Water-soluble conjugated polymers as the platform for protein sensors
Kai
Li
and
Bin
Liu
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117576. E-mail: cheliub@nus.edu.sg; Fax: +65 67791936; Tel: +65 65168049
First published on 21st December 2009
Abstract
The booming development of protein detection requires simple, sensitive, and reliable biosensor systems. Water-soluble conjugated polymers have been widely used for protein sensing due to their distinct optical response in the presence of different analytes. This review summarizes the recent developments in water-soluble conjugated polymer-based protein sensors. Based on the different optical responses of conjugated polymers to proteins, these assays are categorized into three groups. The conformational change of cationic poly(thiophene)s is used to generate unique colorimetric or fluorescent transduction upon interaction with target proteins. Super-quenching of water-soluble poly(phenylene vinylene)s and poly(phenylene ethynylene)s has been developed into fluorescence turn-on and turn-off proteinassays . Energy transfer between poly(fluorene-co-phenylene) derivatives and acceptor dyes is also utilized to develop fluorescence turn-on proteinassays with amplified signal output. The fine-tuning of conjugated polymer structures will benefit the design of versatile optical probes to satisfy the sophisticated requirements for protein sensors.
 Kai Li | Kai Li received his BE degree in Polymer Materials and Engineering from Beijing University of Chemical Technology and his ME degree in Polymer Engineering from Chungnam National University. He is currently pursuing a PhD degree at the National University of Singapore (NUS), under the supervision of Dr Bin Liu. His work focuses on the design and optimization of conjugated polymer-based nanoparticles for biological applications. |
 Bin Liu | Bin Liu received her PhD degree in Chemistry from the National University of Singapore (NUS) in 2001. After post-doctoral training at the University of California Santa Barbara (UCSB), she joined NUS where she is currently an assistant professor in the Department of Chemical and Biomolecular Engineering. Her current research focuses on the design and synthesis of functional water-soluble conjugated polymers and exploration of their applications in chemosensors, biosensors and optoelectronic devices. |
Introduction
Identification and quantification of specific proteins is an important issue in medical and clinical research as many diseases have a specific change in protein expression.1 The most commonly used technique in clinics is enzyme-linked immunosorbent assay (ELISA), which requires specific storage of active enzymes and tedious protein modification.2 Different detection strategies have been developed to simplify the detection procedure, which include fluorescence,3 electrochemistry,4 Raman,5chemiluminescence,6flow cytometry,7 and microfluidic methods.8 However, most of these methods require sophisticated instrumentation and proficient manipulation, which highly motivate the development of simple and reliable protein detection systems.Conjugated polymers (CPs) have been widely used for electronics and biosensor applications due to their highly delocalized backbone structures and unique electronic and optical properties.9CPs could be viewed as a collection of short, conjugated (oligomeric) units kept in close proximity by virtue of the polymer backbone. The chemical structure allows for effective electronic coupling and therefore fast intra- and interchain energy transfer.10CP-based sensors are sensitive to minor perturbations, due to amplification by the collective response, which offer advantages when compared with small molecule counterparts.11 This collective response influences optoelectronic properties, such as absorbance, Förster resonance energy transfer (FRET ), electrical conductivity, and fluorescence efficiency, which can be used to report, or “transduce”, target analyte presence.9
Water-solubility of CPs is essential for their biological applications, which is typically achieved by attaching hydrophilic side-chains or ionic groups to the CP backbone.12 Based on different backbone structures, several types of water-soluble CPs have been reported for protein detection, which include poly(thiophene)s (PTs),13poly(phenylene vinylene)s (PPVs),14poly(phenylene ethynylene)s (PPEs),15 and poly(fluorene-co-phenylene)s (PFPs).16 The chemical structures of some typical water-soluble CPs used for protein detection are shown in Scheme 1. The charges, unique functional groups attached to the polymer side-chains, and hydrophobic backbones allow the polymers to associate with different proteins through versatile interactions, producing specific optical responses to target proteins.

This review focuses on water-soluble CP-based protein sensors and is organized according to the three main mechanisms for protein detection. We start with colorimetric and fluorescent proteinassays based on conformational changes of PTs, which is followed by super-quenching assays with PPVs and PPEs as the probes. At last, we summarize FRET -based assays using PFPs as the signal amplifier. Protein detection based on aggregation-induced fluorescence change of CPs is also included in the super-quenching and FRET categories. As the application of water-soluble CPs for enzyme activity and protein conformation study has been recently reviewed by different research groups,17 these assays are not the focus of the review and will only be briefly mentioned according to their operation mechanisms.
Protein detection based on conformational changes
Cationic PTs were initially reported to undergo conformational change upon association with oligonucleotides, which led to solution color and fluorescence alterations in the presence of single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA).18 As this strategy does not require sophisticated chemical functionalization of polymers or analytes, cationic PTs have been widely used to detect various analytes in solution.19 By taking advantage of specific interaction between an aptamer (5′-GGTTGGTGTGGTTGG-3′) and thrombin, Leclerc's group reported that the cationic PT (P1) could be used for label-free specific protein detection.13 The aqueous solution of P1 is yellow in color, which is related to the random-coil conformation of the polymer chains. In the presence of a nonspecific thrombin aptamer or the nonspecific protein, bovine serum albumin (BSA), P1 formed complex with the aptamer to adopt a planar formation (path B in Fig. 1) and yielded a red-shifted absorption maximum (red–violet color) and weak fluorescence. When the specific aptamer bound to human α-thrombin, a compact unimolecular quadruplex structure was formed. In this case, P1 wrapped around the quadruplex to form a complex which partially hindered the aggregation and planarization of the polymer chains, leading to an orange color and high fluorescence (path A in Fig. 1). The assay allowed for human α-thrombin detection by both colorimetric and fluorescent methods. Using a standard fluorometer, the limit of detection (LOD) for human α-thrombin was calculated to be 2 × 10−15 mol (10−11 M in 200 μL).13 | ||
| Fig. 1 Schematic illustration of the specific detection of human α-thrombin using a ssDNA thrombin aptamer and a cationic PT. Reproduced from ref. 13 with permission. Copyright American Chemical Society 2004. | ||
Subsequently, the same group developed a protein array based on cationic PT–DNA aptamer complexes with improved detection sensitivity (Fig. 2).20 The integrated biochips allowed direct detection of human thrombin in less than one hour at room temperature. First, stoichiometric duplexes were formed between cationic P1 and the Cy3-labeled ssDNAaptamer (5′-NH2-C6-GGTTGGTGTGGTTGG-Cy3-3′) immobilized on the solid support. In the presence of human thrombin, the negatively charged DNA aptamer bound to thrombin to form a folded structure, which increased the Cy3 signal output upon excitation of P1 as compared to that in the absence of thrombin. The signal amplification was reported to result not only from FRET , but also from fluorescence chain reaction (FCR).21 Under the same experimental conditions, the fluorescent signal of Cy3 was quite low when nonspecific proteins (e.g. BSA and immunoglobulin E) were used, indicating good assay specificity. The LOD is ∼1.5 × 107 molecules for human thrombin.
 | ||
| Fig. 2 Schematic illustration of the specific recognition of protein by aptamer–PT aggregates on glass slides. Reproduced from ref. 20 with permission. Copyright Wiley-VCH 2006. | ||
It is noteworthy that cationic PTs are able to directly report the conformational change of certain proteins through distinct fluorescence output.22 Inganäs' group showed that the fluorescence of an amino acid-substituted PT (P2) could be altered by the conformational change of a synthetic peptide.23 The same group further applied cationic PT (P3) to sense Ca2+-induced conformational change of calmodulin.24P3 has shown planar geometry upon complexation with calmodulin, which led to red-shifted absorption (from 421 to 455 nm) and emission spectra (from 565 to 608 nm) with decreased fluorescence intensity. Addition of Ca2+ to the mixture caused blue-shift of the absorption maximum to 440 nm and emission maximum to 601 nm, which was due to a Ca2+-induced calmodulin change that further affected the conformation of P3–protein complexes. A similar strategy was further extended to detect amyloid fibrils in chicken lysozyme25 and protein conformational changes on a poly(dimethylsiloxane) (PDMS) biochip.26 In addition to detecting the conformational change of certain proteins, Aguzzi's group and Leclerc's group reported an advanced strategy to discriminate four distinct prion strains propagated in mice, providing fundamental understanding of protein misfolding disorders over a wide range.27
Protein detection based on fluorescence quenching/dequenching
In 1995, Swager's group demonstrated amplified fluorescence quenching of CPs.28 The greatly enhanced sensitivity of CPs relative to small molecule counterparts in the presence of quenchers is believed to be derived from the delocalized electron structure of CPs, which facilitates efficient energy migration along the polymer backbone. As shown in Fig. 3, a single bound quencher molecule is able to trap almost all the excitons along the polymer backbone. Qualification of CP fluorescence quenching is determined by Stern–Volmer equation:| F0/F = 1 + KSV[Q] |
 | ||
| Fig. 3 Schematic illustration of the mechanism of amplified quenching of CPs upon interaction with an electron transfer quencher . Reproduced from ref. 28 with permission. Copyright American Chemical Society 1995. | ||
The amplified quenching effect allows the design of CP-based protein sensors by monitoring the fluorescence change of CPs in the absence and presence of analytes. The “quencher –tether–ligand” (QTL) approach was one of the earliest proteinassays to operate on CP fluorescence superquenching. The approach involved the use of a biotin-linked cationic quencher (methyl viologenMV2+). The fluorescence of P4 in aqueous solution was efficiently quenched by a very small amount of biotin-linked MV2+ (B–MV). Addition of avidin was able to significantly recover the polymer fluorescence due to specific interactions between biotin and avidin that could pull B–MV away from the vicinity of P4. Similarly, Cao's group reported an anionic polyfluorene-based assay , taking advantage of specific recognition between biotin and avidin as well as electrostatic interactions between an anionic PFP (P11) and avidin.16 The quenching of P11 by a biotin-labeled Lucifer dye was only observed in the presence of avidin, leading to the formation of an avidin–biotin–quencher complex which electrostatically associated with P11. Control experiments using the biotin-labeled Lucifer dye in the presence of nonspecific proteins were performed to prove the assay’s specificity.
In addition to the reported specific interactions, studies from Heeger's and Bazan's groups revealed that nonspecific interactions between proteins and CPs existed in these assays , which could significantly affect protein detection and lead to misinterpretation of results.29,30 To minimize electrostatic interactions between proteins and CPs, the concept of a charge-neutral complex (CNC) was proposed by mixing anionic P5 and a cationic polyelectrolyte at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge ratio.29 Based on the detection scheme shown in Fig. 4, quenching of CNC fluorescence by a negatively charged dinitrophenol (DNP) derivative and specific dequenching upon addition of anti-DNPantibody were demonstrated. In addition, the authors also showed that the quenched fluorescence of CNC could not be recovered when the nonspecific antibody rat IgG or a nonspecific quencher (2,4,6-trinitrophenol, TNP) to anti-DNPantibody was used. The LOD for this assay is 300 nM.
:1 charge ratio.29 Based on the detection scheme shown in Fig. 4, quenching of CNC fluorescence by a negatively charged dinitrophenol (DNP) derivative and specific dequenching upon addition of anti-DNPantibody were demonstrated. In addition, the authors also showed that the quenched fluorescence of CNC could not be recovered when the nonspecific antibody rat IgG or a nonspecific quencher (2,4,6-trinitrophenol, TNP) to anti-DNPantibody was used. The LOD for this assay is 300 nM.
 | ||
| Fig. 4 Schematic illustration of the quenched CNC fluorescence response after addition of specific antibody (A), and nonspecific antibody (B, C) to the quenchers . Reproduced from ref. 29 with permission. Copyright National Academy of Sciences 2002. | ||
Although the initial efforts for CP-based proteinassays were made toward using biospecific receptor–CP complexes for sensing, the fluorescence change of CPs upon nonspecific interaction with different proteins has also attracted great research interest. Heeger et al. found that the fluorescence of an anionic PPV (P5) could be efficiently quenched by cytochrome c (cyt c), an iron-containing protein, which was attributed to electron transfer from the photoexcited polymer to cyt c upon complex formation.31 Subsequently, we studied the fluorescence quenching of an anionic polyfluorene by different proteins, which revealed that cyt c-induced fluorescence quenching was caused by both charge transfer and aggregation of polymer chains.32 In 2006, Huang's group reported the detection of an anionic iron-sulfur-based protein, rubredoxin .33 The fluorescence of cationic P6 was efficiently quenched by rubredoxin due to electron transfer from P6 to the iron center. In contrast, control experiments under the same experimental conditions showed that the quenching of P6 by insulin and cyt c was remarkably less than that of rubredoxin , indicating the good selectivity of the polymer toward rubredoxin even in the absence of specific analyte–receptor pairs.
An important concept in sensor arrays for protein detection, by taking advantage of nonspecific interactions between CPs and proteins, was proposed by Bunz's and Rotello's groups.34 The sensor array contained six non-covalent gold nanoparticle (NP)–CP aggregates. The detection scheme is illustrated in Fig. 5. Initially, the fluorescence of P7 was quenched by the gold NPs.35 These NPs were engineered with different hydrophobic functional groups to tune the interactions between the NPs and polymers or proteins. Upon addition of proteins, the interaction between gold NPs and CPs was disrupted and the displacement of the polymers by proteins on the NP surface led to distinct fluorescence response patterns. These patterns were highly repeatable for each individual protein, providing a robust platform for multiprotein detection with nanomolar sensitivity. The same groups further reported a sensor array containing six functionalized PPE derivatives, each of which possessed distinct charge characteristics and molecular scales to provide different binding diversities upon interaction with proteinanalytes.36 The distinct fluorescence response pattern allowed the identification of 17 different proteins with a high accuracy of 97%.
 | ||
| Fig. 5 Schematic illustration of (a) the displacement of a quenched fluorescent PPE by proteinanalyte (in blue) from gold NPs to recover the fluorescence, and (b) unique fluorescence pattern generation through differential release of PPEs. Reproduced from ref. 34 with permission. Copyright Nature Publishing Group 2007. | ||
In addition to quencher -induced polymer fluorescence quenching via electron transfer, specific recognition-induced polymer aggregation could also lead to polymer fluorescence quenching, which is useful for protein detection. Lectins , a type of protein which plays a crucial role in cell signaling, surface recognition and pathogen docking, have been actively studied using sugar-containing conjugated polymers as the probe.36–46 The most frequently studied lectins are concanavalin A (Con A) and Escherichia coli (E. coli), which can bind to α-mannose-substituted water-soluble CPs and induce polymer aggregation. It should be noted that neutral water-soluble CPs are preferred for lectin detection in order to avoid electrostatic interactions between charged polymers and proteins. In 2004, Swager et al. reported the detection of E. coli strains using a water-soluble mannose-functionalized PPE.37 After addition of the polymer into an E. coli suspension, the mannose-binding bacteria formed clusters and the polymer exhibited a red-shifted emission spectrum, due to π–π stacking interactions between the polymer strands. Meanwhile, Bunz's group reported the synthesis of a mannose-bearing PPE (P8) through well-defined mannose-substituted monomers for Con A detection.38 They further studied the fluorescence quenching mechanism of sugar functionalized PPEs upon complex formation with Con A, suggesting that the assay sensitivity could be improved by increasing the linker length and introducing more complicated sugar units.46Mannose-substituted polymers with backbones of PPV,39poly(p-phenylene) (PPP),40PT,43poly(phenylacetylene) (PPA),44 and PFP47 have also been reported for Con A sensing. It was found that introducing flexible poly(ethylene glycol) as the spacer between the sugar substitution and polymer backbone could greatly increase water solubility of these CPs, leading to reduced nonspecific interactions for Con A sensing.47
In addition to protein detection, polymer quenching/dequenching-based assays have also been widely used for enzyme activity studies.48 A typical approach is to use a quencher -labeled biomolecule complex that could be hydrolyzed by an active enzyme to release the quencher moiety and induce fluorescence change of the polymer solution. Schanze's and Whitten's groups reported several significant studies about CP-based fluorescent assays for enzyme activity study using anionic PPEs in solution or anionic PPE-coated fluorescent microspheres as the probes.49,50 A typical example is shown in Fig. 6.49 The fluorescence turn-on approach included an anionic PPE (P9) and a cationic peptide (the enzyme substrate) labeled with p-nitroanilide as the quencher . Electrostatic association between the anionic P9 and cationic peptide led to fluorescence quenching of the polymer by p-nitroanilide. In the presence of a proteolytic enzyme, peptidase, the quencher -labeled peptide was hydrolyzed and the quencher was released. As the free p-nitroanilide was neutral, it could not associate with the polymer and the solution fluorescence was recovered. On the other hand, the same system can be designed as a turn-off assay by using P7 in combination with the bis-Arg derivative of Rhodamine-100 (Rho-Arg-2). Rho-Arg-2 did not quench the polymer fluorescence, however, the presence of a protease in the mixture led to the monoamide derivative (Rho-Arg), which was able to quench the fluorescence of P7. Based on a similar strategy, more assays have been designed to study the reactivity of different enzymes, which can be found in recent reviews.17
 | ||
| Fig. 6 Schematic illustration of CP-based turn-on and turn-off assays for protease activity study. Reproduced from ref. 49 with permission. Copyright National Academy of Sciences 2004. | ||
Protein detection based on Förster resonance energy transfer (FRET )
A reliable method based on long-range FRET from a CP to a signaling chromophore was first applied to DNA detection, where the polymer serves as the energy donor and the signaling chromophore serves as the energy acceptor.51 The high extinction coefficients and highly delocalized backbone make CPs excellent energy donors in FRET -based assays to amplify the acceptor signal, yielding high sensitivity and selectivity with minimized background signals.52 The strong distance-dependent FRET provides opportunity for sensing as the distance change between the donor and acceptor could be monitored by fluorescence intensity changes. FRET -based biosensors are of growing interest as they have several advantages over colorimetric and quenching/dequenching sensors, which include detection versatility and multi-channel signal collection.53 According to Föster’s theory, short donor–acceptor distance,54 good spectral overlap55 and optimized dipole orientation56 should favor efficient energy transfer. In addition, our recent study revealed that the HOMO and LUMO energy levels of the acceptor should be contained within those of the donor to favor FRET over photoinduced electron transfer (PET).57Following the QTL strategy, by modifying the quencher attached to biotin to a fluorescent dye, Wang et al. reported a fluorescence ratiometric proteinassay for strepavidin detection.58 The assay contains three elements: a cationic PFP (P10), a negatively charged biotinylated fluorescein probe (F1-B), and the target protein (Fig. 7). The emission spectrum of P10 showed a good overlap with the absorption spectrum of F1. In the presence of strepavidin, the biotin moiety of F1-B associated with streptavidin and F1 was buried in the adjacent vacant binding site. This binding prevented the electrostatic interaction between F1 and the positively charged polymer, resulting in weak F1 emission upon excitation of P10. In the presence of a nonspecific protein, such as BSA, the F1-B was not associated with the protein. Electrostatic interaction between F1 and the polymer resulted in efficient energy transfer to amplify the fluorescence intensity of F1. Streptavidin detection is thus achieved by monitoring F1 emission intensity in solution upon excitation of P10.
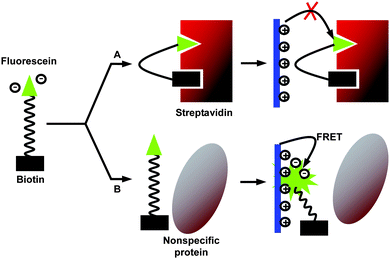 | ||
| Fig. 7 Schematic illustration of streptavidin detection based on the FRET mechanism. Reproduced from ref. 58 with permission. Copyright Wiley-VCH 2006. | ||
In addition to protein detection based on specific lock–key interactions, an array-based FRET assay has also been developed for protein detection based on nonspecific interactions between CP/dye-labeled ssDNA (ssDNA-C*) complexes and proteins.59 The design concept is motivated by the fact that proteins with different charges and hydrophobicities can effectively perturb electron coupling between optical units within a complex of CP/ssDNA-C* and in turn vary FRET -induced fluorescence of both CP (donor) and C* (acceptor). A series of complexes between a cationic oligofluorene and five 6-carboxylfluorescein (FAM)-labeled ssDNA with different sequences and lengths were used to form complexes, which served as FRET probes to generate an array of fluorescence response toward proteins. The corresponding fluorescence generated unique patterns to classify 14 proteins with good accuracy. Although this array cannot be used for protein quantification and is less suitable for mixed samples, it has good reliability and versatility for the detection of purified proteins in aqueous media.
To solve the problem that most homogeneous fluorescent proteinassays can not be operated in biological media, recently we developed a FRET -based homogeneous proteinassay by taking advantage of specific aptamer/protein recognition.60 The detection mechanism is based on surface-charge-switch-induced energy transfer between an anionic CP and a FAM-labeled aptamer. One started with a solution that contains an anionic CP (P11) and the FAM-aptamer. In the presence of lysozyme, the FAM-aptamer bound to lysozyme and the surface charge was switched from negative to positive, inducing electrostatic association between the aptamer–lysozyme complex and the anionic polymer. As a consequence, FRET occurred from P11 to FAM. On the other hand, nonspecific proteins were not able to recognize the FAM-aptamer and the surface charge of the aptamer remained negative, thus no FRET occurred as the distance between the polymer and FAM-aptamer was too far. Noteworthy is that the FAM signal obtained from a urine sample (Fig. 8) was almost the same as that obtained in pure water, indicating that the selectivity and sensitivity were maintained in biological media.
![Normalized fluorescence spectra of P11 incubated with lysozymeaptamer in the absence (black) and presence (red) of lysozyme in urine, and in the presence of lysozyme in water (green). [aptamer] = 5.0 × 10−7 M, [lysozyme] = 26.5 μg mL−1. Reproduced from ref. 60.](/image/article/2010/PY/b9py00283a/b9py00283a-f8.gif) | ||
| Fig. 8 Normalized fluorescence spectra of P11 incubated with lysozymeaptamer in the absence (black) and presence (red) of lysozyme in urine, and in the presence of lysozyme in water (green). [aptamer] = 5.0 × 10−7 M, [lysozyme] = 26.5 μg mL−1. Reproduced from ref. 60. | ||
Subsequently, we developed NP-based proteinassays for goat IgG and thrombin detection by taking advantage of the separation of NPs and light-harvesting properties of CPs to amplify the detection signal.61,62 For goat IgG detection, the prime antibodies were immobilized on the NP surface and the NPs were used to capture antigen and Cy3-labeled secondary antibodies in a sandwich assay format to yield fluorescent NPs (Fig. 9).61 Cationic P12 was then bound to proteins on the NP surface. FRET occurred upon excitation of P12 which led to high sensitivity and selectivity of the assay with a detection limit of 1 ng mL−1. In the presence of nonspecific proteins, such as BSA and thrombin, the signal from Cy3 was very weak. By replacing the antibodies immobilized on the NP surface with aptamers, the assay was further developed for thrombin and lysozyme detection in serum.62 These results suggested that CP-amplified NP-based assays are generic and could be readily applicable to detect various proteins in important biological media.
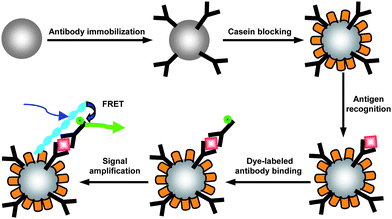 | ||
| Fig. 9 Schematic illustration of the cationic PFP-amplified NP-based fluoroimmunoassay. Figure used with permission. Reproduced from ref. 61 with permission. Copyright Elsevier 2009. | ||
Recently, we also demonstrated that CPs could be used for the naked-eye detection and quantification of proteins. Based on the understanding that intermolecular energy transfer could be more efficient than intramolecular energy transfer,63 we developed multicolor CPs, which contain fluorene segments and 2,1,3-benzothiadiazole (BT) units for naked-eye protein sensing. The operation mechanism was based on aggregation-induced FRET from the fluorene segments to BT units.64 The discrimination of different proteins was illustrated using P13 as a probe, which had a blue emission color in aqueous solution. Addition of lysozyme, BSA, and cyt c to the polymer solution changed the polymer emission color to yellow, green, and dark, respectively. The distinguishable fluorescent color in the presence of different proteins was due to the variation in hydrophobic nature, net charge, and the structure among the proteins. Both energy transfer from the fluorene segments to the BT units upon aggregation and electron transfer between P13 and the metal center in cyt c played an important role in the observed color change. Subsequently, we developed cationic CPs containing BT units as fluorescence ‘light-up’ probes for BSA and DNA sensing based on the understanding that BT emission is sensitive to environmental polarity due to its charge transfer electronic states.65 These polymers have shown very weak fluorescence in aqueous media. Addition of BSA or DNA to the polymer solution significantly enhanced polymer fluorescence due to the increased hydrophobicity of polymers upon complex formation with biomacromolecules. Moreover, the polymers exhibited linear fluorescence enhancement as a function of BSA concentration.
We further designed and synthesized a neutral water-soluble polymer (P14) for naked-eye detection and quantification of Con A by introducing mannose to the polymer side-chain.66 The detection and quantification of Con A was achieved by the linear increase of BT emission intensity upon addition of Con A to the solution of P14. The increased yellow emission was due to polymer aggregation caused by specific interactions between Con A and α-mannose that brought different polymer chains into close proximity (Fig. 10). By varying the concentration of P14, a standard calibration curve was obtained for Con A quantification in the concentration range from 1 to 250 nM. This is the first CP-based Con A assay that shows signal increase upon Con A recognition, which is significantly different from all the previously reported CP-based fluorescence quenching assays for Con A detection.
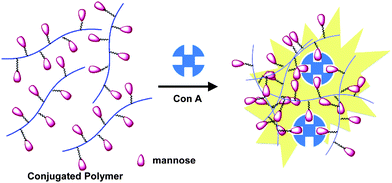 | ||
| Fig. 10 Schematic illustration of aggregation-induced BT emission of the mannose-bearing CP-containing BT units. Figure used with permission. Reproduced from ref. 66 with permission. Copyright Wiley-VCH 2009. | ||
FRET -based assays have also been widely used for enzyme activity studies by monitoring the fluorescence change of reporters.67 To overcome the limitation that the majority of reported fluorescence assays are limited to one specific enzyme in one solution, Wang's group recently reported a simultaneous multiplex detection of nucleases using a cationic PFP (P10) and a Y-shaped DNA labeled at the 5′-termini with fluorescein, TexRed, and Cy5.68 In the absence of enzymes, complex formation between P10 and Y-DNA strands through electrostatic interactions allowed energy transfer from the polymer to the dyes. In the presence of Hae II, Pvu II or EcoRV, the specific recognition of cleavage sites in DNA strands caused release of each individual dye, leading to unique solution fluorescence change upon excitation of the polymer. This study paved a way for the design of simple sensor systems for simultaneous detection of multiplexes of nucleases.
Conclusions
Various protein sensors that take advantage of the sensitive optical responses of water-soluble conjugated polymers have been summarized in this review. These assays operate on either specific lock–key recognition or nonspecific interactions between CPs and proteins, which do not require sophisticated instrumentation. The reported array-based protein sensors open up new opportunities to achieve fast, sensitive, and reliable multiprotein detection, and have great potential for clinical applications. However, it should be noted that most CP-based fluorescence assays only work with purified protein samples; nonspecific interactions between CPs and different components in biological media remain a problem to be solved. Further optimization of chemical structures and optical properties of water-soluble CPs will boost their applications in future biosensors with improved sensitivity and selectivity.Acknowledgements
The authors are grateful to the National University of Singapore (R-279-000-234-123), the Singapore Ministry of Education (R-279-000-255-112) and the Temasek Defense Systems Institute (R-279-000-268-422, R-279-000-268-592, and R-279-000-268-232) for financial support.References
- S. Fields, Science, 2001, 291, 1221–1224 CrossRef CAS; R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS; D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS; C. J. Johnson, N. Zhukovsky, A. E. G. Cass and J. M. Nagy, Proteomics, 2008, 8, 715–730 CrossRef CAS.
- T. Kodadek, Chem. Biol., 2001, 8, 105–115 CrossRef CAS.
- E. Benito-Peña, M. C. Moreno-Bondi, G. Orellana, K. Maquieira and A. V. Amerongen, J. Agric. Food Chem., 2005, 53, 6635–6642 CrossRef CAS.
- J. Wang, G. D. Liu, M. H. Engelhard and Y. H. Lin, Anal. Chem., 2006, 78, 6974–6979 CrossRef CAS.
- T. Li, L. P. Guo and Z. X. Wang, Biosens. Bioelectron., 2008, 23, 1125–1130 CrossRef CAS.
- G. F. Jie, J. J. Zhang, D. C. Wang, C. Cheng, H. Y. Chen and J. J. Zhu, Anal. Chem., 2008, 80, 4033–4039 CrossRef CAS.
- C. P. Soman and T. D. Giorgio, Langmuir, 2008, 24, 4399–4404 CrossRef CAS.
- L. S. Wan, B. B. Ke and Z. K. Xu, Enzyme Microb. Technol., 2008, 42, 332–339 CrossRef CAS.
- S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS.
- J. E. Guillet, Polymer Photophysics and Photochemistry, Cambridge University Press, Cambridge, 1985 Search PubMed; H. F. Kauffmann, Photochemistry and Photophysics, ed. J. E. Radek, CRC, Boca Raton, 1990, vol. 2 Search PubMed; G. D. Scholes and K. P. Ghiggino, J. Chem. Phys., 1994, 101, 1251–1261 Search PubMed; S. E. Webber, Chem. Rev., 1990, 90, 1469–1483 Search PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS.
- M. R. Pinto and K. S. Schanze, Synthesis, 2002, 1293–1309 CrossRef CAS.
- H. A. Ho and M. Leclerc, J. Am. Chem. Soc., 2004, 126, 1384–1387 CrossRef CAS.
- L. Chen, D. W. McBranch, H. L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12287–12292 CrossRef CAS.
- S. Kumaraswamy, T. Bergstedt, X. Shi, F. Rininsland, S. Kushon, W. Xia, K. Ley, K. Achyuthan, D. McBranch and D. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7511–7515 CrossRef CAS.
- F. Huang, X. Wang, D. Wang, W. Yang and Y. Cao, Polymer, 2005, 46, 12010–12015 CrossRef CAS.
- K. E. Achyuthan, T. S. Bergstedt, L. Chen, R. M. Jones, S. Kumaraswamy, S. A. Kushon, K. D. Ley, L. Lu, D. McBranch, H. Mukundan, F. Rininsland, X. Shi, W. Xia and D. G. Whitten, J. Mater. Chem., 2005, 15, 2648–2656 RSC; A. Herland and O. Inganäs, Macromol. Rapid Commun., 2007, 28, 1703–1713 CrossRef CAS; F. Feng, F. He, L. L. An, S. Wang, Y. L. Li and D. B. Zhu, Adv. Mater., 2008, 20, 2959–2964 CrossRef CAS; H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS; L. L. An, S. Wang and D. B. Zhu, Chem.–Asian J., 2008, 3, 1601–1606 CrossRef CAS.
- H. A. Ho, M. Boissinot, M. G. Bergeron, G. Corbeil, K. Doré, D. Boudreau and M. Leclerc, Angew. Chem., Int. Ed., 2002, 41, 1548–1551 CrossRef CAS.
- H. A. Ho, A. Najari and M. Leclerc, Acc. Chem. Res., 2008, 41, 168–178 CrossRef CAS.
- M. Béra Abérem, A. Najari, H. A. Ho, J. F. Gravel, P. Nobert, D. Boudreau and M. Leclerc, Adv. Mater., 2006, 18, 2703–2707 CrossRef.
- H. A. Ho, K. Doré, M. boissinot, M. G. Bergeron, R. M. Tanguay, D. Boudreau and M. Leclerc, J. Am. Chem. Soc., 2005, 127, 12673–12676 CrossRef CAS.
- A. Herland and O. Inganäs, Macromol. Rapid Commun., 2007, 28, 1703–1713 CrossRef CAS.
- K. P. R. Nilsson, J. Rydberg, L. Baltzer and O. Inganäs, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10170–10174 CrossRef CAS.
- K. P. R. Nilsson and O. Inganäs, Macromolecules, 2004, 37, 9109–9113 CrossRef CAS.
- K. P. R. Nilsson, A. Herland, P. Hammarström and O. Inganäs, Biochemistry, 2005, 44, 3718–3724 CrossRef CAS.
- P. Åsberg, K. P. R. Nilsson and O. Inganäs, Langmuir, 2006, 22, 2205–2211 CrossRef CAS.
- C. J. Sigurdson, K. P. R. Nilsson, S. Hornemann, G. Manco, M. Polymenidou, P. Schwarz, M. Leclerc, P. Hammarström, K. Wüthrich and A. Aguzzi, Nat. Methods, 2007, 4, 1023–1030 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 7017–7018 CrossRef.
- D. L. Wang, X. Gong, P. S. Heeger, F. Rininsland, G. C. Bazan and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 49–53 CrossRef CAS.
- S. J. Dwight, B. S. Gaylord, J. W. Hong and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 16850–16859 CrossRef CAS.
- C. Fan, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2002, 124, 5642–5643 CrossRef CAS.
- Y. Zhang, B. Liu and Y. Cao, Chem.–Asian J., 2008, 3, 739–745 CrossRef CAS.
- F. Cheng, G. W. Zhang, X. M. Lu, Y. Q. Huang, Y. Chen, Y. Zhou, Q. L. Fan and W. Huang, Macromol. Rapid Commun., 2006, 27, 799–803 CrossRef CAS.
- C. C. You, O. R. Miranda, B. Gider, P. S. Ghosh, I. B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Bunz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 318–323 CrossRef CAS.
- C. H. Fan, S. Wang, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS.
- O. R. Miranda, C. C. You, R. Phillips, I. B. Kim, P. S. Ghosh, U. H. F. Bunz and V. R. Rotello, J. Am. Chem. Soc., 2007, 129, 9856–9857 CrossRef CAS.
- M. D. Disney, J. Zheng, T. M. Swager and P. H. Seeberger, J. Am. Chem. Soc., 2004, 126, 13343–13346 CrossRef CAS.
- I. B. Kim, J. N. Wilson and U. H. F. Bunz, Chem. Commun., 2005, 1273–1275 RSC.
- A. Takasu, K. Iso, T. Dohmae and T. Hirabayashi, Biomacromolecules, 2006, 7, 411–414 CrossRef CAS.
- C. H. Xue, S. P. Jog, P. Murthy and H. Y. Liu, Biomacromolecules, 2006, 7, 2470–2474 CrossRef CAS.
- T. L. Kelly, M. C. W. Lam and M. O. Wolf, Bioconjugate Chem., 2006, 17, 575–578 CrossRef CAS.
- C. H. Xue, V. R. R. Donuru and H. Y. Liu, Macromolecules, 2006, 39, 5747–5752 CrossRef CAS.
- C. H. Xue, F. T. Luo and H. Y. Liu, Macromolecules, 2007, 40, 6863–6870 CrossRef CAS.
- I. Otsuka, T. Hongo, H. Nakade, A. Narumi, R. Sakai, T. Satoh, H. Kaga and T. Kakuchi, Macromolecules, 2007, 40, 8930–8937 CrossRef CAS.
- R. L. Phillips, I. B. Kim, L. M. Tolbert and U. H. F. Bunz, J. Am. Chem. Soc., 2008, 130, 652–6954.
- R. L. Phillips, I. B. Kim, B. E. Carson, B. Tidbeck, Y. Bai, T. L. Lowary, L. M. Tolbert and U. H. F. Bunz, Macromolecules, 2008, 41, 7316–7320 CrossRef CAS.
- C. H. Xue, S. Velayudham, S. Johnson, R. Saha, A. Smith, W. Brewer, P. Murthy, S. T. Bagley and H. Y. Liu, Chem.–Eur. J., 2009, 15, 2289–2295 CrossRef CAS.
- J. H. Wosnick, C. M. Mello and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 3400–3405 CrossRef CAS; L. L. An, L. B. Liu and S. Wang, Biomacromolecules, 2009, 10, 454–457 CrossRef CAS.
- M. R. Pinto and K. S. Schanze, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7505–7510 CrossRef CAS.
- F. Rininsland, W. Xia, S. Wittenburg, X. Shi, C. Stankewicz, K. Achyuthan, D. McBranch and D. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15295–15300 CrossRef CAS.
- B. S. Gaylord, A. J. Heeger and G. C. Bazan, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10954–10957 CrossRef CAS.
- K. Y. Pu and B. Liu, Biosens. Bioelectron., 2009, 24, 1067–1073 CrossRef CAS.
- B. Liu and G. C. Bazan, Chem. Mater., 2004, 16, 4467–4476 CrossRef CAS.
- B. Liu, B. S. Gaylord, S. Wang and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 6705–6714 CrossRef CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 1942–1943 CrossRef CAS.
- B. Liu, S. Wang, G. C. Bazan and A. Mikhailovsky, J. Am. Chem. Soc., 2003, 125, 13306–13307 CrossRef CAS; B. Liu, T. T. T. Dan and G. C. Bazan, Adv. Funct. Mater., 2007, 17, 2432–2438 CrossRef CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2006, 128, 1188–1196 CrossRef CAS.
- L. L. An, Y. L. Tang, S. Wang, Y. L. Li and D. B. Zhu, Macromol. Rapid Commun., 2006, 27, 993–997 CrossRef CAS.
- H. P. Li and G. C. Bazan, Adv. Mater., 2009, 21, 964–967 CrossRef CAS.
- J. Wang and B. Liu, Chem. Commun., 2009, 2284–2286 RSC.
- Y. Y. Wang and B. Liu, Biosens. Bioelectron., 2009, 24, 3293–3298 CrossRef CAS.
- Y. Y. Wang and B. Liu, Langmuir, 2009, 25, 12787–12793 CrossRef CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 1942–1943 CrossRef CAS; B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS.
- D. Y. Yu, Y. Zhang and B. Liu, Macromolecules, 2008, 41, 4003–4011 CrossRef CAS.
- K. Y. Pu, L. P. Cai and B. Liu, Macromolecules, 2009, 42, 5933–5940 CrossRef CAS.
- J. B. Shi, L. P. Cai, K. Y. Pu and B. Liu, Chem.–Asian J., 2009 DOI:10.1002/asia.200900297.
- H. M. Joong, P. MacLean, W. McDaniel and L. F. Hancock, Chem. Comm., 2007, 4910–4912 RSC; Y. Zhang, Y. Y. Wang and B. Liu, Anal. Chem., 2009, 81, 3731–3737 CrossRef CAS.
- X. L. Feng, X. R. Duan, L. B. Liu, F. D. Feng, S. Wang, Y. L. Li and D. B. Zhu, Angew. Chem., Int. Ed., 2009, 48, 5316–5321 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |