Direct RAFT polymerization of an unprotected isocyanate-containing monomer and subsequent structopendant functionalization using “click”-type reactions†
Joel D.
Flores
a,
Junghwan
Shin
a,
Charles E.
Hoyle
ab and
Charles L.
McCormick
*ab
aDepartment of Polymer Science, The University of Southern Mississippi, 118 College Drive #10076, Hattiesburg, MS 39406, USA. E-mail: charles.mccormick@usm.edu; Fax: +1 601 266 5504; Tel: +1 601 266 4872
bDepartment of Chemistry and Biochemistry, The University of Southern Mississippi, 118 College Drive #10076, Hattiesburg, MS 39406, USA
First published on 22nd December 2009
Abstract
The controlled reversible addition–fragmentation chain transfer (RAFT) homopolymerization of an unprotected isocyanate-containing monomer, in this case 2-(acryloyloxy)ethylisocyanate (AOI), is reported. The polymerization conditions were optimized in terms of the choice of RAFT chain transfer agent (CTA), polymerization temperature and the reaction medium. Direct RAFT polymerization of AOI requires a neutral CTA, and relatively low reaction temperature to yield AOI homopolymers with low polydispersities. Efficient side-chain functionalization of poly(2-(acryloyloxy)ethylisocyanate) (PAOI) homopolymers was achieved via reaction with model amine, thiol and alcohol compounds yielding urea, thiourethane and urethane derivatives, respectively. Reactions with amines and thiols (in the presence of base) were rapid, quantitative and efficient. However, the reaction with alcohols catalyzed by dibutyltin dilaurate (DBTDL) was relatively slow but proceeded to completion. Selective reaction pathways for the addition of difunctional ethanolamine and mercaptoethanol were also investigated.
Introduction
Applications of (co)polymers in areas including nanomedicine, biotechnology, and electronics require macromolecules with controllable structures and compositions. Thus considerable effort has been devoted over the past two decades toward rational design of polymer architectures with specific physico-chemical characteristics.1,2 Recently, there has been increased activity in this arena, primarily due to the establishment of the free radical polymerization techniques that allow for the synthesis of (co)polymers that have controlled molecular weights and narrow molecular weight distributions.3 Additionally, extensive work has been dedicated toward preparing polymer “scaffolds” with reactive functionality capable of further, highly efficient reactions.4 The direct homopolymerization of monomers with structopendant reactive groups assures that each repeating unit possesses the desired functionality. However, functional groups such as free thiols and amines which can act as catalyst deactivators or chain transfer agents are only rarely tolerated by the usual polymerization methods. While protection–deprotection chemistries have been utilized, these approaches often require multiple synthetic and purification steps and are often less efficient. Given these limitations, an attractive route to post-polymerization derivatization is the combination of direct, controlled radical polymerization (CRP) yielding reactive pendant functionality followed by highly specific and efficient “click”-type reactions.5To date a number of chemical functional groups have been utilized to demonstrate the efficiency possible with post-polymerization modification.4–6 These facile chemical reactions generally require only a single step, often with no by-products, and proceed with high yield and selectivity. The most common include the substitution reactions of activated esters,7–15 addition to carbon–carbon multiple bonds (e.g. thiol–ene/yne, Michael-addition type reactions),14,16–21 disulfide exchange,11,21–24 addition to aldehydes/ketones13,25–27 and cycloaddition of unsaturated species (e.g. Diels–Alder transformations, Huisgen 1,3-dipolar cycloaddition).27–31 Controlled polymerization techniques such as atom-transfer radical polymerization (ATRP),8,22,25,29 ring-opening metathesis polymerization (ROMP),27 stable free radical polymerization (SFRP)32–35 and reversible addition–fragmentation chain transfer (RAFT)9,10,15,19,23,26,36–39 polymerization have been employed to synthesize well-defined (co)polymers containing a wide range of reactive functional groups that can be utilized in post-polymerization modification.
(Co)polymers synthesized by the RAFT technique may be designed to contain structopendant and/or structoterminal reactive functional groups that can be used in post-polymerization modifications.9,10,15,19,23,30,31,36,38–43 The trithiocarbonate or dithioester moieties in RAFT synthesized (co)polymers can be cleaved to thiols after polymerization and then utilized in subsequent reactions to prepare telechelic (co)polymers.11,17,20,21,24,44,45 Recently, there have been an increasing number of reports of transformations of RAFT-generated (co)polymers via thiol–ene and thiol–yne reactions.14,17,20,21,45 Conjugation of biomolecules through disulfide exchange after conversion of the CTA moieties into disulfides has been successfully demonstrated as well.21,43 The RAFT polymerization technique arguably has proven to be the most versatile of the CRP methods when it comes to the choice of monomer. The chemical groups that are tolerated by RAFT include fluorines, alcohols, amines, zwitterions, sulfonates, carboxylic acids and thiiranes among others.46 In addition, the RAFT method has also been employed in the polymerization of monomers containing activated ester,9,10,14,26 disulfide,19,23 acetal26 and epoxy groups.47
It is well-known that the reactions between isocyanates and active hydrogen containing functional groups (i.e. alcohols, thiols, and amines) are efficient and quantitative under certain reaction conditions. In contrast to reaction with alcohols, isocyanates react with amines and thiols (in the presence of a base catalyst) rapidly in an efficient often selective manner.5,48–54 As such these reactions exhibit many of the attributes described for “click” chemistry.5,55 Base-catalyzed reactions of thiols and isocyanates have recently been utilized in the chain end functionalization of thiol-terminated polymers prepared by RAFT.56 While the reactions of isocyanates and alcohols are not always fast and efficient, these can be quantitative depending on the reaction conditions. Reports of free radical polymerization of isocyanate-bearing monomers are limited.37,38,57–61 Barner et al. reported surface grafting of styrene and m-isopropenyl-α,α′-dimethylbenzylisocyanate onto a polypropylene solid support via γ-initiated RAFT polymerization.37 Statistical RAFT copolymers containing a limited number of isocyanate pendant groups for efficient cross-linking were very recently reported in a collaborative effort between the Hawker and Wooley groups.38 The copolymers, which were statistical copolymers of 2-(methacryloyloxy)ethylisocyanate (5–20%) and methyl methacrylate, were reacted with diamines in dilute solutions to afford intramolecularly cross-linked nanoparticles.
