density increases, a single chain can be subdivided into ‘blobs’ where each blob contains a number of monomers. At high grafting the chains stretch to avoid excluded volume effects and the number of monomers per blob is much less than the total number of monomers in the chain. Each blob acts as a hard sphere. Therefore, the chain acts as a linear string of blobs stretched normal to the wall. Binder12 later argued that this image is too regular. Some chains can turn backwards, chain ends can be found anywhere in the brush, and the chains undergo non-uniform stretching. The result of this is that the blob diameter gradually increases in the direction normal from the surface, and the final blob is much larger in size accounting for a significant portion of the brush height. When brushes are fully stretched in good solvents the end-monomer density profiles changes so that there is exposure of the end-groups at the brush–solvent interface.13
Polymer brush height is typically in the size regime of 10–100 nm. This is the depth that is typically considered as a ‘surface-layer’ in materials science and probed by surface analytical techniques. The polymer brush community typically relies on ellipsometry, contact angle analysis and ATR FTIR spectroscopy where the information obtained is often restricted to bulk properties and global measurements. These techniques are more available and easier to execute than other ‘principle’ surface techniques, e.g.X-ray photoelectron spectroscopy (XPS) (also called electron spectroscopy for chemical analysis (ESCA)), NEXAFS, scattering experiments, and SIMS.14XPS is commonly used to accurately determine the elemental composition of polymer brush surfaces. This is a powerful surface technique that can be used to follow surface transformations. High-resolution region scans of particular elemental binding energies afford not only the fraction of that element present, but also individual binding environments (e.g. C–C vs. C–O vs. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). As a result of only photoelectrons from the surface layer of atoms (∼10 nm) being able to escape, XPS characterization is confined to obtaining information at the surface of a material. Near-edge X-ray absorption fine structure (NEXAFS) microscopy provides quantitative analytical capabilities at the mesoscopic length scale.15 This has been developed into a powerful polymer characterization tool, combining atomic specific soft X-ray spectroscopy with real-space techniques. NEXAFS has been used to show that RNAase A does not immobilize to poly(acrylic acid) brushes in a preferred orientation, and that semifluorinated side chains in thick styrenic polymer brushes orientate themselves towards the surface–air interface.16,17NEXAFS has been used with spectroscopic ellipsometry to follow a gradient of coverage of poly(acrylic acid) (PAA) brushes across a surface by focusing on the 1s → π*CO transition at 531 eV.18Secondary ion mass spectrometry (SIMS) is the mass spectrometry of ionized particles that are emitted when a surface is bombarded with energetic particles. The emitted or ‘secondary’ particles may be electrons, neutral species or ions. The majority of the species that are emitted are neutral particles, but it is the secondary ions that are detected. Examples of SIMS analysis of polymer brushes include depth profiling of poly(methacrylic acid) brushes with regard to RGD motif functionalization for cell adhesion applications,19 observing initiator grafting on mica substrates20 and depth profiling a diblock copolymer brush prepared by reversible addition fragmentation chain transfer (RAFT) SIP.21 Ivanov and coworkers used SIMS to characterize boronate-containing polymer brushes that were examined for mammalian cell attachment and ‘on-demand’ detachment.22 Ga+ ions were used as the primary ions, resulting in both positive and negative secondary ions being formed. SIMS was able to demonstrate the chemical functionalization of the substrate and subsequent coverage by the polymer brush. Neutron scattering has been used to study the polymerization of n-butyl methacrylate (nBMA) from spherical silica particles where it was shown that colloidal stability was conserved throughout the polymerization process with very little particle aggregation.23Neutron reflectivity has been used to examine stimuli-responsive brushes.24
O). As a result of only photoelectrons from the surface layer of atoms (∼10 nm) being able to escape, XPS characterization is confined to obtaining information at the surface of a material. Near-edge X-ray absorption fine structure (NEXAFS) microscopy provides quantitative analytical capabilities at the mesoscopic length scale.15 This has been developed into a powerful polymer characterization tool, combining atomic specific soft X-ray spectroscopy with real-space techniques. NEXAFS has been used to show that RNAase A does not immobilize to poly(acrylic acid) brushes in a preferred orientation, and that semifluorinated side chains in thick styrenic polymer brushes orientate themselves towards the surface–air interface.16,17NEXAFS has been used with spectroscopic ellipsometry to follow a gradient of coverage of poly(acrylic acid) (PAA) brushes across a surface by focusing on the 1s → π*CO transition at 531 eV.18Secondary ion mass spectrometry (SIMS) is the mass spectrometry of ionized particles that are emitted when a surface is bombarded with energetic particles. The emitted or ‘secondary’ particles may be electrons, neutral species or ions. The majority of the species that are emitted are neutral particles, but it is the secondary ions that are detected. Examples of SIMS analysis of polymer brushes include depth profiling of poly(methacrylic acid) brushes with regard to RGD motif functionalization for cell adhesion applications,19 observing initiator grafting on mica substrates20 and depth profiling a diblock copolymer brush prepared by reversible addition fragmentation chain transfer (RAFT) SIP.21 Ivanov and coworkers used SIMS to characterize boronate-containing polymer brushes that were examined for mammalian cell attachment and ‘on-demand’ detachment.22 Ga+ ions were used as the primary ions, resulting in both positive and negative secondary ions being formed. SIMS was able to demonstrate the chemical functionalization of the substrate and subsequent coverage by the polymer brush. Neutron scattering has been used to study the polymerization of n-butyl methacrylate (nBMA) from spherical silica particles where it was shown that colloidal stability was conserved throughout the polymerization process with very little particle aggregation.23Neutron reflectivity has been used to examine stimuli-responsive brushes.24
Foster at the University of Akron has used several techniques to obtain molecular level information of polymer brushes. His group has shown that the interfaces a diblock poly(methyl acrylate-block-deuterated styrene) (PMA-b-dPS) brush can be correlated over a range of thicknesses using X-ray reflectometry (XR).25 Reflectivity and longitudinal scans resulting from the interference of beams reflected from the air–film and film–substrate interfaces were in phase, indicating that the interfaces were correlated. This correlation existed despite the measurement occurring above the Tg for bulk PMA. Swelling of the brush by a good solvent was found to perturb this correlation however. Swelling leads to several changes in the brush, including an increased stretching of the individual chains, weakening of the layered character of the brush through elimination of a ‘sharp’ interface between blocks, alteration of the interfacial tension, and increasing the mobility of the chains. Later, in collaboration with Rühe's group, Foster's group studied thicker homopolymer brushes of PS and poly(n-butyl acrylate) (P(nBA)) prepared using free-radical SIP.26 They showed the surface of the brush followed a self-affine (self-similar) roughness model. This result agreed with their earlier conclusions. An interesting finding from this study was that tethered P(nBA)chains have very different surface dynamics from those of a viscous liquid of untethered P(nBA)chains. Additionally, the PS brush exists as a static surface structure, which is very different from the case of untethered PS chains. The concept of dry polymer brushes possessing static surface structures was further shown using X-ray photon correlation spectroscopy in a later article.27 These findings are interesting to compare to theoretical studies that showed slight variations in polydispersity of the chains could result in dramatic effects regarding whether that chain was stretched out of the bulk brush or collapsed and trapped within the brush.28 The result of such a phenomenon would result in an extra ‘polydispersity’ of the chains comprising the polymer brush.
Atomic force microscopy (AFM) has been commonly used to visualize polymer brush surface morphology and in some instances film thicknesses.29–31AFM has also been shown to visualize the formation of metal complexes with polymer brushes, phase transitions in poly(N-isopropylacrylamide)PNIAAmpolymer brushes and probe the micromechanical properties of polymer brushes.32–34AFM has been proven ideally suited for determining nanometre-size features within brushes that possess internal structure. For example, 2D and 3D height images of poly(methoxy-PEG-methacrylate) P(MePEGMA) brushes showed resolved boundaries between spherulite domains and lamellae structures.35 Helical polyisocyanidepolymer brushes have been shown to undergo changes in molecular organization upon solvent treatment using AFM (Fig. 2).36 As the film thickness increased the surface features diminished. This was argued to be a result of the film thickness becoming larger than the persistence length of the macromolecular chains.
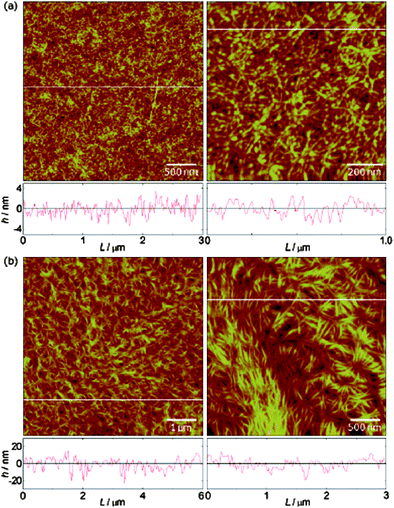 | ||
| Fig. 2 AFM images showing the morphology of poly(L-isocyanoalanyl-L-alanine methyl ester) brushes grown on APTS-modified silicon wafers (a) 5 nm thick and (b) 19 nm thick brushes. Reprinted with permission from ref. 36. Copyright 2008 American Chemical Society. | ||
A clearer picture of the molecular structure of polymer brushes has emerged from these experiments (and others omitted for brevity) and these support – and are supported by – a large volume of theoretical considerations of polymer brushes.37
A persistent question remains however: While polymer brushes are fascinating surface-confined polymer architectures, what are the relevant and transferable applications for this technology? It is this question that this review seeks to address in the next two sections.
3. Polymer brushes are increasingly used at the interface of materials science and biology
Biomaterials are typically considered in medical applications; however they are also used to grow cells in culture, in assays in clinical laboratories, in equipment for processing biomolecules and in diagnostic gene array chips.38 In terms of medical devices, biomaterials have been defined as “a non viable material used in a medical device intended to interact with biological systems”.39 Related to this, biocompatibility has been defined as “the ability of a material to perform with an appropriate host response in a specific application”.40 Medical devices are now ubiquitous, and despite many advances, a significant percentage of these become colonized by bacteria and the focus of a device-related infection.38 In device-related infections bacteria are present on the biomaterial surface and form a biofilm. All biofilms form with a basic sequence of events:411. A conditioning film of proteins and other organic molecules forms on the biomaterial surface prior to microbial deposition.
2. Microorganisms are transported towards the surface.
3. Initial microbial adhesion occurs.
4. Microorganisms strongly attach and anchor to the surface through production of an extracellular polymeric substance (EPS) matrix.
5. Adherent microorganisms grow on the surface and produce more EPS.
6. Isolated fragments of the biofilm detach and migrate.
Device-related infection and biofilm formation can therefore cause severe problems, from loss of function of the device to lethal sepsis of the patient. The only effective remedy at this stage of infection is removal of the infected device at considerable healthcare burden and patient discomfort. A more elegant approach is prevention of microbial adhesion and subsequent proliferation onto the implanted biomaterial. Poly(ethylene glycol) (PEG) has been used for this strategy. These brushes are typically prepared by “grafting to” however and so will only briefly be considered here.41 Leckband et al.42 described how PEG brushes are used similarly to prevent protein adsorption. The adsorbed biomolecule induces ‘overcrowding’ within the brush and the resulting osmotic penalty counterbalances the adsorption free energy of the biomolecule on the surface. Furthermore, the polymer brush increases the effective viscosity encountered by the biomolecule. Finally, the polymer brush can form an incompressible layer, preventing the biomolecule from reaching the surface. While it can be argued that PEGylated surfaces have become the ‘first choice’ strategy for surface coatings, concern exists that PEGylated surfaces can both increase complement activation and PEG chains can induce an anti-PEG IgM response.43–47
Poly(acrylamide) (PAAm) polymer brushes have been prepared on silicon oxide wafers as a model substrate for siliconerubber using SIP.48 Deposition and adherence of S. aureus, S. salivarius, and C albicans was shown to be greatly reduced on brush coated wafers compared to non-coated controls (Fig. 3). A reduction in bacterial adhesion of 98% was seen in some instances. The system was later transferred to silicon rubber and shown to reduce adhesion of the same bacterial strains and be stable in reconstituted saliva for one month.49
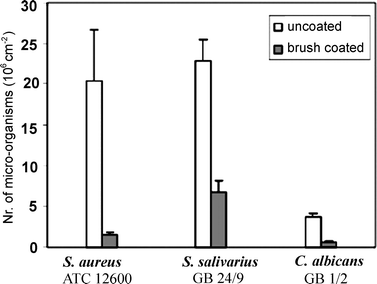 | ||
| Fig. 3 Adhesion of microorganisms to uncoated and PAAm-coated silicon wafer surfaces after 4 h in a parallel plate flow chamber at room temperature. All error bars represent the average standard deviation obtained from three separate experiments. Reprinted with permission from ref. 48. Copyright 2007 American Chemical Society. | ||
Huck's group has investigated sulfopropyl methacrylate brushes containing Ag+ ions.50 While this system was effective at inhibiting bacterial colonization, a subsequent paper showed that effective silver concentrations were also cytotoxic towards mammalian cells.51 The same group have conjugated the antibacterialpeptide magainin I to methacryloyl polymer brushes containing ethylene oxide side-chains and shown it to be effective against colonization by two strains of Gram negative bacteria.52
Quaternary ammonia polymer brushes have been examined for biocidal applications.53–57Polycations are often used as they are thought to disrupt the integrity of the bacterial cell membrane. Russell and co-workers58 recently investigated if this is primarily due to a cell penetration mechanism. They observed that surfaces with a dry brush thickness of 10 nm could kill bacteria cells. A dense, low polydispersity polymer brush of this thickness could not extend through the E. coli bacterial cell envelope used in the study. By studying the effect of surface density it was revealed that kill efficacy increased with charge density. The conclusions from this study were that for high grafting density polymer brushes the biocidal ability is not dependent on cell penetration and that charge density is a critical parameter.
Polymer brushes can be designed to suppress non specific adhesion of biomolecules to surfaces, or promote specific attachment of cell types to surfaces.59–64 Proposed applications for these biomaterials include diagnostics and cell culture. Poly(poly(ethylene glycol) methacrylate) (PPEGMA) brushes have been tested for stability in cell culture medium at 37 °C. Detachment of the polymer brush was observed under these conditions.65 This is an important result as it serves to demonstrate that observing a polymer brush behavior in highly controlled media may not be informative if the proposed application is in complex milieu (i.e. serum).
A popular strategy for cell attachment to polymer brush coated surfaces is to use the integrin bindingpeptide sequence arginine–glycine–aspartic acid (RGD).66,67RGD functional materials have recently been reviewed.68 Examples of SIPpolymer brushes that use the RGD sequence for cell adherence include poly(methacrylic acid)PMAA brushes for 3T3 fibroblast69 and human osteoblast MG63 cell lines ,19 PPEGMA brushes for 3T3 cell attachment,70 and HUVEC attachment to RGD-functionalized poly(2-hydroxyethyl methacrylate) (PHEMA) and PPEGMA brushes.71 It will be interesting to follow the progress of RGD-functional biomaterials in in vivo studies. While promising examples exist,72 other reports73 showed RGD-functional quantum dots being taken up primarily through the liver, spleen and bone-marrow rather than tumors that should over express RGD receptors. Furthermore, it is well known that platelets also bind to the RGD sequence through glycoprotein receptors.74,75
Other techniques for the attachment of cells and proteins to surfaces using polymer brushes include using boronic acid22 or azalactone76 containing polymer brushes, using DCC coupling of biomolecules to carboxybetaine brushes,77 using fibronectin adhesion to gradient PHEMA surfaces,78 and using a polymer brush comprising of 2-gluconamidoethyl methacrylate conjugated to a cell adhesionpeptide sequence (Fig. 4).79
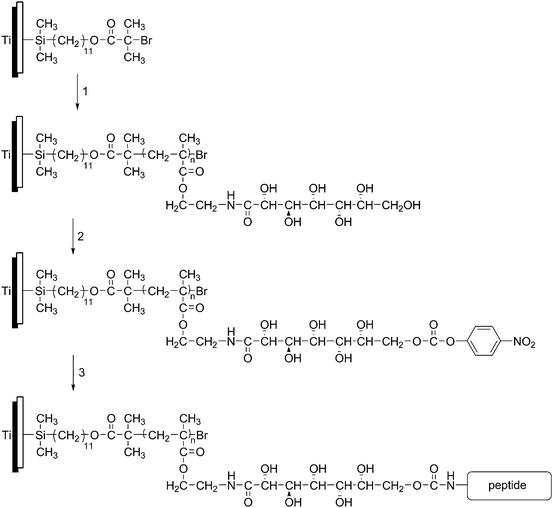 | ||
| Fig. 4 Attachment of a cell-adhesive peptide to a titanium surface through (1) SIPATRP of 2-gluconamidoethyl methacrylate, (2) modification with nitrophenyl carbonate and (3) attachment of peptide. Figure adapted from ref. 78. | ||
Finally, thermoresponsive polymer brushes of PNIPAAm have been used for cell attachment and release.80,81 Although strictly outside the parameters of this review, Okano has used cell culture dishes coated with PNIPAAm to culture a variety of cells and release them as cell sheets (i.e. ECM intact) for regenerative medicine.82
Dormidontova and co-workers83 used Monte Carlo simulations to study binding between a functionalized planar polymer layer and a cell surface decorated with mobile receptors. Increasing the polymer grafting density from the mushroom to the brush regime increased ligand availability and maximum binding distance resulting in a larger attractive force. There was also a concomitant higher free energy minimum due to the smaller tolerance for compression within the polymer brush. Furthermore, polymer layers with smaller grafting density, shorter chain length, and larger degree of functionalization possessed a larger affinity to the cell surface. The authors concluded that designing polymer layers with high binding affinity and high binding specificity is the ultimate goal. This could be best accomplished by using polymer layers with an intermediate degree of affinity to the cell as this lead to higher specificity of interaction.
Creating surfaces that prevent protein and platelet adsorption from plasma has been a long-held goal, both to stop non-specific adsorption in clinical assays and for blood-contacting indwelling devices.84,85 This is particularly challenging considering the diverse protein population present in human plasma over a large dynamic range. Plasma contains proteins secreted by solid tissues, immunoglobulins , receptor ligands, temporary passengers, tissue leakage products, and potential aberrant secretions and foreign proteins.86 Currently, the synthesis of zwitterionic surfaces and PEGylated surfaces are attracting attention with surface-initiated polymerization surface modification techniques.87–91 For example, polymer brushes of (oligoethylene glycol) methacrylate and sulfo- and carboxybetaines were tested against human serum and human plasma with some non-specific adsorption observed for all brushes.92 Other studies have shown low protein adsorption and platelet adhesion onto these types of polymer brushes, although recalcified plasma clotting times were not altered compared to polystyrene controls.93 The zwitterionic surfaces prevent protein adsorption by creating a strong hydration layer.94 Disruption of this hydration layer leads to a net repulsive force on proteins. The charged polymer layers have strong ionic hydration where water molecules around the betaine exchange slowly with the bulk, increasing the affinity of the water to the polymer layer.95 Therefore, only water molecules that are in contact with or near the polymer chain are removed from the overall H-bond network of the bulk water.96
4. Polymer brushes are used in areas of nanotechnology including sensors, conjugated films, and electrochemistry
While nanotechnology is a widely used term with seemingly many definitions and uses, as polymer brushes posses a dimension under 100 nm (namely thickness) they can be considered as falling under the realm of ‘nanoscience’. Indeed, it has been argued that surface and interface science was the precursor to nanoscience, where increasingly complex surfaces with increasing numbers of dimensions under 100 nm have been studied and characterized for the last 50 years and more.97 This section of the review seeks to highlight recent contributions of polymer brushes to nanotechnology.Polymer brushes, acting as well defined and highly regular thin films , have been used to enhance sensors. The polymer brush can either act as the sensing element, or improve the performance of the sensor.98 Examples include humidity sensors99 and photochromic ion sensors.100 The photochromic ion sensor used spiropyran-functionalized monomers copolymerized with methyl methacrylate (MMA) from glass substrates. Upon complexation with different metal ions there was a shift in the absorbance of the film that was ion dependant and easily discriminated visually (Fig. 5).
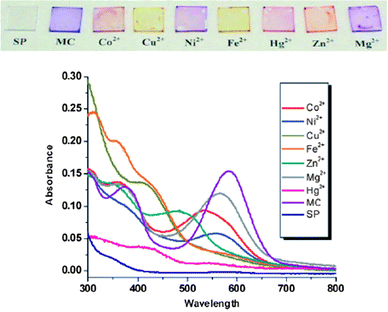 | ||
| Fig. 5 UV–vis spectra of the polymer brush film complexed with different ions. Reproduced from ref. 100. | ||
In addition to these examples, surface plasmon resonance (SPR) and quartz crystal microbalance (QCM) have been used as platforms to follow the synthesis and properties of surface-initiated polymer brushes. It is easy to envision polymer brushes being incorporated into these devices, in addition to other platforms such as surface-enhanced Raman spectroscopy (SERS), to fabricate novel chemical and biological sensing elements. Similarly, a recent example by Wong and co-workers demonstrates ionomeric polymer brushes being used for the fractionation of biologic samples prior to MALDI-MS analysis.101 Currently, ‘grafting to’ brushes are predominately used in sensing applications, for example as enzyme logic gate sensors.102,103
Electrochemistry has been used to study ionomeric polymer brushes, where the oxidation state of transition metal complexes trapped within brushes can be reversibly changed. Polymer brushes containing quaternary ammonium functionality104 and ionic liquid moieties105 have been examined this way recently.
Advincula's group106 used cyclic voltammetry to crosslink a poly(vinyl carbazole)polymer brush, forming the basis of a polymer light-emitting diode. The current increased with each cycle indicating the electropolymerization of the carbazole units. Distinct differences in both the onset of oxidation and the shift of the oxidation peak were seen in the polymer brush compared to a spin-coated sample. The differences were ascribed to increased chain density and homogenous film morphology in the brush over the spin-coated film. Conjugated polymers are a promising class of materials with applications in solar cells, LEDs, memory storage and sensors. Kamuda-type catalysts have been used for surface-initiated polycondensation reactions producing poly(thiophene) and poly(p-phenylene)polymer brushes.107 Suzuki polycondensations have also been used for the surface-initiated synthesis of polymer brushes.31 Surface-initiated Yamamoto polycondensation of 2,7-dibromo-9,9-dihexylfluorene has been reported by Carter's group. The conjugated polymer gave a strong blue optical fluorescence when placed under a UV lamp.108
Rühe reported polymer brushes with liquid crystal side groups some time ago.109 These systems are thought to be promising candidates for display technologies. Huck later synthesized polymer brushes containing the reactive mesogen RM488 and constructed liquid crystal cells from polymer-brush-coated glass slides.110 Interactions between the liquid crystalline polymer brush and liquid crystal material within the cell were clearly visible. Using monomers with liquid crystalline mesogenic side-groups has proven a popular strategy, with several reports detailing this approach.111–113 An alternative route is to polymerize poly(ε-caprolactone)polymer brushes from gold surfaces.114 Nematic 4-cyano-4′-pentylbiphenyl is then anchored to the polymer brush. With increasing thickness of the brush, the orientation of the liquid crystal changed from a uniform to a non-uniform orientation.
Polymer brushes can be used to stabilize colloidal particles.115,116 This has been extended to control and stabilize the formation of colloidal crystals.117,118Colloidal crystals are materials with periodic structures prepared by self-assembly of sub-micrometre colloidal particles. For example, P(nBA)polymer brushes have been prepared from SiO2 nanoparticles using nitroxide-mediated polymerization.119 Other groups have used PMMA brushes prepared using ATRP.120Colloidal crystals have potential applications as photonic crystals in optoelectronic materials such as telecommunication devices, lasers and sensors.
Polymer brushes have been used in several areas to support metallic nanoparticles; due to surface area considerations these have typically been performed from spherical substrates.121 However, metallic nanoparticles were shown to form within planar polyelectrolytepolymer brushes.122Gold nanoparticles have been prepared in polymer brushes,123,124 and recently Huck's group125 has studied how gold nanoparticles infiltrate and organize within block copolymerpolymer brushes. Santer and Rühe have shown how stimuli-responsive diblock copolymer brushes can be used to induce motion of nanoparticles on the surface of polymer brush films.126 Lower dimension materials are now being studied as substrates for polymer brushes. For example, palladium nanoparticles have been prepared by reducing PdCl2−4 in a quaternized poly(2-(dimethylamino)ethyl methacrylate) (poly(DMAEMA)) brush that was polymerized from TiO2nanowires.127Palladium nanoparticles immobilized within polymer brushes have been used to perform Heck reactions.128
Inorganic magnetic nanoparticles have been developed that possess applications in storage media, biosensors , and medical applications.129–132 The surfaces of these magnetic nanoparticles can be modified using polymer brushes. Polystyrene brushes were synthesized from magnetic nanoparticles to improve the dispersion of the nanoparticles within a polystyrene-block-polybutadiene-block-polystyrenecopolymer.133 Hydrophilic copolymers of PEGMA and PNIPAAm have been prepared from magnetic nanoparticles.134 In an alternative approach, PNIPAAm brushes were grown from SiO2 particles and Fe3O4nanoparticles prepared inside the brush.135 Gill and co-workers polymerized a modified polystyrene brushes from magnetic nanoparticles.136 The styrene was modified to possess Co(III)salen catalysts or piperazine bases. The Co(III)salen brushes were shown to be effective for the hydrolytic kinetic resolution of epichlorohydrin, achieving >99% ee after 60 min. The piperazine-functionalized brush displayed ≥99% conversion in a Knoevenagel reaction after 3 cycles. The catalytic nanoparticles were easily recovered with application of a magnetic field.
Polymer brushes acting as ultra-thin films have been used in a variety of ways to modify the surfaces and pores of membranes.137 The membrane material can either be organic or inorganic. For example, organic membranes of polypropylene,138polysulfone,139nylon,140,141polyethylene terephthalate (PET),142 and polycarbonate143 have all been modified using polymer brushes to affect surface chemistry and properties. Polymer brush coated membranes have found promising uses in biotechnology applications.144Glycidal methacrylate (GMA) has been polymerized from porous glass and modified to act as an ion exchange material that could absorb BSA from a solution.145 The same polymer brushes have also been used to immobilize lipase onto polyethylene hollow-fiber membranes with retention of enzymatic activity.146 The NTA-His6proteinpurification technique has been adapted to poly(HEMA)polymer brush coated membranes to create high affinity substrates.147 Membranes coated with polymer brushes have attracted attention for applications as ion channels, in roles such as fuel cells and mimics of biological channels. For example, Smith and Zharov have prepared sulfonated methacrylate and sulfonated styrene polymer brushes from nanoporous silica materials and demonstrated their potential as proton conducting membranes.148 Yameen and co-workers149 have used sulfonated- and PEGylated-monomers to fabricate proton conducting membranes with conductivity values above 10−2 S cm−1, over a wide range of humidity. The same group showed that ion channels coated with poly(vinyl pyridine) brushes could be cycled between “on” and “off” states with pH.150 Recently, polyzwitterionic brushes on mesoporous silica have been shown to modulate the transport properties of both negatively charges Fe(CN)3−6 and positively charged Ru(NH3)3+6 ions (Fig. 6).151
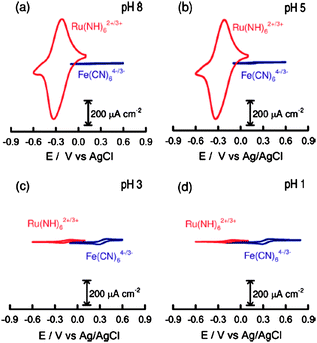 | ||
| Fig. 6 Cyclic voltammograms corresponding to a polyzwitterionic-brush-modified mesoporous silica film deposited on an ITO electrode in the presence of 1 mM Ru(NH3)3+6 (red trace) and 1 mM Fe(CN)3−6,respectively, under different pH conditions: (a) 8, (b) 5, (c) 3, (d) 1. Reprinted with permission from ref. 149. Copyright 2009 American Chemical Society. | ||
It is evident that the use of highly functional, high grafting density, covalently attached surface layers is conceptually attractive. However, a question that often arises from polymer brushes being employed in different applications is “is a polymer brush really necessary?” An alternative way of looking at this is to consider that if the application relies on modification of surface properties, what does polymer brush architecture gain over thin films or small molecule adlayers? As polymer brushes find increased use in a variety of areas, the gain in performance from using a brush will most likely be application dependent. For example, the grafting density of PNIPAAm brushes changed the separation properties of columns probing elution times of steroid molecules.152 Alternatively, liquid crystalline films showed different performance depending on if they were polymer brushes or spun-cast films.109Polymer brushes offered superior performance to monolayers as oligonucleotide microarrays.153 Conversely, Madkour and co-workers154 recently showed that polymer brushes could be highly antimicrobial, killing 100% of S. aureus and E. coli within less than 5 min. However, the killing ability was independent of polymer layer thickness and density. As the “tool box” of surface modifications increases, it is likely that polymer brushes will be applied selectively and appropriately. For example, the chemical etch resistance of brushes of different grafting density has been used to prepare hierarchically structured thin films .155
5. Conclusions
In summary, the field of surface-initiated polymerization remains vibrant, with polymer brushes being leveraged into many applied areas, a few of which have been highlighted in this review. Encouragingly, these applications appear to be driving a large part of the academic endeavor, rather than scientists making polymer brush systems we find intriguing and then trying to envision a use. Moreover, polymer brushes are gaining increasing traction in areas of chemistry, surface science, materials science, nanotechnology, biomedical engineering and biology. Indeed, opportunities exist for polymer brushes to have a synergistic effect at the interfaces of traditional disciplines. Many challenges persist however, including routine characterization of polymer brushes on a molecular level, increased long term stability of these thin films , determining the behavior of polymer brushes in complex milieu (i.e. whole serum) and the potential toxicity and biocompatibility of polymer brush coated materials in vivo. As we move forward, the need to extend polymer brushes from surfaces other than silicon oxide and gold will become more persistent. This review has highlighted materials such as metal oxides, polymers, and tissues culture vessels that have all been surface modified with brushes. Finally, the question of if the brush architecture results in performance gain will not readily go away, and I suspect the polymer brush community will constantly have to be prepared to answer this with control experiments.References
- R. Ducker, A. Garcia, J. Zhang, T. Chen and S. Zauscher, Soft Matter, 2008, 4, 1774–1786 RSC.
- S. Edmondson and S. P. Armes, Polym. Int., 2009, 58, 307–316 CrossRef CAS.
- W. T. S. Huck, Mater. Today, 2008, 11, 24–32 CrossRef CAS.
- I. Luzinov, S. Minko and V. V. Tsukruk, Soft Matter, 2008, 4, 714–725 RSC.
- R. Toomey and M. Tirrell, Annu. Rev. Phys. Chem., 2008, 59, 493–517 CrossRef CAS.
- G. Zhang and C. Wu, Macromol. Rapid Commun., 2009, 30, 328–335 CrossRef CAS.
- F. Zhou and W. T. S. Huck, Phys. Chem. Chem. Phys., 2006, 8, 3815–3823 RSC.
- Y. Lu, A. Wittemann and M. Ballauff, Macromol. Rapid Commun., 2009, 30, 806–815 CrossRef CAS.
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS.
- S. Alexander, J. Phys. (Paris), 1977, 38, 983 CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- K. Binder, Eur. Phys. J. E, 2002, 9, 293–298 CrossRef CAS.
- I. Coluzza and J.-P. Hansen, Phys. Rev. Lett., 2008, 100, 016104 CrossRef.
- J. C. Vickerman, ed., Surface Analysis – The Principal Techniques, John Wiley & Sons, Ltd, Chichester, 1997 Search PubMed.
- H. Ade and A. P. Hitchcock, Polymer, 2008, 49, 643–675 CrossRef CAS.
- L. Andruzzi, A. Hexemer, X. Li, C. K. Ober, E. J. Kramer, G. Galli, E. Chiellini and D. A. Fischer, Langmuir, 2004, 20, 10498–10506 CrossRef CAS.
- S. P. Cullen, X. Liu, I. C. Mandel, F. J. Himpsel and P. Gopalan, Langmuir, 2008, 24, 913–920 CrossRef CAS.
- T. Wu, P. Gong, I. Szleifer, P. Vlcek, V. Subr and J. Genzer, Macromolecules, 2007, 40, 8756–8764 CrossRef CAS.
- M. Navarro, E. M. Benetti, S. Zapotoczny, J. A. Planell and G. J. Vancso, Langmuir, 2008, 24, 10996–11002 CrossRef CAS.
- B. Lego, W. G. Skene and S. Giasson, Langmuir, 2008, 24, 379–382 CrossRef CAS.
- Q. Peng, D. M. Y. Lai, E. T. Kang and K. G. Neoh, Macromolecules, 2006, 39, 5577–5582 CrossRef CAS.
- A. E. Ivanov, J. Eccles, H. Ahmad Panahi, A. Kumar, M. V. Kuzimenkova, L. Nilsson, B. Bergenstaahl, N. Long, G. J. Phillips, S. V. Mikhalovsky, I. Y. Galaev and B. Mattiasson, J. Biomed. Mater. Res., Part A, 2009, 88, 213–225 CrossRef.
- G. Carrot, A. El Harrak, J. Oberdisse, J. Jestin and F. Boue, Soft Matter, 2006, 2, 1043–1047 RSC.
- J. Zhang, T. Nylander, R. A. Campbell, A. R. Rennie, S. Zauscher and P. Linse, Soft Matter, 2008, 4, 500–509 RSC.
- B. Akgun, W. J. Brittain, X. Li, J. Wang and M. D. Foster, Macromolecules, 2005, 38, 8614–8616 CrossRef CAS.
- B. Akgun, D. R. Lee, H. Kim, H. Zhang, O. Prucker, J. Wang, J. Rühe and M. D. Foster, Macromolecules, 2007, 40, 6361–6369 CrossRef CAS.
- B. Akgun, G. Ugur, Z. Jiang, S. Narayanan, S. Song, H. Lee, W. J. Brittain, H. Kim, S. K. Sinha and M. D. Foster, Macromolecules, 2009, 42, 737–741 CrossRef CAS.
- H. Merlitz, G.-L. He, C.-X. Wu and J.-U. Sommer, Macromolecules, 2008, 41, 5070–5072 CrossRef CAS.
- B. Lego, M. Francois, W. G. Skene and S. Giasson, Langmuir, 2009, 25, 5313–5321 CrossRef CAS.
- Y. Zou, N. A. A. Rossi, J. N. Kizhakkedathu and D. E. Brooks, Macromolecules, 2009, 42, 4817–4828 CrossRef CAS.
- T. Beryozkina, K. Boyko, N. Khanduyeva, V. Senkovskyy, M. Horecha, U. Oertel, F. Simon, M. Stamm and A. Kiriy, Angew. Chem., Int. Ed., 2009, 48, 2695–2698 CrossRef CAS.
- N. Ishida and S. Biggs, Langmuir, 2007, 23, 11083–11088 CrossRef CAS.
- K. Sha, D. S. Li, Y. Li, S. Wang and J. Wang, J. Mater. Sci., 2007, 42, 4916–4925 CrossRef CAS.
- J. Zhao, M. Chen, Y. An, J. Liu and F. Yan, Appl. Surf. Sci., 2008, 255, 2295–2302 CrossRef CAS.
- Y. Zheng, M. L. Bruening and G. L. Baker, Macromolecules, 2007, 40, 8212–8219 CrossRef CAS.
- E. Lim, G. Tu, E. Schwartz, J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and W. T. S. Huck, Macromolecules, 2008, 41, 1945–1951 CrossRef CAS.
- A. Naji, C. Seidel and R. R. Netz, Adv. Polym. Sci., 2006, 198, 149–183 CAS.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen, J. E. Lemonsand Editors, Biomaterials Science, an Introduction to Materials in Medicine, Elsevier Academic Press, California, USA, 2nd Edition, 2004 Search PubMed.
- D. F. Williams, J. Mater. Sci., 1987, 22, 3421–3445 CrossRef CAS.
- A. Remes and D. F. Williams, Biomaterials, 1992, 13, 731–743 CrossRef CAS.
- A. Roosjen, W. Norde, H. C. van der Mei and H. J. Busscher, Prog. Colloid Polym. Sci., 2006, 132, 138–144 CAS.
- D. Leckband, S. Sheth and A. Halperin, J. Biomater. Sci., Polym. Ed., 1999, 10, 1125–1147 CrossRef CAS.
- Y. Arima, M. Toda and H. Iwata, Biomaterials, 2008, 29, 551–560 CAS.
- I. Hamad, A. C. Hunter, J. Szebeni and S. M. Moghimi, Mol. Immunol., 2008, 46, 225–232 CrossRef CAS.
- T. Ishida, S. Kashima and H. Kiwada, J. Controlled Release, 2008, 126, 162–165 CAS.
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354, 56–62 CrossRef CAS.
- A. Kidane and K. Park, J. Biomed. Mater. Res., 1999, 48, 640–647 CrossRef CAS.
- I. Cringus-Fundeanu, J. Luijten, H. C. Van der Mei, H. J. Busscher and A. J. Schouten, Langmuir, 2007, 23, 5120–5126 CrossRef CAS.
- I. Fundeanu, H. C. van der Mei, A. J. Schouten and H. J. Busscher, Colloids Surf., B, 2008, 64, 297–301 CrossRef CAS.
- M. Ramstedt, N. Cheng, O. Azzaroni, D. Mossialos, H. J. Mathieu and W. T. S. Huck, Langmuir, 2007, 23, 3314–3321 CrossRef CAS.
- M. Ramstedt, B. Ekstrand-Hammarstroem, A. V. Shchukarev, A. Bucht, L. Oesterlund, M. Welch and W. T. S. Huck, Biomaterials, 2009, 30, 1524–1531 CrossRef CAS.
- K. Glinel, A. M. Jonas, T. Jouenne, J. Leprince, L. Galas and W. T. S. Huck, Bioconjugate Chem., 2009, 20, 71–77 CrossRef CAS.
- G. Cheng, H. Xue, Z. Zhang, S. Chen and S. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831–8834 CrossRef CAS.
- M. Ignatova, S. Voccia, S. Gabriel, B. Gilbert, D. Cossement, R. Jerome and C. Jerome, Langmuir, 2009, 25, 891–902 CrossRef CAS.
- M. Ignatova, S. Voccia, B. Gilbert, N. Markova, D. Cossement, R. Gouttebaron, R. Jerome and C. Jerome, Langmuir, 2006, 22, 255–262 CrossRef CAS.
- L. Li, Z. Ke, G. Yan and J. Wu, Polym. Int., 2008, 57, 1275–1280 CrossRef CAS.
- S. J. Yuan, F. J. Xu, S. O. Pehkonen, Y. P. Ting, K. G. Neoh and E. T. Kang, Biotechnol. Bioeng., 2009, 103, 268–281 CrossRef CAS.
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS.
- L. Lavanant and H.-A. Klok, Chimia, 2008, 62, 793–798 CrossRef CAS.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- A. Hucknall, A. J. Simnick, R. T. Hill, A. Chilkoti, A. Garcia, M. S. Johannes, R. L. Clark, S. Zauscher and B. D. Ratner, Biointerphases, 2009, 4, FA50–FA57 Search PubMed.
- J. E. Raynor, J. R. Capadona, D. M. Collard, T. A. Petrie and A. J. Garcia, Biointerphases, 2009, 4, FA3–FA16 Search PubMed.
- J. Wang, M. I. Gibson, R. Barbey, S.-J. Xiao and H.-A. Klok, Macromol. Rapid Commun., 2009, 30, 845–850 CrossRef CAS.
- Z. Zhang, H. VaisocherovaÌ, G. Cheng, W. Yang, H. Xue and S. Jiang, Biomacromolecules, 2008, 9, 2686–2692 CrossRef CAS.
- S. Tugulu and H.-A. Klok, Biomacromolecules, 2008, 9, 906–912 CrossRef CAS.
- M. Ebara, M. Yamato, T. Aoyagi, A. Kikuchi, K. Sakai and T. Okano, Adv. Mater., 2008, 20, 3034–3038 CrossRef CAS.
- M. Ebara, M. Yamato, T. Aoyagi, A. Kikuchi, K. Sakai and T. Okano, Biomaterials, 2008, 29, 3650–3655 CrossRef CAS.
- L. Perlin, S. MacNeil and S. Rimmer, Soft Matter, 2008, 4, 2331–2349 RSC.
- B. P. Harris, J. K. Kutty, E. W. Fritz, C. K. Webb, K. J. L. Burg and A. T. Metters, Langmuir, 2006, 22, 4467–4471 CrossRef CAS.
- N. Singh, X. Cui, T. Boland and M. Husson Scott, Biomaterials, 2007, 28, 763–771 CrossRef CAS.
- S. Tugulu, P. Silacci, N. Stergiopulos and H.-A. Klok, Biomaterials, 2007, 28, 2536–2546 CrossRef CAS.
- P. Schaffner, J. Meyer, M. Dard, R. Wenz, B. Nies, S. Verrier, H. Kessler and M. Kantlehner, J. Mater. Sci.: Mater. Med., 1999, 10, 837–839 CrossRef CAS.
- W. Cai, K. Chen, Z.-B. Li, S. S. Gambhir and X. Chen, J. Nucl. Med., 2007, 48, 1862–1870 CrossRef CAS.
- T. A. Springer, J. Zhu and T. Xiao, J. Cell Biol., 2008, 182, 791–800 CrossRef CAS.
- J. G. Zhang, O. B. Krajden, R. K. Kainthan, J. N. Kizhakkedathu, I. Constantinescu, D. E. Brooks and M. I. C. Gyongyossy-Issa, Bioconjugate Chem., 2008, 19, 1241–1247 CrossRef CAS.
- S. P. Cullen, I. C. Mandel and P. Gopalan, Langmuir, 2008, 24, 13701–13709 CrossRef CAS.
- Z. Zhang, S. Chen and S. Jiang, Biomacromolecules, 2006, 7, 3311–3315 CrossRef CAS.
- R. R. Bhat, B. N. Chaney, J. Rowley, A. Liebmann-Vinson and J. Genzer, Adv. Mater., 2005, 17, 2802–2807 CrossRef CAS.
- J. E. Raynor, T. A. Petrie, K. P. Fears, R. A. Latour, A. J. Garcia and D. M. Collard, Biomacromolecules, 2009, 10, 748–755 CrossRef CAS.
- A. Mizutani, A. Kikuchi, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2008, 29, 2073–2081 CrossRef CAS.
- F. J. Xu, S. P. Zhong, L. Y. L. Yung, E. T. Kang and K. G. Neoh, Biomacromolecules, 2004, 5, 2392–2403 CrossRef CAS.
- N. Matsuda, T. Shimizu, M. Yamato and T. Okano, Adv. Mater., 2007, 19, 3089–3099 CrossRef CAS.
- M. C. Hagy, S. Wang and E. E. Dormidontova, Langmuir, 2008, 24, 13037–13047 CrossRef CAS.
- N. Ayres, D. J. Holt, C. F. Jones, L. E. Corum and D. W. Grainger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7713–7724 CrossRef CAS.
- B. D. Ratner, Biomaterials, 2007, 28, 5144–5147 CrossRef CAS.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CrossRef CAS.
- Y. Chang, S.-C. Liao, A. Higuchi, R.-C. Ruaan, C.-W. Chu and W.-Y. Chen, Langmuir, 2008, 24, 5453–5458 CrossRef CAS.
- C. Rodriguez Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. Bologna Alles, Langmuir, 2009, 25, 6328–6333 CrossRef CAS.
- G. Li, H. Xue, G. Cheng, S. Chen, F. Zhang and S. Jiang, J. Phys. Chem. B, 2008, 112, 15269–15274 CrossRef CAS.
- W. Yang, S. Chen, G. Cheng, H. Vaisocherova, H. Xue, W. Li, J. Zhang and S. Jiang, Langmuir, 2008, 24, 9211–9214 CrossRef CAS.
- J. N. Kizhakkedathu, J. Janzen, Y. Le, R. K. Kainthan and D. E. Brooks, Langmuir, 2009, 25, 3794–3801 CrossRef CAS.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS.
- Z. Zhang, M. Zhang, S. Chen, T. A. Horbett, B. D. Ratner and S. Jiang, Biomaterials, 2008, 29, 4285–4291 CrossRef CAS.
- J. C. Hower, M. T. Bernards, S. Chen, H.-K. Tsao, Y.-J. Sheng and S. Jiang, J. Phys. Chem. B, 2009, 113, 197–201 CrossRef CAS.
- Y. He, J. Hower, S. Chen, T. Bernards Matthew, Y. Chang and S. Jiang, Langmuir, 2008, 24, 10358–10364 CrossRef CAS.
- H. Kitano, S. Tada, T. Mori, K. Takaha, M. Gemmei-Ide, M. Tanaka, M. Fukuda and Y. Yokoyama, Langmuir, 2005, 21, 11932–11940 CrossRef CAS.
- M. A. Van Hove, Surf. Sci., 2009, 603, 1301–1305 CrossRef CAS.
- H. Vaisocherová, W. Yang, Z. Zhang, Z. Cao, G. Cheng, M. Piliarik, J. Homola and S. Jiang, Anal. Chem., 2008, 80, 7894–7901 CrossRef CAS.
- B. Bilen, Y. Skarlatos, G. Aktas, M. N. Inci, T. Dispinar, M. M. Kose and A. Sanyal, J. Appl. Phys., 2007, 102, 073534 CrossRef.
- K. Fries, S. Samanta, S. Orski and J. Locklin, Chem. Commun., 2008, 6288–6290 RSC.
- V. N. Wong, G. Fernando, A. R. Wagner, J. Zhang, G. R. Kinsel, S. Zauscher and D. J. Dyer, Langmuir, 2009, 25, 1459–1465 CrossRef CAS.
- M. Pita, T. K. Tam, S. Minko and E. Katz, ACS Appl. Mater. Interfaces, 2009, 1, 1166–1168 Search PubMed.
- J. Zhou, T. K. Tam, M. Pita, M. Ornatska, S. Minko and E. Katz, ACS Appl. Mater. Interfaces, 2009, 1, 144–149 Search PubMed.
- E. Spruijt, E.-Y. Choi and W. T. S. Huck, Langmuir, 2008, 24, 11253–11260 CrossRef CAS.
- B. Yu, F. Zhou, H. Hu, C. Wang and W. Liu, Electrochim. Acta, 2007, 53, 487–494 CrossRef CAS.
- T. M. Fulghum, P. Taranekar and R. C. Advincula, Macromolecules, 2008, 41, 5681–5687 CrossRef CAS.
- S. K. Sontag, N. Marshall and J. Locklin, Chem. Commun., 2009, 3354–3356 RSC.
- S. B. Jhaveri, J. J. Peterson and K. R. Carter, Langmuir, 2009, 25, 9552–9556 CrossRef CAS.
- B. Peng, D. Johannsmann and J. Rühe, Macromolecules, 1999, 32, 6759–6766 CrossRef CAS.
- P. J. Hamelinck and W. T. S. Huck, J. Mater. Chem., 2005, 15, 381–385 RSC.
- T. Uekusa, S. Nagano and T. Seki, Langmuir, 2007, 23, 4642–4645 CrossRef CAS.
- T. Uekusa, S. Nagano and T. Seki, Macromolecules, 2009, 42, 312–318 CrossRef CAS.
- V. N. Vasilets, G. A. Shandryuk, G. N. Savenkov, A. M. Shatalova, G. N. Bondarenko, R. V. Talroze and N. A. Plate, Macromolecules, 2004, 37, 3685–3688 CrossRef CAS.
- Y. Gu, F. Nederberg, R. Kange, R. R. Shah, C. J. Hawker, M. Moller, J. L. Hedrick and N. L. Abbott, ChemPhysChem, 2002, 3, 448–451 CrossRef CAS.
- B. Radhakrishnan, R. Ranjan and W. J. Brittain, Soft Matter, 2006, 2, 386–396 RSC.
- R. Ranjan and W. J. Brittain, Macromol. Rapid Commun., 2008, 29, 1104–1110 CrossRef CAS.
- C. R. Han, J. G. Cao, L. W. Zhou, X. C. Pang, J. L. Huang, L. F. Zhang and J. P. Huang, Phys. Lett. A, 2009, 373, 3174–3181 CrossRef CAS.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143–9150 CrossRef CAS.
- C. Deleuze, M. H. Delville, V. Pellerin, C. Derail and L. Billon, Macromolecules, 2009, 42, 5303–5309 CrossRef CAS.
- T. Morinaga, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2008, 41, 3620–3626 CrossRef CAS.
- M. Schrinner, F. Polzer, Y. Mei, Y. Lu, B. Haupt, M. Ballauff, A. Goeldel, M. Drechsler, J. Preussner and U. Glatzel, Macromol. Chem. Phys., 2007, 208, 1542–1547 CrossRef CAS.
- S. G. Boyes, B. Akgun, W. J. Brittain and M. D. Foster, Macromolecules, 2003, 36, 9539–9548 CrossRef CAS.
- O. Azzaroni, A. A. Brown, N. Cheng, A. Wei, A. M. Jonas and W. T. S. Huck, J. Mater. Chem., 2007, 17, 3433–3439 RSC.
- R. R. Bhat and J. Genzer, Appl. Surf. Sci., 2006, 252, 2549–2554 CrossRef CAS.
- R. Oren, Z. Liang, J. S. Barnard, S. C. Warren, U. Wiesner and W. T. S. Huck, J. Am. Chem. Soc., 2009, 131, 1670–1671 CrossRef CAS.
- S. Santer and J. Rühe, Polymer, 2004, 45, 8279–8297 CrossRef CAS.
- Q. Ye, X. Wang, H. Hu, D. Wang, S. Li and F. Zhou, J. Phys. Chem. C, 2009, 113, 7677–7683 CrossRef CAS.
- Y. B. Malysheva, A. V. Gushchin, Y. Mei, Y. Lu, M. Ballauff, S. Proch and R. Kempe, Eur. J. Inorg. Chem., 2008, 379–383 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- A. S. Teja and P.-Y. Koh, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22–45 CrossRef CAS.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 Search PubMed.
- I. García, A. Tercjak, N. E. Zafeiropoulos, M. Stamm and I. Mondragon, Macromol. Rapid Commun., 2007, 28, 2361–2365 CrossRef CAS.
- S. Wang, Y. Zhou, W. Guan and B. Ding, Appl. Surf. Sci., 2008, 254, 5170–5174 CrossRef CAS.
- S. Bi, X. Wei, N. Li and Z. Lei, Mater. Lett., 2008, 62, 2963–2966 CrossRef CAS.
- C. S. Gill, W. Long and C. W. Jones, Catal. Lett., 2009, 131, 425–431 CrossRef CAS.
- M. L. Bruening, D. M. Dotzauer, P. Jain, O. Lu and G. L. Baker, Langmuir, 2008, 24, 7663–7673 CrossRef CAS.
- F. Yao, G.-D. Fu, J. Zhao, E.-T. Kang and K. G. Neoh, J. Membr. Sci., 2008, 319, 149–157 CrossRef CAS.
- H.-B. Dong, Y.-Y. Xu, Z. Yi and J.-L. Shi, Appl. Surf. Sci., 2009, 255, 8860–8866 CrossRef CAS.
- F. J. Xu, J. P. Zhao, E. T. Kang, K. G. Neoh and J. Li, Langmuir, 2007, 23, 8585–8592 CrossRef CAS.
- Z. B. Zhang, X. L. Zhu, F. J. Xu, K. G. Neoh and E. T. Kang, J. Membr. Sci., 2009, 342, 300–306 CrossRef CAS.
- A. Friebe and M. Ulbricht, Langmuir, 2007, 23, 10316–10322 CrossRef CAS.
- I. Lokuge, X. Wang and P. W. Bohn, Langmuir, 2007, 23, 305–311 CrossRef CAS.
- P. Jain, G. L. Baker and M. L. Bruening, Annu. Rev. Anal. Chem., 2009, 2, 387–408 Search PubMed.
- H. Kawakita, H. Masunaga, K. Nomura, K. Uezu, I. Akiba and S. Tsuneda, J. Porous Mater., 2007, 14, 387–391 CrossRef CAS.
- M. Goto, T. Okubo, H. Kawakita, K. Uezu, S. Tsuneda, K. Saito, M. Goto, M. Tamada and T. Sugo, Biochem. Eng. J., 2007, 37, 159–165 CrossRef CAS.
- P. Jain, L. Sun, J. Dai, G. L. Baker and M. L. Bruening, Biomacromolecules, 2007, 8, 3102–3107 CrossRef CAS.
- J. J. Smith and I. Zharov, Chem. Mater., 2009, 21, 2013–2019 CrossRef CAS.
- B. Yameen, A. Kaltbeitzel, A. Langer, F. Mueller, U. Goesele, W. Knoll and O. Azzaroni, Angew. Chem., Int. Ed., 2009, 48, 3124–3128 CrossRef CAS.
- B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Nano Lett., 2009, 9, 2788–2793 CrossRef CAS.
- A. Calvo, B. Yameen, F. J. Williams, G. J. A. A. Soler-Illia and O. Azzaroni, J. Am. Chem. Soc., 2009, 131, 10866–10868 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2008, 24, 511–517 CrossRef CAS.
- G. Pirri, M. Chiari, F. Damin and A. Meo, Anal. Chem., 2006, 78, 3118–3124 CrossRef CAS.
- A. E. Madkour, J. M. Dabkowski, K. Nusslein and G. N. Tew, Langmuir, 2009, 25, 1060–1067 CrossRef CAS.
- M. Wang, J. E. Comrie, Y. Bai, X. He, S. Guo and W. T. S. Huck, Adv. Funct. Mater., 2009, 19, 2236–2243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |