Methyl acrylate polymerizations in the presence of a copper/N3S3 macrobicyclic cage in DMSO at 25 °C†
Craig A.
Bell
a,
Qiao
Sun
a,
Hong
Zhang
a,
Sean C.
Smith
a,
Paul V.
Bernhardt
b and
Michael J.
Monteiro
*a
aAustralian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane QLD 4072, Australia. E-mail: m.monteiro@uq.edu.au
bSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane QLD 4072, Australia
First published on 22nd December 2009
Abstract
Macrobicyclic hexa-amine cage ligands are known to completely encapsulate transition metals inside the ligand cavity. Recent work has shown that a five-coordinate bromido complex [Cu(AMME-N3S3sar)Br]+ was observed, featuring a novel tetradentate (N2S2) coordinated form of the cage ligand in DMSO. Reduction to its monovalent state results in no change in the geometry of the complex. The electrochemistry of this complex in the presence of an alkyl halide showed that this complex has a low activation to convert these alkyl halides to their corresponding incipient radicals. Further polymerization kinetics with methyl acrylate showed that the molecular weight was uncontrolled, implying that deactivation was negligible. Therefore, this copper/ligand complex with an alkyl halide initiator only acts as an initiation source. This led us to use this Cu/AMME-N3S3sar complex to initiate MA polymerizations at room temperature in the presence of a RAFT agent. The results showed that the molecular weight distribution was controlled and followed ideal ‘living’ radical behavior, in which the molecular weight polydispersity for a range of different molecular weight targets was less than 1.1.
Introduction
Copper complexes of multidentate polyamine ligands are very effective catalysts in ‘living’ radical polymerizations.1–3 The monovalent form of the catalyst (Cu(I)L) activates an alkyl halide (RX) through a partial oxidative addition releasing the alkyl radical (Rʹ) while the halide ion (X−) is coordinated to Cu(II). The reverse (deactivation) reaction is also important in controlling the concentration of free-radicals (eqn (1)).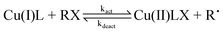 | (1) |
In general, a more negative Cu(II/I) redox potential (E′) of the complex has been correlated with higher activity in atom radical transfer polymerizations (ATRP). Most nitrogen based macrocyclic ligands are highly active compared to their acyclic bi-, tri- or tetradentate analogues, with the highest known catalytic activity found for a bicyclic cyclam macrocycle bearing a strap adjoining opposite N-donors.4 On the other hand, macrocyclic ligands with mixed donor atoms (N and S) are less studied,5 and the ability of S-donors to stabilize Cu(I) would be expected to lower catalytic activity according to current models.2
Macrobicyclic hexa-amine cage ligands6 are known to completely encapsulate transition metals inside the ligand cavity, and once inside, the metal is extremely difficult to remove. In previous work, the macrobicyclic mixed donor (N3S3) encapsulating ligand (see Scheme 1), 1-methyl-8-amino-3,13,16-trithia-6,10,19-triazabicyclo[6.6.6]eicosane (AMME-N3S3sar) complexed with CuBr in toluene did not effectively control the polymerization of styrene (STY) at 60 or 100 °C, but acted only as an initiation source while deactivation by CuBr2/AMME-N3S3sar was found to be negligible.5 The coupling reaction between α,ω-PSTY (i.e. Br-PSTY-Br) using CuBr/AMME-N3S3sar and excess Cu(0) in toluene at 100 °C gave no loss of the starting Br-PSTY-Br.5 However, changing the solvent to DMSO and decreasing the temperature to 60 °C led to 87% consumption of starting polymer in only 10 min. with the concomitant formation of high molecular weight multiblock copolymer. Since the complex showed no disproportionation of Cu(I) to Cu(0) and Cu(II) in DMSO, we do not believe that nascent Cu(0) plays a role in this reaction, as shown to be the case in single electron transfer-living radical polymerization (SET-LRP).7