The objectives of the research reported in this manuscript were: (a) to develop a direct procedure for controlled radical polymerization of an isocyanate-containing monomer, in this case 2-(acryloyloxy)ethylisocyanate (AOI), without the need of protecting groups; (b) to determine the reaction parameters including reagent stoichiometry and solvent yielding the best control over molecular weight, polydispersity, and maintenance of pendant isocyanate functionality; and (c) to demonstrate facile side-chain reactions exhibiting the attributes of “click”-type chemistry utilizing model amine, alcohol, and thiol compounds. Herein, we demonstrate that by maintaining anhydrous conditions, moderate temperature and by judicious choice of stoichiometry, CTA, and solvent, reasonable control of reaction kinetics, molecular weight, and polydispersity can be attained in the RAFT polymerization of 2-(acryloyloxy)ethylisocyanate. To our knowledge, this is the first literature study of homopolymerization of an isocyanate-based monomer by a CRP technique. Additionally, we demonstrate facile post-polymerization modification with model amines, alcohols and thiols, as well as selective reaction pathways for the addition of difunctional ethanolamine and mercaptoethanol.
Experimental
Materials
2-(Acryloyloxy)ethylisocyanate (AOI) was obtained from Showa Denko K.K. (Tokyo, Japan), purified by distillation under reduced pressure and stored in a freezer until use. The RAFT chain transfer agents (2-(ethylsulfanylthiocarbonylsulfanyl)-2-methylpropionic acid (EMP),62,63 4-cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic acid (CEP),64 4-cyanopentanoic acid dithiobenzoate (CTP)65 and N,N-dimethyl-S-thiobenzoylthiopropionamide (TBP)66) were synthesized according to literature procedures. 2,2′-Azobis(2,4-dimethylvaleronitrile) (ADVN) was purchased from Wako Chemicals and recrystallized from ethanol. Other reagents and solvents were obtained from Sigma-Aldrich at the highest purity available and used as received unless otherwise stated.General procedure for RAFT polymerization
The AOI monomer (0.41 g, 2.9 mmol), TBP (8.6 mg, 0.034 mmol) and ADVN (1.7 mg, 0.0068 mmol) were dissolved in anhydrous dioxane (3.2 mL) in a septum-sealed vial. The solution was purged with nitrogen gas for an hour at room temperature. After purging, the vial was placed in a preheated reactor station (STEM Electrothermal RS6000) and polymerization reaction was allowed to proceed for a fixed time. For polymerization kinetics, the reaction was carried out in a rubber septum-sealed round bottom flask heated in an oil bath. Aliquots were taken from the polymerization mixture using degassed syringes at predetermined time intervals. The aliquots were cooled to room temperature and briefly exposed to the atmosphere to quench the reaction. The polymerization mixture was reacted with methanol for size exclusion chromatography (SEC) analysis. To ensure that all isocyanate was completely reacted, the absorbance at 2280 cm−1 was monitored using FT-IR spectroscopy.Chain extension
AOI monomer (0.34 g, 2.4 mmol), TBP (8.6 mg, 0.034 mmol) and ADVN (1.7 mg, 0.0069 mmol) were dissolved in anhydrous dioxane (2.5 mL). The solution was purged with N2 gas for 45 min. Polymerization was carried out in a reactor station (STEM Electrothermal RS6000) preheated at 50 °C. After 12 h (conversion ∼40%), the reaction was quenched by cooling the mixture in an ice bath followed by brief exposure to air. Aliquots (1 mL) were taken for analysis. Into the remaining mixture, AOI (0.15 g, 1.1 mmol), ADVN (0.75 mg, 0.003 mmol) and anhydrous dioxane (1 mL) were added. The mixture was purged for 45 min with N2 gas and allowed to react at 50 °C for an additional 8 h. After quenching the reaction, the mixture was reacted with methanol for GPC analysis.Side-chain functionalization
The functional agent (1.5 equivalents of alcohol with 0.1 wt% (1.6 mM) of dibutyltin dilaurate (DBTDL), 1 equivalent of amine or 1 equivalent of thiol with 0.5 wt% (50 mM) triethylamine (TEA)) was added to the polymerization mixture containing 0.83 M of isocyanate (NCO) groups. The mixture was allowed to react overnight and the completion of the reaction of the NCO groups was monitored by FT-IR spectroscopy. After the reaction, the mixtures were subjected to SEC to determine polymer molecular weight and polydispersity index. The polymerization mixture became decolorized, when amine and thiol agents were used, indicating the reaction of the CTA moiety as well.Determination of monomer conversion
The monomer conversion was determined using either UV-visible or 1H NMR spectroscopy. For UV-visible spectroscopy, 25 µL of polymerization mixture were diluted with 2.5 mL of tetrahydrofuran (THF). Absorbances at 250 nm due to the vinyl groups normalized to the absorbance of CTA at 303 nm were used to calculate the conversion of the AOI monomer. In the polymerization using solvents other than dioxane, UV absorbance due to the solvent significantly overlapped with that of the vinyl groups. In this case, 1H NMR spectroscopy was used to calculate monomer conversion. Briefly, 100 µL of the polymerization mixture were added into 500 µL of acetone-d6 and the decreases in the normalized peak areas due to the vinyl protons were correlated to the conversion of the monomer.Characterization
Monomer conversion was determined using a Varian 500 Scan UV-Vis-NIR spectrophotometer and a Varian INOVA 300 MHz NMR spectrometer. FT-IR spectra were recorded using a modified Bruker 88 spectrometer. Samples were placed between two sodium chloride salt plates. Each spectrum was collected over 32 scans. The data were analyzed with the Bruker OPUS/IR version 4.0 software. Molecular weights and polydispersities of poly(2-(acryloyloxy)ethylisocyanate) (PAOI) after reaction with specified functional reagents were determined by size exclusion chromatography (SEC) using 0.02 M lithium bromide in DMF as eluent at a flow rate of 1.0 mL min−1 at 35 °C. This instrument was equipped with Viscotek I-Series Mixed Bed low-Mw (exclusion limit > 20K PS) and mid-Mw (exclusion limit > 200K PS) columns, Viscotek-TDA (302 nm RI, viscosity, 7 mW 90° and 7° true low angle light scattering detectors (670 nm)). The dn/dc of PAOI capped with methanol (0.059 mL g−1) in the above eluent was determined separately using a Bausch and Lomb refractometer.Results and discussion
RAFT polymerization
Our initial screening experiments in preparing homopolymers of 2-(acryloyloxy)ethylisocyanate (AOI) for further reaction, as illustrated in Scheme 1, involved RAFT polymerization utilizing a number of dithioester and trithiocarbonate CTAs at temperatures in the 40 to 60 °C range. Trithiocarbonates, EMP and CEP, as well as the dithioester, CTP, yielded polymers with broad molecular weight distributions while TBP was successful in yielding homopolymers with narrow molecular weight distributions. Examples of SEC traces from the polymerization of AOI using these four CTAs are shown in Fig. 1. These four chain transfer agents have been previously utilized in the RAFT polymerization of (meth)acrylamides, (meth)acrylates and styrenics.46,67 However, it is apparent that the carboxylic acid functionality present in the first three interferes with controlled polymerization. Carboxylic acid groups catalyze or directly react with isocyanates,68–75 the latter usually occurring at temperatures higher than the 50–60 °C range utilized in our initial experiments. Nonetheless, possible coupling of a few CTA carboxylic acid groups with pendant isocyanate groups could easily explain the broadening of the molecular weight distributions. Acceptable control was attained utilizing TBP; therefore, it was chosen in further experiments for optimization of polymerization conditions. | ||
| Scheme 1 Direct RAFT polymerization of 2-(acryloyloxy)ethylisocyanate (AOI) utilizing TBP as the chain transfer agent (CTA) and subsequent functionalization through reaction with alcohols, amines and thiols. | ||
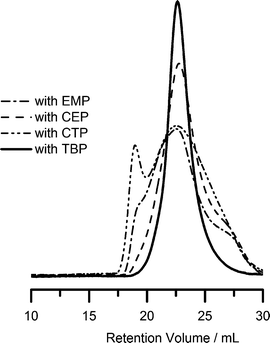 | ||
| Fig. 1 SEC traces from the RAFT polymerization of AOI using EMP, CEP, CTP and TBP as chain transfer agents in dioxane at 50 °C (monomer–CTA–initiator = 67 : 1 : 0.2). The polymers were reacted with methanol prior to SEC analysis. | ||
Table 1 shows data from the RAFT polymerization of AOI under selected reaction conditions. The possibility of side reactions might be expected at elevated temperatures, and hence, polymerizations were conducted at temperatures ranging from 40–60 °C. For a fixed amount of initiator, increasing polymerization temperature also increases the rate of radical generation which, in effect, favors radical–radical coupling reactions that can eventually alter the control of the RAFT process. To have good control in RAFT polymerization, a delicate balance between the rates of radical generation, monomer addition (i.e. propagation) and termination reactions must be maintained.46,76 In addition, there must be a constant radical flux to sustain the polymerization to completion.77 For fixed CTA to initiator ratios, polydispersities of PAOI increase with temperature (Table 1, entries 1 and 3) and for a fixed temperature, polydispersities increase with decreasing CTA to initiator ratios (i.e. increasing the amount of initiator) (Table 1, entries 3–7). Very low monomer conversion was observed with polymerization at 40 °C (Table 1, entry 11) which we attribute to the low rate of initiator decomposition at 40 °C. These results indicate that the polymerization temperature has an effect on the control of the RAFT homopolymerization of AOI monomer using TBP as the CTA and the best control was achieved for polymerizations at 50 °C.
| Entry | Temperature/°C | Monomer–CTA–initiator | Solvent | Time/h | Conversion (%) | M n (theo.)a | M n (exptl)b | PDI |
|---|---|---|---|---|---|---|---|---|
| a These values were calculated using methyl carbamate side-chain. b These values were determined from SEC after reacting the polymerization mixture with methanol. | ||||||||
| 1 | 60 | 67 : 1 : 0.2 | Dioxane | 12 | 64 | 7671 | 9600 | 1.72 |
| 2 | 60 | 67 : 1 : 1.0 | Dioxane | 12 | 70 | 8367 | 8800 | 2.26 |
| 3 | 50 | 74 : 1 : 0.2 | Dioxane | 13 | 40 | 5341 | 9300 | 1.13 |
| 4 | 50 | 67 : 1 : 0.3 | Dioxane | 12 | 33 | 4078 | 9200 | 1.30 |
| 5 | 50 | 276 : 1 : 0.2 | Dioxane | 15 | 59 | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 424 424 |
36000 |
1.31 |
| 6 | 50 | 276 : 1 : 0.3 | Dioxane | 15 | 58 | 27947 |
34600 |
1.41 |
| 7 | 50 | 276 : 1 : 1.0 | Dioxane | 15 | 72 | 34632 |
33800 |
1.63 |
| 8 | 50 | 79 : 1 : 0.2 | NMP | 13 | 45 | 6435 | 7300 | 1.35 |
| 9 | 50 | 79 : 1 : 0.2 | DMF | 13 | 45 | 6435 | 10200 |
1.31 |
| 10 | 50 | 82 : 1 : 0.2 | Acetonitrile | 12 | 20 | 3102 | 7900 | 1.09 |
| 11 | 40 | 67 : 1 : 0.3 | Dioxane | 12 | <3 | — | — | — |
RAFT polymerization can be conducted in a wide range of reaction media including protic and aprotic organic solvents, water, and even in less common systems such as ionic liquids and supercritical carbon dioxide.46 Owing to the highly reactive pendant isocyanate groups, RAFT polymerizations in selected solvents were also performed. The lowest polydispersities were obtained with acetonitrile and dioxane; however, very low conversion was attained with the former. The more polar solvents (NMP and DMF) yielded homopolymers with larger polydispersities.
Kinetics of RAFT polymerization
Polymerization kinetic experiments were conducted to determine if the control of RAFT polymerization varies with monomer conversion. The kinetic profile of AOI polymerization in dioxane at 50 °C was investigated using TBP and ADVN as CTA and free radical initiator, respectively. As can be ascertained from the resulting chromatograms shown in Fig. 2A, the evolution of molecular weight with conversion was well-controlled. In addition, the respective chromatographic traces from successive aliquots in the polymerization mixture at predetermined time intervals shifted well toward lower elution volume with conversion (or polymerization time). The corresponding kinetic plot is shown in Fig. 2B. The polymerization at 50 °C is slow, reaching only about 35% monomer conversion after 15 h. The rate of polymerization decreases slightly at higher conversion as indicated by the changes in slope (kp[P˙]) of the kinetic plot. This behavior is often observed for RAFT polymerization mediated by dithioester-based chain transfer agents which can require relatively long polymerization times.78–80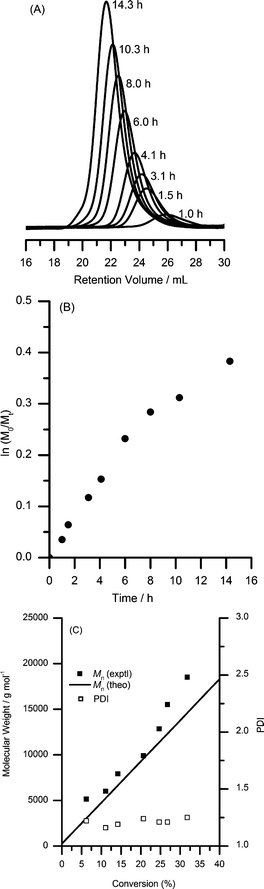 | ||
| Fig. 2 (A) SEC chromatograms (RI detector), (B) kinetic plots and (C) molecular weight and PDI as a function of monomer conversion for AOI RAFT homopolymerization using TBP in dioxane at 50 °C (3 : 1 CTA to initiator ratio). Aliquots were reacted with methanol prior SEC analysis. | ||
The molecular weights increase linearly with conversion; however, in the later stages of the reaction, the experimental molecular weights deviate from the theoretical values (refer to Fig. 2C). With the polymerization at 50 °C, a positive deviation in the molecular weights was observed at longer polymerization times. This molecular weight overshoot is likely due to CTA loss and/or irreversible termination reactions of the intermediate radicals.78,79 For the synthesis of higher molecular weight polymers (requiring higher [AOI]/[CTA]) and with longer polymerization times, broader PDI values were observed (see Table 1, entries 5–7). Based on these results, the polymerization of AOI monomer using TBP as the RAFT chain transfer agent appears to be best controlled at 50 °C using dioxane as the reaction medium and allowing the reaction to proceed to moderate conversions.
Chain extension of PAOI macroCTA
In chain extension or block copolymer formation utilizing the RAFT technique, it is important to choose reaction conversions at which primary chain coupling and thus loss of thiocarbonylthio groups are minimal. In our experiments, we prepared a PAOI macroCTA (Mn = 6600 and PDI = 1.17) by quenching the reaction at ∼40% monomer conversion. Without purification, the reaction mixture was charged with more monomer, free radical initiator and solvent, and the polymerization was restarted to produce chain extended polymer. The chromatograms before and after chain extension are shown in Fig. 3. The chain extended polymer has a slightly higher polydispersity. The absence of a low molecular weight shoulder or significant tailing indicates that all the chains contained the CTA moiety and had been successfully chain extended (self-blocking).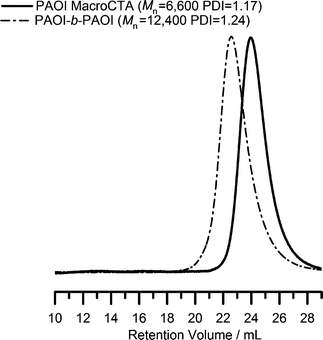 | ||
| Fig. 3 SEC chromatograms from the chain extension (self-blocking) of poly(2-(acryloyloxy)ethylisocyanate) (PAOI). | ||
Side-chain functionalization
The pendant isocyanate groups can be reacted with compounds containing active hydrogens such as alcohols, amines and thiols under appropriate conditions making PAOI a versatile polymer backbone for side-chain conjugation and functionalization. Urea formation from the reaction of amines and isocyanates is one of the “click” reactions previously described in literature.5 Recently, two reports have demonstrated that the base-catalyzed reactions of isocyanates with thiols also exhibit the characteristics of “click” reactions and suggested that they can be considered as such.55,56 With this in mind, we investigated the efficiency and selectivity of such reactions by performing model studies using cyclohexylisocyanate (CHI) and small molecule amines, thiols and alcohols. Details can be found in the accompanying ESI† of this manuscript. Briefly, we found that the reactions of amines and thiols (in the presence of a basic catalyst) with isocyanate are fast and efficient. By contrast, the reactions of isocyanates and alcohols are slower but can proceed to completion with dibutyltin dilaurate (DBTDL) as a catalyst. However, to our great surprise, we also observed the formation of thiourethane groups (especially in the absence of or at low levels of catalyst) when the difunctional mercaptoethanol was used instead of ethanol.We then studied the reactions of the structopendant isocyanate group of PAOI homopolymers with agents containing hydroxyl, amine and thiol groups to demonstrate utility in the synthesis of functional polymers. Without further purification, the polymerization mixture containing PAOI homopolymer (0.83 M NCO) was reacted with hexanol, hexylamine and hexanethiol. DBTDL (0.1 wt%, 1.6 mM) was added as catalyst for the reaction with alcohol. In the reaction with hexanethiol, triethylamine (TEA) (0.5 wt%, 50 mM) was utilized as a base. The base deprotonates the thiol generating a thiolate anion which reacts rapidly with the isocyanate. The reaction of isocyanate with primary or secondary amines does not require catalysis. The completeness of the reaction was followed by FT-IR spectroscopy. Fig. 4 shows the disappearance of the isocyanate peak at 2280 cm−1 after the reactions. The SEC traces for PAOI reaction products with hexanol, hexylamine and hexanethiol are shown in Fig. 6. The SEC traces are unimodal and the corresponding PDI values remain low. Additionally, the formation of respective urea (NCO + amine), urethane (NCO + OH) and thiourethane (NCO + SH) bonds was confirmed by inspection of the carbonyl region of the FT-IR spectra of the products (see Fig. 5A). The carbonyl absorbances for urea, urethane and thiourethane are centered at 1670 cm−1, 1724 cm−1 and 1680 cm−1, respectively. These positions are in accordance with a previous report in literature.51 The urethane peak at 1724 cm−1 overlaps the ester peak of the AOI repeating unit.
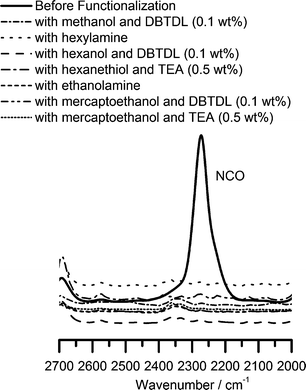 | ||
| Fig. 4 FT-IR spectra showing the disappearance of the NCO absorbance after the reaction of PAOI with alcohols, amines or thiols. | ||
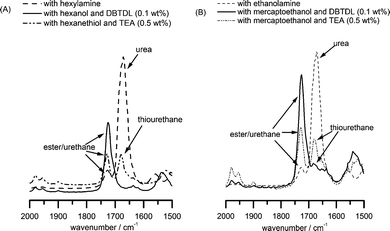 | ||
| Fig. 5 FT-IR spectra showing the carbonyl region of PAOI after reaction with (A) monofunctional and (B) difunctional agents. | ||
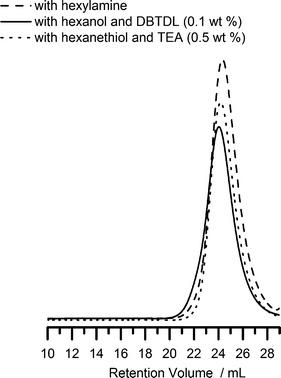 | ||
| Fig. 6 SEC chromatograms of PAOI after reaction with hexylamine (Mn = 12300 and PDI = 1.16), hexanol with 0.1 wt% DBTDL (Mn = 9100 and PDI = 1.23), and hexanethiol with 0.5 wt% TEA (Mn = 12200 and PDI = 1.26). | ||
As described above, the reactions of isocyanate with amines and thiols (in the presence of a base catalyst) are fast and efficient while those of alcohols are slower but can proceed quantitatively under certain reaction conditions. Herein, the selectivity of the reaction between isocyanate/amine and isocyanate/alcohol and between isocyanate/thiol and isocyanate/alcohol is illustrated using ethanolamine and mercaptoethanol, respectively (Scheme 2). The reaction of the amine functionality and isocyanate (NCO : NH2 : OH = 1 : 1 : 1) is faster than that of alcohol group as demonstrated in the case of ethanolamine and the reaction does not require a catalyst. The isocyanate/alcohol reaction could not be made selective over the isocyanate/amine reaction since primary or secondary amines are much more reactive than alcohols. With the stoichiometric amounts of the reacting groups (NCO : SH : OH = 1 : 1 : 1), the base-catalyzed reaction of thiol and isocyanate is extremely fast such that the NCO group is exclusively converted into a thiourethane. By contrast to the reaction with hexanol discussed previously, the isocyanate/alcohol reaction using mercaptoethanol (catalyzed by DBTDL) resulted in competitive formation of urethane and thiourethane linkages. The thiourethane formation, however, could be minimized by increasing the concentration of DBTDL catalyst. From previous literature reports and from our model studies, thiols are relatively unreactive toward isocyanates under neutral conditions and even in the presence of DBTDL catalyst.81,82 This is not surprising as free thiols are less nucleophilic compared to hydroxyl groups. Once deprotonated, however, the thiolate anion becomes an excellent nucleophile and reacts immediately with isocyanate (refer to the ESI† for experimental results and discussion of the model reactions).
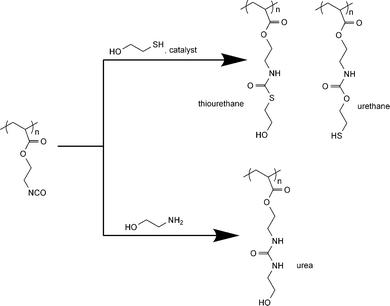 | ||
| Scheme 2 Side-chain functionalization of poly(2-(acryloyloxy)ethylisocyanate) (PAOI) with mercaptoethanol and ethanolamine. | ||
The FT-IR spectra of the carbonyl region of the PAOI after reaction with ethanolamine and mercaptoethanol are illustrated in Fig. 5B. Successful side-chain functionalization was observed based on the characteristic bands for urea, urethane and thiourethane groups. However, the presence of small shoulder peak due to the thiourethane in the reaction of PAOI and mercaptoethanol catalyzed by DBTDL was also observed. The corresponding SEC chromatograms of the PAOI homopolymers after the reaction with ethanolamine and mercaptoethanol are shown in Fig. 7. The resulting homopolymer from the reaction of PAOI with ethanolamine exhibited low polydispersity and was soluble in water. With mercaptoethanol and TEA, the polydispersity was also low but the functionalized homopolymer was not completely soluble in water. A very broad distribution, however, was obtained from the reaction of PAOI and mercaptoethanol with added DBTDL. Initially, we speculated that disulfide bridges formed from the oxidation of the pendant thiol groups caused this broad polydispersity. Attempts to break the linkages by adding reducing agents including dithiothreitol were unsuccessful. From the model reactions, we postulate that this increase in polydispersity is likely due to the reaction of both the alcohol and the thiol groups in mercaptoethanol when DBTDL is used as the catalyst. In our initial experiments, we utilized only 0.01 wt% DBTDL and a functionalized polymer with a PDI of 1.58 was obtained (see Fig. 7A). We increased the amount of DBTDL catalyst to 0.1 wt% and a better polydispersity was achieved, however, it was still higher as compared to the homopolymers functionalized with ethanolamine and mercaptoethanol utilizing TEA as the catalyst (see Fig. 7B). While thiourethane formation in reactions using small molecules was minimized or possibly prevented with 0.1 wt% DBTDL catalyst, this was not completely avoided in the reaction with PAOI homopolymers. Lastly, it should be noted that gelation was not observed in the reaction of PAOI and mercaptoethanol utilizing this catalyst.
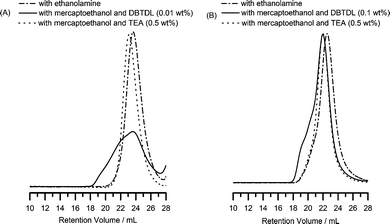 | ||
| Fig. 7 SEC chromatograms of PAOI after reaction with ethanolamine and mercaptoethanol. In the reaction with mercaptoethanol, (A) 0.01 wt% and (B) 0.1 wt% of DBTDL catalyst were used. (A) and (B) are from different polymerizations and have different degrees of polymerization. Their reactions with ethanolamine and mercaptoethanol are shown for comparison. The following are the corresponding Mw and PDI values for (A) with ethanolamine (Mn = 12600 and PDI = 1.26), with mercaptoethanol and 0.01 wt% DBTDL (Mn = 47800 and PDI = 1.58), and with mercaptoethanol and 0.5 wt% TEA (Mn = 14900 and PDI = 1.20); and for (B) with ethanolamine (Mn = 20500 and PDI = 1.24), with mercaptoethanol and 0.1 wt% DBTDL (Mn = 28900 and PDI = 1.46), and with mercaptoethanol and 0.5 wt% TEA (Mn = 23200 and PDI = 1.23). | ||
Conclusions
Successful direct RAFT homopolymerization of 2-(acryloyloxy)ethylisocyanate (AOI) has been accomplished. The polymerization conditions were optimized utilizing TBP as the chain transfer agent by varying stoichiometry of monomer/CTA and varying the polymerization temperature and the solvent. Direct RAFT polymerization of AOI requires a neutral CTA and relatively low reaction temperature to yield AOI homopolymers with good polydispersities. Efficient side-chain functionalization of AOI homopolymers can be achieved through reactions with amines, thiols and alcohols resulting in urea, thiourethane and urethane linkages, respectively. Reactions with amines and thiols (in the presence of base) are fast, quantitative and efficient. However, the reaction with alcohols utilizing dibutyltin dilaurate (DBTDL) catalyst is relatively slow but can proceed to completion. Selective reaction pathways for the side-chain derivatization of PAOI homopolymers using difunctional ethanolamine and mercaptoethanol were identified. Work is now underway in our laboratories to extend these model studies of structopendant isocyanate “click”-type reactions of RAFT-based polymers to thiol, hydroxyl and amine terminated synthetic and biological (macro)molecules.Acknowledgements
We dedicate this paper to our beloved colleague, Dr Charles Hoyle, a long time faculty member in the Departments of Polymer Science and Chemistry and Biochemistry, a dedicated teacher and mentor of student researchers. We gratefully acknowledge the financial support provided by the MRSEC program of the National Science Foundation (DMR-0213883).Notes and references
- J. F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. F. Lutz and H. Schlaad, Polymer, 2008, 49, 817–824 CrossRef CAS.
- E. Smith, J. Bai, C. Oxenford, J. Yang, R. Somayaji and H. Uludag, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3989–4000 CrossRef CAS.
- P. Theato, J. U. Kim and J. C. Lee, Macromolecules, 2004, 37, 5475–5478 CrossRef CAS.
- A. Favier, F. D'Agosto, M. T. Charreyre and C. Pichot, Polymer, 2004, 45, 7821–7830 CrossRef CAS.
- J. J. Vosloo, M. P. Tonge, C. M. Fellows, F. D'Agosto, R. D. Sanderson and R. G. Gilbert, Macromolecules, 2004, 37, 2371–2382 CrossRef CAS.
- A. W. York, C. W. Scales, F. Q. Huang and C. L. McCormick, Biomacromolecules, 2007, 8, 2337–2341 CrossRef CAS.
- S. Y. Wong and D. Putnam, Bioconjugate Chem., 2007, 18, 970–982 CrossRef CAS.
- D. Samanta, S. McRae, B. Cooper, Y. Hu, T. Emrick, J. Pratt and S. A. Charles, Biomacromolecules, 2008, 9, 2891–2897 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889–1895 CrossRef CAS.
- A. W. York, Y. L. Zhang, A. C. Holley, Y. L. Guo, F. Q. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936–943 CrossRef CAS.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Polymer, 2005, 46, 12057–12064 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- Z. Hordyjewicz-Baran, L. C. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef CAS.
- Z. F. Jia, L. J. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106–3113 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
- L. J. Wong, C. Boyer, Z. F. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS.
- C. Boyer, J. Q. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830–847 CrossRef CAS.
- R. C. Li, R. M. Broyer and H. D. Maynard, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5004–5013 CrossRef CAS.
- J. Y. Hwang, R. C. Li and H. D. Maynard, J. Controlled Release, 2007, 122, 279–286 CrossRef CAS.
- S. K. Yang and M. Weck, Macromolecules, 2008, 41, 346–351 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180–4183 CrossRef CAS.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- R. G. Jones, S. Yoon and Y. Nagasaki, Polymer, 1999, 40, 2411–2418 CrossRef CAS.
- D. C. Tully, M. J. Roberts, B. H. Geierstanger and R. B. Grubbs, Macromolecules, 2003, 36, 4302–4308 CrossRef CAS.
- M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS.
- X. W. Xu, A. E. Smith, S. E. Kirkland and C. L. McCormick, Macromolecules, 2008, 41, 8429–8435 CrossRef CAS.
- L. Barner, S. Perera, S. Sandanayake and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 857–864 CrossRef CAS.
- J. B. Beck, K. L. Killops, T. Kang, K. Sivanandan, A. Bayles, M. E. Mackay, K. L. Wooley and C. J. Hawker, Macromolecules, 2009, 42, 5629–5635 CrossRef CAS.
- Y. T. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- X. P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474–481 CrossRef CAS.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- X. P. Qiu and F. M. Winnik, Macromolecules, 2007, 40, 872–878 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- J. Zhu, D. Zhou, X. Zhu and G. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2558–2565 CrossRef CAS.
- E. Dyer, H. A. Taylor, S. J. Mason and J. Samson, J. Am. Chem. Soc., 1949, 71, 4106–4109 CrossRef CAS.
- J. W. Baker and J. Gaunt, J. Chem. Soc., 1949, 9–18 RSC.
- E. Dyer and J. F. Glenn, J. Am. Chem. Soc., 1957, 79, 366–369 CrossRef CAS.
- J. F. Smith and E. C. Friedrich, J. Am. Chem. Soc., 1959, 81, 161–163 CrossRef CAS.
- Y. Iwakura and H. Okada, Can. J. Chem., 1960, 38, 2418–2424 CAS.
- E. Dyer, J. F. Glenn and E. G. Lendrat, J. Org. Chem., 1961, 26, 2919–2925 CrossRef CAS.
- E. Klemm and C. Stockl, Makromol. Chem., 1991, 192, 153–158 CrossRef CAS.
- J. Shin, H. Matsushima, J. W. Chan and C. E. Hoyle, Macromolecules, 2009, 42, 3294–3301 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. Hoyle and A. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- D. Beyer, W. Paulus, M. Seitz, G. Maxein, H. Ringsdorf and M. Eich, Thin Solid Films, 1995, 271, 73–83 CrossRef CAS.
- M. Dörr, R. Zentel, R. Dietrich, K. Meerholz, C. Bräuchle, J. Wichern, S. Zippel and P. Boldt, Macromolecules, 1998, 31, 1454–1465 CrossRef.
- H. S. Wu, M. H. Chuang and J. W. Hwang, J. Appl. Polym. Sci., 1999, 73, 2763–2770 CrossRef CAS.
- L. Barner, C. Barner-Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1064–1074 CrossRef CAS.
- J. Barruet, C. Gaillet and J. Penelle, Macromol. Rapid Commun., 2007, 28, 2007–2011 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123–4132 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- W. R. Sorenson, J. Org. Chem., 1959, 24, 978–980 CrossRef CAS.
- S. Motoki, T. Saito and H. Kagami, Bull. Chem. Soc. Jpn., 1974, 47, 775–776 CrossRef CAS.
- P. A. Berlin, M. A. Levina, Y. N. Chirkov, R. P. Tiger and S. G. Entelis, Kinet. Catal., 1993, 34, 568–572.
- H. Xiao, H. X. Xiao, K. C. Frisch and N. Malwitz, High Perform. Polym., 1994, 6, 235–239 CrossRef CAS.
- K. Schwetlick and R. Noack, J. Chem. Soc., Perkin Trans. 2, 1995,(2), 395–402 RSC.
- A. C. Draye and J. J. Tondeur, J. Mol. Catal. A: Chem., 1999, 138, 135–144 CrossRef CAS.
- A. C. Draye and J. J. Tondeur, J. Mol. Catal. A: Chem., 1999, 140, 31–40 CrossRef CAS.
- C. Gurtler and K. Danielmeier, Tetrahedron Lett., 2004, 45, 2515–2521 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, WeinHeim, Germany, 2008 Search PubMed.
- D. B. Thomas, A. J. Convertine, L. J. Myrick, C. W. Scales, A. E. Smith, A. B. Lowe, Y. A. Vasilieva, N. Ayres and C. L. McCormick, Macromolecules, 2004, 37, 8941–8950 CrossRef CAS.
- Y. Kwak, A. Goto, Y. Tsujii, Y. Murata, K. Komatsu and T. Fukuda, Macromolecules, 2002, 35, 3026–3029 CrossRef CAS.
- F. M. Calitz, J. B. McLeary, J. M. McKenzie, M. P. Tonge, B. Klumperman and R. D. Sanderson, Macromolecules, 2003, 36, 9687–9690 CrossRef CAS.
- M. L. Coote and L. Radom, J. Am. Chem. Soc., 2003, 125, 1490–1491 CrossRef CAS.
- R. P. Houghton and A. W. Mulvaney, J. Organomet. Chem., 1996, 518, 21–27 CrossRef CAS.
- K. K. Majumdar, A. Kundu, I. Das and S. Roy, Appl. Organomet. Chem., 2000, 14, 79–85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results (FT-IR and 13C NMR spectra) and discussions of model reactions utilizing cyclohexylisocyanate and small molecule thiol, amine and alcohol compounds. See DOI: 10.1039/b9py00294d |
| This journal is © The Royal Society of Chemistry 2010 |