 | ||
| Scheme 1 Structures of high activating ligands and minimized geometries of AMME-N3S3sar ligand calculated by MP2. | ||
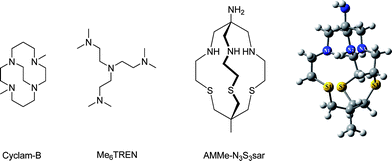
We therefore, carried out an in-depth study8 into the nature of this phenomenon by examining the coordination chemistry of the [Cu(AMME-N3S3sar)Br]+ complex in DMSO. A five-coordinate bromido complex [Cu(AMME-N3S3sar)Br]+ was observed featuring a novel tetradentate (N2S2) coordinated form of the cage ligand. This copper(II) complex was characterized by X-ray crystallography and with solution spectroscopy. The kinetics of the interconversion between these six- and five-coordinate Cu(II) complexes were resolved (Scheme 2). These processes were reversible. When bromide concentrations were very low, [Cu(AMME-N3S3sar)Br]+ reverted to the six-coordinate complex [Cu(AMME-N3S3sar)]2+ while [Cu(AMME-N3S3sar)]2+ reacted cleanly with excess bromide ions in DMSO to generate five-coordinate [Cu(AMME-N3S3sar)Br]+. Electrochemistry of [Cu(AMME-N3S3sar)Br]+ in DMSO shows that upon reduction to the monovalent state, a five-coordinate tetradentate conformation formed exclusively regardless of whether a six- or five-coordinate copper(II) complex was the precursor. This shows that in DMSO the bromide ion can be removed resulting in the hexadentate coordination of Cu(II) within the cavity of the cage ligand. We also find that Cu(I) resided on the face of the ligand (Scheme 2), and even if encapsulated initially as Cu(II) will rapidly rearrange to be located on the face upon reduction.
 | ||
| Scheme 2 Interconversion of Cu(II)/AMME-N3S3sar complexes in DMSO. | ||
In this paper, we provide further evidence using cyclic voltammetry on the activity of [Cu(AMME-N3S3sar)Br]+ in DMSO as a polymerization catalyst. In addition, the kinetics for the polymerization of methyl acrylate (MA) in DMSO using CuBr/AMME-N3S3sar and ethyl 2-bromoisobutyrate (EBiB) at 25 °C were evaluated, and the external orders of reaction determined for Cu(I), Cu(II) and ligand. This unique redox system was then used to initiate reversible addition-fragmentation chain transfer (RAFT)9 polymerization of MA at 25 °C using a highly reactive chain transfer agent (CTA), resulting in the production of polymer with well controlled molecular weight and narrow molecular weight distributions (MWDs).
Experimental
Materials
Ethanol (Ajax, 99.8%) was dried with magnesium (Reidel-de Haën, 99.5%) and iodine (Univar, 99.8%), and distilled. Sodium metal (Riedel-de Haën, 99%), cysteamine hydrochloride (Fluka, 97%), cobalt(II) nitrate hydrate (UniLab, 96%), sodium carbonate anhydrous (Pronalys, 99.8%), formaldehyde (Univar, 37% in H2O), nitromethane (Aldrich, 96%), zinc powder (Analar, 90%), hydrogen peroxide (Univar, 30% in H2O), hydrochloric acid (Ajax, 32% in H2O), methanol (Univar, 98%), sodium cyanide (Sigma, 99.8%), potassium hydroxide (Chem Supply, 85%), sodium sulfate (Univar, 99%), chloroform (CHCl3, Pronalys, 99%), dichloromethane (CH2Cl2, Labscan, AR grade), diethyl ether (Et2O, Pronalys, AR grade), tetrahydrofuran (THF, HPLC grade, LABSCAN, 99.8%), toluene (HPLC, LABSCAN, 99.8%), dimethyl sulfoxide (DMSO, LABSCAN, 99.8%), formic acid (Univar, 90%), butanethiol (Aldrich, 99%), triethylamine (Et3N, Fluka, purum), carbon disulfide (CS2, Riedel-de Haën, 99%), methyl 2-bromopropionate (MBP, Aldrich, 98%), ethyl 2-bromoisobutyrate (EBiB, Aldrich, 98%), cuprous bromide (CuBr, Aldrich, 99.999%), and cupric bromide (CuBr2, Aldrich, 99%) were all used as received. Methyl acrylate (MA, Aldrich, 99%, 100 ppm monomethyl ether hydroquinone inhibitor) was purified by passage through a column of activated basic alumina (Aldrich, Brockmann I, standard grade, ∼150 mesh, 58 Å).Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pentane–ethyl acetate, second band) gave the desired product.
:1 pentane–ethyl acetate, second band) gave the desired product.
1H NMR (CDCl3) δ 0.92 (tr, J = 7.5 Hz, 3H, CH3), 1.43 (mult, J = 7.5 Hz, 2H, CH2), 1.62 (d, J = 7.5 Hz, 3H, CH3), 1.65 (quin, J = 7.5 Hz, 2H, CH2), 3.36 (tr, J = 7.5 Hz, 2H, CH2), 3.73 (s, 3H, CH3), 4.84 (quad, J = 7.5 Hz, 1H, CH); 13C NMR (CDCl3) δ 13.55, 16.91, 22.02, 29.89, 36.94, 47.68, 52.82, 171.63 (CH–C(= O)–O), 221.99 (S–C(= S)–S)
Techniques
Results and discussion
Cyclic voltammetry of AMME-N3S3sar/CuBr
The objective of the electrochemical measurements was to provide insight into the redox properties of Cu complexes of AMME-N3S3sar that form upon mixing each component together in solution as in a typical polymerization experiment, and correlating this to other copper/ligand complexes know to undergo ATRP. Fig. 1 shows the voltammograms starting with a mixture of CuBr2 and AMME-N3S3sar in DMSO at ambient temperature. On the cathodic sweep two peaks were found associated with reduction of the mixture of five-coordinate [Cu(AMME-N3S3sar)Br]+ (higher potential) and six-coordinate [Cu(AMME-N3S3sar)]2+ (lower potential) each to their monovalent forms. It should be noted that these assignments are based on our previous studies with a crystalline (and crystallographically characterized) sample of [Cu(AMME-N3S3sar)Br]Br; the results are indistinguishable.8 The reverse sweep shows a single anodic peak at −620 mV, representing a common Cu(I) species bound in a tetradentate fashion on the face of the ligand. This illustrates that reduction of either six-coordinate Cu(II) (inside the cage) or five coordinate Cu(II) (partially coordinated) leads to the same tetradentate coordinated Cu(I) complex through rapid equilibration after reduction.![Cyclic voltammograms of [Cu(AMME-N3S3sar)]2+/+ in DMSO. Complex concentrations are 1 mM and all voltammograms are recorded at 100 mV s−1 scanning rate in 0.1 M Et4NClO4 and are referenced to Ag/AgNO3.](/image/article/2010/PY/b9py00315k/b9py00315k-f1.gif) | ||
| Fig. 1 Cyclic voltammograms of [Cu(AMME-N3S3sar)]2+/+ in DMSO. Complex concentrations are 1 mM and all voltammograms are recorded at 100 mV s−1 scanning rate in 0.1 M Et4NClO4 and are referenced to Ag/AgNO3. | ||
The redox potentials, ΔEp, of CuBr/AMME-N3S3sar complex in DMSO relative to the ferrocene/ferrocenium external standard were, in the case of a mixture of Cu(I/II)/AMME-N3S3sar, slightly higher than 60 mV but in line with what was previously observed for a wide range of copper/ligand complexes in MeCN.3,11 The non-aqueous solution resistance is most likely responsible for this feature as no compensation for iR drop was made. The average peak potentials (of the anodic and higher potential cathodic peaks) provide insight into the reducing power of the tetradentate coordinated Cu(I) species. A linear correlation between the Cu(II/I) redox potential and the equilibrium constant (where Keqapp = kact/kdeact, see eqn (1)) determined from the kinetics of polymerization was found from previous literature.3,11
The redox potential for the copper/AMME-N3S3sar complex was similar to the values found in the literature3,11 for other copper/ligand complexes (e.g. CuBr/Me6TREN in MeCN), suggesting that the AMME-N3S3sar complex should also have a similar activation capability of reducing the dormant initiating and polymeric halide species R-X or P-X, respectively, to their corresponding radicals. Voltammograms of the CuBr2/AMME-N3S3sar complex mixture formed in situ in the presence of increasing concentrations of a conventional ATRP initiator, ethyl 2-bromoisobutyrate (EBiB), allowed a qualitative investigation of the catalytic activity of the Cu(I) complex toward radical activation (Fig. 2 and Fig. S2 in the ESI†). As the potential was swept in the negative direction, the Cu(I) form is present exclusively within the diffusion layer at the electrode surface and should be able to react with EBiB to generate an incipient radical. If activation is fast, the Cu(I) complex is consumed in this chemical reaction before it can be reoxidised electrochemically and a small (or no) anodic peak will be observed on the reverse sweep (a so called EC mechanism). Furthermore, if this coupled chemical reaction is fast, there will be an amplification of the cathodic current as the system becomes catalytic and the Cu(II) product of the activation is regenerated at the electrode (a so called ECcat mechanism). The concentration of radicals will depend on the activation by the CuBr/AMME-N3S3sar complex. A greater activation rate coefficient will result in a higher radical concentration close to the electrode with a greater rate of bimolecular termination, and thus a greater build up of Cu(II) species.
![Cyclic voltammograms of [Cu(AMME-N3S3sar)]2+/+ (a) without EBiB and (b) with EBiB (100 mM) in DMSO. Complex concentrations are 1 mM and all voltammograms are recorded at 100 mV s−1 scanning rate in 0.1 M Et4NClO4 and are referenced to Ag/AgNO3.](/image/article/2010/PY/b9py00315k/b9py00315k-f2.gif) | ||
| Fig. 2 Cyclic voltammograms of [Cu(AMME-N3S3sar)]2+/+ (a) without EBiB and (b) with EBiB (100 mM) in DMSO. Complex concentrations are 1 mM and all voltammograms are recorded at 100 mV s−1 scanning rate in 0.1 M Et4NClO4 and are referenced to Ag/AgNO3. | ||
An ECcat mechanism was seen when Me6TREN was used as the Cu binding ligand (see Fig. S3 in the ESI†); the asymmetry of the voltammograms as well as the amplification of the cathodic current with increasing amounts of EBiB are apparent. The full mechanistic details of the Cu/Me6TREN system are not the focus of this paper and they will be published separately. It can be seen for the Cu/AMME-N3S3sar system that regardless of the concentration of EBiB (Fig. 2 and Fig. S2†) there is little change in the voltammograms, suggesting that activation by the CuBr/AMME-N3S3sar complex was much slower than for the CuBr/Me6TREN complex. This was in agreement with kinetic data found for the polymerization of styrene in toluene at 100 °C,5 and will be discussed below for the kinetic polymerizations carried out in DMSO at 25 °C
Kinetics of MA activated with CuBr/AMME-N3S3sar in DMSO at 25 °C
Polymerizations of MA were carried out varying the CuBr concentration (Fig. 3). The conversion-time data shows that the maximum conversion was 50% for the lowest CuBr concentration (curve a in Fig. 3A) and 75% conversion for the higher concentrations (curve c). The rate increased with the concentration of CuBr and was obviously non-first order (Fig. 3B). This suggests that the equilibrium between activating and deactivating species was not established in this system, leading to non-living radical behavior. The molecular weight distribution (MWD) data (Fig. 3C and D) confirms the non-living behavior with high number-average molecular weight (Mn) and polydispersity index (PDI) values. These polymerizations are more indicative of conventional free-radical polymerizations.![Kinetic data for the polymerization of methyl acrylate (3.67 M) in DMSO (9.36 M) at 25 °C with EBiB (1.83 × 10−2 M), CuBr and AMME-N3S3sar. (A) conversion versus time, (B) ln[M]0/[M] versus time (C) number-average molecular weight (Mn,SEC) versus conversion, and (D) polydispersity index versus conversion. The concentration ratios of reactants [MA] : [EBiB] : [CuBr] : [AMME-N3S3sar] are (a) 200 : 1 : 0.1 : 0.1, (b) 200 : 1 : 0.5 : 0.5, and (c) 200 : 1 : 1 : 1.](/image/article/2010/PY/b9py00315k/b9py00315k-f3.gif) | ||
| Fig. 3 Kinetic data for the polymerization of methyl acrylate (3.67 M) in DMSO (9.36 M) at 25 °C with EBiB (1.83 × 10−2 M), CuBr and AMME-N3S3sar. (A) conversion versus time, (B) ln[M]0/[M] versus time (C) number-average molecular weight (Mn,SEC) versus conversion, and (D) polydispersity index versus conversion. The concentration ratios of reactants [MA]:[EBiB]:[CuBr]:[AMME-N3S3sar] are (a) 200:1:0.1:0.1, (b) 200:1:0.5:0.5, and (c) 200:1:1:1. | ||
The external orders of the polymerization in [CuBr], [CuBr2] and [AMME-N3S3sar] for MA in DMSO were given in Fig. 4, and provides information on the effect of AMME-N3S3sar on deactivation and whether AMME-N3S3sar has an effect on the kinetics. The external order of the reaction in [CuBr] is 0.42, which was close to 0.5 for activation only by CuBr. However, the order for [AMME-N3S3sar] was −2.08 suggesting that the ligand retards the rate contrary to what is found for the styrene system (order = 0.47).5 This was seen more clearly in the rate data given in the ESI (Fig. S4).† For CuBr2, the order was very low and close to zero, supporting the postulate that deactivation using the cage ligand was negligible and in accord with what was found5 from the styrene data.
![Determination of the external order of reaction in [Cu(i)]0, [Cu(ii)]0 and [AMME-N3S3sar]0 for the CuBr/AMME-N3S3sar -catalyzed polymerization of methyl acrylate (MA) in DMSO at 25 °C initiated with ethyl 2-bromoisobutyrate (EBiB). [MA] : [EBiB] = 200 : 1; (A) ln kpappversus ln [CuBr]0, (B) ln kpappversus ln [CuBr2]0, (C) ln kpappversus ln [AMME-N3S3sar]0. The concentration of ligand was equal to the total concentration of copper (i.e. CuBr and CuBr2) used in each experiment except for ligand order reactions.](/image/article/2010/PY/b9py00315k/b9py00315k-f4.gif) | ||
| Fig. 4 Determination of the external order of reaction in [Cu(I)]0, [Cu(II)]0 and [AMME-N3S3sar]0 for the CuBr/AMME-N3S3sar -catalyzed polymerization of methyl acrylate (MA) in DMSO at 25 °C initiated with ethyl 2-bromoisobutyrate (EBiB). [MA]:[EBiB] = 200:1; (A) ln kpappversus ln [CuBr]0, (B) ln kpappversus ln [CuBr2]0, (C) ln kpappversus ln [AMME-N3S3sar]0. The concentration of ligand was equal to the total concentration of copper (i.e. CuBr and CuBr2) used in each experiment except for ligand order reactions. | ||
RAFT-mediated polymerization of MA initiated with EBiB and AMME-N3S3sar/CuBr in DMSO at 25 °C
The kinetic and uncontrolled MWD data of MA polymerizations above shows that the EBiB in the presence of CuBr/AMME-N3S3sar complex can be used as a unique redox couple to initiate polymerizations at low temperatures. The advantage of this redox couple is the little or no deactivation of the polymeric chain end with Cu(II), and can be used to initiate RAFT-mediated polymerizations at low temperatures with high chain-end fidelity with RAFT moieties. Fig. 5 shows the RAFT-mediated polymerization of MA with varying concentrations of a highly reactive CTA (MCEBTTC, a trithioester RAFT agent). The monomer concentration in all cases was kept constant, and therefore the polymerizations become slower with increased [CTA]o as a result of the lower [CuBr]o. The Mn for all polymerizations increased linearly with conversion and was close to theory for an ideal ‘living’ polymerization. The polydispersity index decreases with conversion down to below 1.1, showing that well-defined polymers were synthesized. Carrying out polymerizations at this low temperature significantly reduces side reactions that would otherwise produce short and long chain branches or crosslinks at temperatures normally used to polymerize MA.![Kinetic data for the polymerisation of methyl acrylate (3.67 M) in DMSO (9.36 M) at 25 °C with EBiB, MCEBTTC, CuBr and AMME-N3S3sar at various degrees of polymerization. (A) Conversion vs. time, (B) number-average molecular weight (Mn,SEC) vs. conversion, and (C) polydispersity index vs. conversion. The concentration ratios of reactants [MA] : [EBiB] : [CuBr] : [AMME-N3S3sar] : [MCEBTTC] are (a) 200 : 0.1 : 0.1 : 0.1 : 1, (b) 400 : 0.1 : 0.1 : 0.1 : 1, and (c) 800 : 0.1 : 0.1 : 0.1 : 1. (--- represents Mn,theory).](/image/article/2010/PY/b9py00315k/b9py00315k-f5.gif) | ||
| Fig. 5 Kinetic data for the polymerisation of methyl acrylate (3.67 M) in DMSO (9.36 M) at 25 °C with EBiB, MCEBTTC, CuBr and AMME-N3S3sar at various degrees of polymerization. (A) Conversion vs. time, (B) number-average molecular weight (Mn,SEC) vs. conversion, and (C) polydispersity index vs. conversion. The concentration ratios of reactants [MA]:[EBiB]:[CuBr]:[AMME-N3S3sar]:[MCEBTTC] are (a) 200:0.1:0.1:0.1:1, (b) 400:0.1:0.1:0.1:1, and (c) 800:0.1:0.1:0.1:1. (--- represents Mn,theory). | ||
Conclusion
The redox potential of the copper/N3S3 cage ligand complex was found to be similar to many of the nitrogen based ligands (e.g. Me6TREN in acteonitrile). Even though it has been found that the greater the redox potential the greater the activation capacity of the complex for nitrogen-based ligands, the Cu/AMME-N3S3sar system showed little or no activation of the alkyl halide. This low activation capability of the Cu/AMME-N3S3sar was confirmed from the very small change in the voltammograms when an alkyl halide was introduced into the electrochemical cell. The polymerization kinetic data also supported this conclusion, and further showed that deactivation was negligible. This led us to use this Cu/AMME-N3S3sar complex to initiate MA polymerizations at room temperature in the presence of a RAFT agent. The results showed that the molecular weight distribution was controlled and followed ideal ‘living’ radical behavior.Acknowledgements
M.J.M acknowledges financial support from the ARC Discovery grant (DP0987315).References
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101(9), 2921–2990 CrossRef CAS; M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101(12), 3689–3745 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- W. Tang, Y. Kwak, W. A. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, W. Tang, S. J. Brooks, K. Matyjaszewski, G. R. Weisman and E. H. Wong, J. Mol. Catal. A: Chem., 2006, 257, 132–140 CrossRef CAS.
- C. A. Bell, M. R. Whittaker, L. R. Gahan and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(1), 146–154 CrossRef CAS.
- (a) I. I. Creaser, J. M. Harrowfield, A. J. Herlt, A. M. Sargeson, J. Springborg, R. J. Geue and M. R. Snow, J. Am. Chem. Soc., 1977, 99, 3181–3182 CrossRef CAS; (b) R. J. Geue, T. W. Hambley, J. M. Harrowfield, A. M. Sargeson and M. R. Snow, J. Am. Chem. Soc., 1984, 106(19), 5478–5488 CrossRef CAS.
- (a) V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128(43), 14156–14165 CrossRef CAS; (b) V. Percec, A. V. Popov, E. Ramirez-Castillo, M. J. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124(18), 4940–4941 CrossRef CAS.
- C. A. Bell, P. V. Bernhardt, L. R. Gahan, M. Martinez, M. J. Monteiro, C. Rodriguez and C. A. Sharrad, Chem–Eur. J. Search PubMed , accepted.
- (a) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- (a) G. Johnston-Hall and M. J. Monteiro, Macromolecules, 2008, 41(3), 727–736 CrossRef CAS; (b) M. R. Whittaker and M. J. Monteiro, Langmuir, 2006, 22(23), 9746–9752 CrossRef CAS; (c) M. R. Whittaker, C. N. Urbani and M. J. Monteiro, Langmuir, 2007, 23(15), 7887–7890 CrossRef CAS.
- J. Qiu, K. Matyjaszewski, L. Thouin and C. Amatore, Macromol. Chem. Phys., 2000, 201, 1625–1631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanisms for AMME-N3S3sar mediated initiation and RAFT mediated polymerizations, redox potentials of CuBr2/AMME-N3S3sar, CuBr2/Me6TREN and ferrocene, cyclic voltammograms of [Cu(AMME-N3S3sar)]2+/+, cyclic voltammograms of [Cu(Me6TREN)]2+/+, kinetic data for the polymerization of methyl acrylate, and size exclusion chromatograms of MCEBTTC mediated polymerization of methyl acrylate. See DOI: 10.1039/b9py00315k |
| This journal is © The Royal Society of Chemistry 2010 |