Synthesis of well-defined primary amine-based homopolymers and block copolymers and their Michael addition reactions with acrylates and acrylamides
Elizabeth S.
Read
,
Kate L.
Thompson
and
Steven P.
Armes
*
Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, UK S3 7HF. E-mail: s.p.armes@sheffield.ac.uk
First published on 8th January 2010
Abstract
A series of well-defined primary amine-based AB diblock copolymers were synthesised via atom transfer radical polymerisation (ATRP) using 2-aminoethyl methacrylate hydrochloride (AMA), with the other block comprising the following comonomers: 2-(diisopropylamino)ethyl methacrylate (DPA), 2-hydroxypropyl methacrylate (HPMA) and 2-(methacryloyloxy)ethyl phosphorylcholine (MPC). These copolymers were prepared with reasonably narrow polydispersities (Mw/Mn ≈ 1.1–1.4) in either 80 : 20 or 95 : 5 2-propanol–water mixtures at 50 °C using a 2-(N-morpholino)ethyl isobutyryl bromide initiator. The chain extension efficiency of PAMA was also investigated using a further charge of AMA. Unfortunately, such ‘self-blocking’ was problematic due to both catalyst deactivation and termination of the living chain ends. Nevertheless, low polydispersity all-methacrylic diblock copolymers were obtained in high yields via sequential monomer addition, provided that AMA was used as the second monomer in such syntheses. PAMA49 homopolymer and selected PAMA-based copolymers were reacted with various acrylates and acrylamides in aqueous solution at pH 9, with mean degrees of functionalisation being determined by 1H NMR spectroscopy. This facile Michael addition chemistry provides access to a library of novel functional water-soluble homopolymers and diblock copolymers.
Introduction
Over the last decade or so, the emergence of living radical polymerisation techniques such as atom transfer radical polymerisation (ATRP)1–4 and reversible addition–fragmentation transfer (RAFT)5–9 polymerisation has resulted in a paradigm shift in the synthesis of functional polymers of controlled architectures. Thousands of papers have described facile syntheses of relatively well-defined vinyl polymers, including many examples based on water-soluble/hydrophilic monomers.10–16 Nevertheless, there are still relatively few studies that have exploited ATRP or RAFT for the synthesis of controlled-structure primary amine-based vinyl polymers.17–252-Aminoethyl methacrylate hydrochloride (AMA) is a commercially available primary amine-based methacrylic monomer. However, it is both relatively expensive and only available as a technical grade of ∼90% purity. Fortunately, AMA is readily prepared by reacting methacryloyl chloride with molten 2-aminoethanol hydrochloride at 95 °C.26 It has been previously used as a comonomer for the preparation of cationic latexes27,28 and has also been oligomerised using catalytic chain transfer polymerisation to afford polydisperse AMA-based macromonomers.29 Narain et al. reported that AMA can be reacted with lactobionolactone in order to prepare a new cyclic sugar methacrylate.21,26
Recently we demonstrated the successful polymerisation of AMA directly in its hydrochloride form via both RAFT and ATRP.20 Linear PAMA homopolymers and AB diblock copolymers [where A = AMA and B = 2-(diisopropylamino)ethyl methacrylate [DPA]] were prepared with narrow polydispersities (Mw/Mn ≈ 1.2–1.3) at 70 °C using cumyl dithiobenzoate (CDB) as a RAFT chain transfer agent. In addition, ATRP was used to prepare a range of PAMA homopolymers in alcohol–water mixtures and also several examples of well-defined PAMA-based diblock copolymers using poly(ethylene oxide) macro-initiators. In its neutral form, AMA monomer is rapidly converted into 2-hydroxyethyl methacrylamide. However, this internal rearrangement can be completely suppressed by ensuring that the AMA monomer is maintained in its hydrochloride salt form. PAMA is much more stable than AMA monomer, but slow degradation occurs via at least two pathways in alkaline media.18
Michael addition chemistry benefits from mild reaction conditions, no by-products, high functional group tolerance, high conversions and favourable reaction rates.30 Typical ‘Michael donors’ include amines, enolates, thiols and phosphines. These can react with a wide range of ‘Michael acceptors’, including acrylates, acrylamides, acrylonitriles, maleimides, methacrylates, cyanoacrylates and vinyl sulfones.30 Michael addition has been exploited in step growth polymerisation to prepare biocompatible poly(amido amines),31–33 poly(amino esters)34 and poly(aspartamides).35 There are also numerous literature examples of the modification of polymers using Michael addition. For example, it has also been used to conjugate synthetic polymers to proteins by reacting vinyl sulfone-terminated poly(ethylene oxide) chains with the primary amine groups on either lysine or cysteine groups to produce useful ‘pegylated’ proteins for therapeutic applications.36 Michael addition has also been used to modify surfaces,37 produce cross-linked networks,38,39 gels40 and prepare other types of bioconjugates41–43 and, more recently, model branched copolymers.22
Herein we report the synthesis of well-defined PAMA-based diblock copolymers and the attempted chain extension of PAMA homopolymers via sequential monomer addition. Moreover, post-polymerisation functionalisation of these homopolymers and PAMA-based diblock copolymers via Michael addition has been conducted in aqueous solution using various acrylates and acrylamides to yield a library of novel copolymers with identical molecular weight distributions (see Scheme 1).
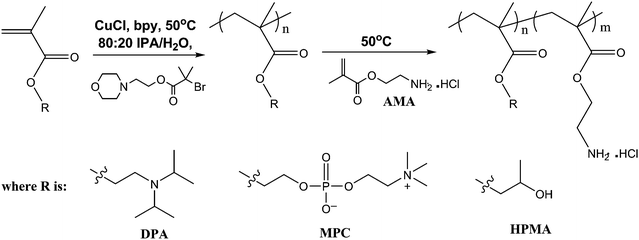 | ||
| Scheme 1 Schematic representation of the synthesis of all-methacrylic PAMA-based diblock copolymers by using either 2-(diisopropylamino)ethyl methacrylate (DPA), 2-(methacryloyloxy)ethyl phosphorylcholine (MPC) or 2-hydroxypropyl methacrylate (HPMA) in the first-stage polymerisation. | ||
Experimental
Materials
2-Hydroxypropyl methacrylate (HPMA, >97%) was donated by Cognis Performance Chemicals Ltd (Hythe, UK). 2-(Methacryloyloxy)ethyl phosphorylcholine (MPC, 99.9%) was donated by Biocompatibles, UK. These two monomers were used without further purification. 2-(Diisopropylamino)ethyl methacrylate (DPA) was purchased from Scientific Polymer Products (USA); inhibitor was removed by passing through a basic alumina column prior to use. Methacryloyl chloride (>97%), ethanolamine hydrochloride (>98%), hydroquinone (99%), Cu(I)Cl (99.995%), 2,2′-bipyridine (bpy, 99%), N,N,N′,N″,N″-pentamethyldiethylenetriamine (99%), 4-(2-hydroxyethyl)morpholine, poly(ethylene glycol) methyl ether acrylate (PEGA, 99%), 2-(dimethylamino)ethyl acrylate (DMAA, 98%), N,N-dimethylacrylamide (DMAc, 99%), sodium acrylamido-2-methyl-1-propanesulfonate (AMPS, 50 w/w% in water), (3-acrylamidopropyl)trimethylammonium chloride (ATAC, 75 w/w% in water), 4-acryloylmorpholine (AM, 97%), sodium acrylate (NaAA, 99%), acrylamidoglycolic acid monohydrate (AGA, 96%), sodium vinyl sulfonate (SVS, 98%), methyl acrylate (MA, 99%), D2O, NaOD and DCl were all purchased from Aldrich and were used as received. Methanol and 2-propanol (IPA) were purchased from Fisher and were used as received. De-ionised water was used in all experiments involving aqueous or mixed aqueous solvents. NMR solvents (CD3OD and CDCl3) and regenerated cellulose dialysis membrane (Spectra/Por 6, molecular weight cut-off 1000) were all purchased from Aldrich and were used as received.AMA monomer synthesis
AMA monomer was synthesised using a previously reported protocol.26 2-Aminoethanol hydrochloride (65 g, 0.68 mol), methacryloyl chloride (100 mL, 0.96 mol), and hydroquinone (0.50 g) were added to a three-necked round-bottomed flask fitted with a condenser. The molten 2-aminoethanol hydrochloride salt was esterified in the melt at 90–95 °C under a nitrogen atmosphere for 1 h and the reaction was maintained at 70–75 °C for a further 2 h. The HCl gas evolved during this reaction was neutralised using an aqueous NaOH solution connected to the reaction vessel. The crude product was cooled to 40 °C and diluted with THF (50 mL) to form a slurry. This slurry was then precipitated into excess n-pentane (500 mL). The resulting creamy white precipitate was isolated by filtration, washed thoroughly with n-pentane (500 mL), and dried under vacuum. It was then recrystallised using a 7 : 3 ethyl acetate–IPA mixture at −25 °C. After recrystallisation, the crude product was washed on a frit with a minimum amount of THF. The final product was dried under vacuum to produce an overall yield of 66% (>99% purity as judged by 1H NMR). The 2-(4-morpholino)ethyl 2-bromoisobutyrate (ME–Br) ATRP initiator, PEO-based macro-initiator (PEOn–Br where n = 45) and a PPO-based macro-initiator (PPO43–Br) were synthesised according to the literature.12,44,45Homopolymerisation of AMA
The homopolymerisation of AMA was performed using ME–Br initiator in 80 : 20 IPA–H2O, as described previously.20 In a typical ATRP synthesis, AMA (1.47 g, 9.0 mmol), ME–Br (50 mg, 0.18 mmol, target DPn = 50) and bpy (56 mg, 0.36 mmol) were added to a round-bottomed flask. After sealing the flask with a rubber septum, three vacuum/nitrogen cycles were performed to remove oxygen. Degassed 80 : 20 IPA–H2O (3.1 mL, such that [AMA]o = 33 w/v%) was introduced into the flask and this stirred solution was immersed in an oil bath at 50 °C. On addition of Cu(I)Cl catalyst (18 mg, 0.18 mmol) the reaction solution turned dark brown, indicating the onset of polymerisation. After 6 h, the polymerising solution was diluted with water and dialysed against water (Spectrapor 6, molecular weight cut-off 1000 Da) for three days to remove the spent blue Cu(II) catalyst. During dialysis, the blue/green polymer solution gradually became colourless. The aqueous solution was then freeze-dried from water overnight to give a white polymer. The attempted ‘self-blocking’ chain extension of PAMA50 involved essentially the same protocol: a second charge of AMA monomer was added at high conversion (as judged by 1H NMR). In the case of the attempted diblock copolymer chain extension experiments, either MPC, HPMA or DPA was added in situ to PAMA homopolymer (see Table 1 for further synthesis conditions).| Target copolymer composition | 1st Block | 2nd Block | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent | Time/h | Conversiona (%) | M n | M w/Mnb | Solvent | Time/h | Conversiona (%) | M n | M w/Mnb | |
| a Conversions were determined using 1H NMR spectroscopy by comparing the area of the vinyl proton resonances to the total methyl signal. b As determined by aqueous GPC at pH 3 using a series of poly(2-vinylpyridine) calibration standards. c Determined by DMF GPC after modification of AMA units with excess methyl acrylate in the presence of NEt3. d 50 w/v% monomer concentration. | ||||||||||
| PAMA30–PAMA30 | 80 : 20 IPA–H2O | 3 | 96 | 7800 | 1.11 | 80 : 20 IPA–H2O | 24 | 36 | 8200 | 1.14 |
| PAMA30–PHPMA30 | 80 : 20 IPA–H2O | 3 | 99 | — | — | 80 : 20 IPA–H2O | 24 | 69 | 8600c | 1.40c |
| PAMA30–PDPA30 | 80 : 20 IPA–H2O | 2.5 | 98 | 6700 | 1.19 | 80 : 20 IPA–H2O | 15.5 | 52 | Bimodal | |
| PHPMA30–PAMA30 | 80 : 20 IPA–H2O | 5.5 | 98 | — | — | 80 : 20 IPA–H2O | 24 | 95 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200c 200c |
1.40c |
| PMPC30–PAMA30 | Methanol, 20 °Cd | 1.3 | 100 | 8700 | 1.12 | 80 : 20 IPA–H2O | 24 | 99 | 11200 |
1.11 |
| PDPA30–PAMA30 | IPAd | 5 | 99 | 9900 | 1.29 | 80 : 20 IPA–H2O | 24 | 93 | 11900 |
1.28 |
| PDPA50–PAMA30 | IPAd | 7.5 | 98 | 12100 |
1.21 | 80 : 20 IPA–H2O | 15.5 | 96 | 13200 |
1.28 |
AMA polymerisation using macro-initiators
Homopolymerisation of AMA was also performed in turn using two ATRP macro-initiators, PEO43–Br and PPO45–Br in either 95 : 5 IPA–H2O or 80 : 20 IPA–H2O mixtures. The protocol for each AMA polymerisation was essentially the same, apart from the choice of initiator and solvent. In a typical ATRP synthesis, PEO45–Br (1.00 g, 0.47 mmol) and bpy (147 mg, 0.94 mmol) were added to a flask under a nitrogen atmosphere. Three vacuum/nitrogen cycles were performed to remove oxygen before a deoxygenated solution of AMA (2.31 g, 14.0 mmol) in 95 : 5 IPA–H2O (6.6 mL, 33%) was added. On addition of Cu(I)Cl catalyst (46 mg, 0.47 mmol) the reaction solution turned dark brown, indicating the onset of polymerisation. After 6 h stirring at 50 °C, the polymerising solution was diluted with water and dialysed against water (molecular weight cut-off 1000 Da) for three days to remove the Cu(II) catalyst. During dialysis, the blue/green copolymer solution gradually became colourless. The aqueous copolymer solution was then freeze-dried from water.Chain extension and triblock copolymer syntheses were attempted using essentially the same protocol as that used for the homopolymerisation of AMA using macro-initiators. Thus, a second charge of AMA was added at high conversion (as judged by 1H NMR) for self-blocking experiments (e.g. targeting PEO–PAMA–PAMA) and a new charge of DPA was employed for the PEO–PAMA–PDPA triblock copolymer syntheses. A typical attempted synthesis of PEO45–PAMA30–PDPA50 was as follows. In a 25 mL flask, PEO45–Br (0.50 g, 0.23 mmol), bpy (74 mg, 0.46 mmol) and AMA (1.16 g, 7.0 mmol) were added and sealed with a rubber septum. Three vacuum/nitrogen cycles were performed to remove the oxygen before degassed IPA (3.3 mL, 33%) followed by CuCl (23 mg, 0.23 mmol) were introduced into the flask. After 2.5 h, the conversion was greater than 95% and a deoxygenated solution of DPA (2.49 g, 11.6 mmol) in 80 : 20 IPA–H2O (2.5 mL, 50 w/v%) was added under a positive nitrogen pressure. Purification was carried out using the same protocol as that for the PEO–PAMA diblock copolymers.
Synthesis of all-methacrylic PAMA-based diblock copolymers
A typical synthesis of PDPA30–PAMA30 was as follows. In a 25 mL flask, ME–Br (50 mg, 0.18 mmol), bpy (56 mg, 0.36 mmol) and DPA (1.143 g, 5.4 mmol) were added and sealed with a rubber septum. Three vacuum/nitrogen cycles were performed to remove oxygen before degassed IPA (1.2 mL, 50 w/v%) followed by CuCl (18 mg, 0.18 mmol) were introduced into the flask. After 5 h the conversion was greater than 95% and a deoxygenated solution of AMA (0.89 g, 5.4 mmol), in 80 : 20 IPA–H2O (1.8 mL, 33 w/v %), was added under a positive nitrogen pressure. After 24 h stirring at 50 °C, the polymerising solution was diluted with water and dialysed against water (molecular weight cut-off 1000 Da) for three days to remove the Cu(II) catalyst. During dialysis, the blue/green copolymer solution gradually became colourless. The aqueous copolymer solution was then freeze-dried from water. AMA was also added as the second block to MPC and HPMA homopolymerisations.Michael addition to PAMA homopolymer
In a typical Michael addition, PAMA49 (0.04 g, 0.24 mmol of AMA units) was dissolved in 0.80 g of D2O to produce a 5.0 w/w% solution. To this solution, PEGA was added (0.12 g, 0.26 mmol, 1.1 molar equivalents based on AMA). The pH of this PAMA49 solution was adjusted to pH 9 using NaOD. The extent of PAMA49 modification was monitored by 1H NMR for 6 h at 50 °C. Reactions were also carried out on a larger scale in H2O. Typically, PAMA49 (0.50 g, 3.0 mmol of AMA units) was dissolved in 10.0 g H2O to produce a 5.0 w/w% solution. To this solution, DMAA was added (4.62 g, 3.30 molar equivalents based on AMA). The pH of this PAMA49 solution was adjusted to pH 9 using NaOH. After stirring for 24 h at 50 °C, solutions were dialysed against water to remove any excess unreacted acrylate, followed by freeze-drying. The overall extent of the modification was calculated using 1H NMR from a purified sample.Characterisation techniques
000 g mol−1) were used for calibration. Data were analysed using PL Cirrus GPC software (version 2.0) supplied by Polymer Laboratories.
000 g mol−1) were used for calibration. The data were analysed using Viscotek TriSEC 3.0 software. Prior to analysis, the AMA groups on these copolymers were derivatised with excess methyl acrylate in DMF containing triethylamine at 20 °C for 24 h.
Results and discussion
We have previously reported the homopolymerisation of AMA in its HCl salt form via ATRP.20 Various reaction conditions were investigated, including polymerisation temperature and solvent composition. Empirically, it was found that very high monomer conversions and relatively narrow molecular weight distributions (Mw/Mn < 1.25) were obtained within 3 h at 50 °C using an 80 : 20 IPA–water mixture and an [AMA]o of 33 w/v%. These optimised conditions were also employed in the present work for the first-stage polymerisation of AMA, with very similar results being obtained (see entries 1–3 in Table 1). However, one aspect not investigated in our earlier study was the living character of the PAMA chains at the end of such polymerisations, i.e. to what extent can further chain growth be achieved by the addition of a second monomer charge? This important question is addressed in the present work. Chain extension experiments were conducted to assess the living character of PAMA homopolymers synthesised via ATRP. For example, a second AMA monomer charge was added to a PAMA30 synthesis at 96% AMA conversion (see entry 1 in Table 1). However, only 36% monomer conversion was achieved for the second-stage polymerisation after 24 h. As a result, only a relatively small increase in molecular weight was observed by GPC. Two further chain extension experiments were also performed using HPMA and DPA monomer, respectively. Somewhat higher conversions were achieved (52% for DPA and 69% for HPMA) (see entries 2 and 3 in Table 1). However, the GPC trace for HPMA chain extension was broad (Mw/Mn = 1.40) and for the DPA chain extension the GPC trace was both broad and bimodal.As previously reported, a series of PEO–PAMA diblock copolymers may be readily prepared by ATRP.20 PEO45–PAMA30 proved to be fully soluble in the 80 : 20 IPA–H2O reaction solution, but adding a second charge of AMA monomer (target DP = 30) to this reaction once the first-stage polymerisation had reached high conversion (96%) only led to 63% conversion for the second-stage polymerisation. This incomplete conversion was accompanied by only a relatively modest increase in molecular weight (Mn) from 6800 to 9300 as judged by aqueous GPC.
There are two possible explanations for the incomplete chain extension of PAMA in these ‘self-blocking’ experiments: (1) chain-end termination has occurred, resulting in removal of the terminal bromine and hence preventing further propagation; (2) the catalyst has become deactivated, possibly by interaction with the primary amine groups on PAMA or by reacting with the hydrochloric acid associated with each AMA residue. To investigate the second possibility further, the chain extension of PAMA was repeated but this time, on addition of the second monomer charge (this time at 98% PAMA conversion), further catalyst and ligand were added (doubling the overall catalyst content) in order to compensate for any catalyst deactivation during the first-stage polymerisation. In this case, the second PAMA block reached high conversion (∼100%) and the Mn increased from 4100 to 7900 indicating successful chain extension, see Fig. 1. This significant improvement in conversion seems to support the hypothesis of catalyst deactivation. However, there was also an increase in Mw/Mn from 1.12 to 1.40. This broadening of the molecular weight distribution was accompanied with the appearance of a low molecular weight shoulder in the GPC trace of the chain-extended homopolymer. This suggests that some premature chain-end termination also occurs in these ‘self-blocking’ experiments.
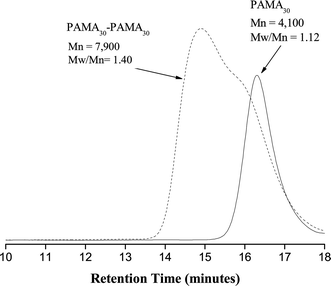 | ||
| Fig. 1 Aqueous GPC traces obtained for the chain extension of PAMA: PAMA30–PAMA30 diblock copolymer (dashed line) and PAMA30 (black line) prepared by ATRP. | ||
Previously, we reported a macro-initiator approach to prepare PAMA-based diblock copolymers via ATRP20 using two PEO-based macro-initiators with mean degrees of polymerisation (DP) of 45 and 113, respectively. Homopolymerisation of AMA (DP = 30) with either PEO45–Br or PEO113–Br at 50 °C in 95 : 5 IPA–H2O proceeded to high conversion within 6 h, whereas a target DP of 60 required 15 h for complete conversion in an 80 : 20 IPA–H2O mixture; these purified diblock copolymers had polydispersities of 1.26 or lower. A PPO43–PAMA60 diblock copolymer was successfully prepared using a 80 : 20 IPA–H2O solvent mixture by ATRP. This solvent composition also gave reasonably good control (Mw/Mn = 1.30, Mn = 23900) and high conversions (99%, within 48 h); it appears to be close to optimal for such syntheses.
Unfortunately, the attempted chain extension of PEO45–PAMA30 with a further AMA charge was also unsuccessful. A high conversion (>98%) was obtained for the second block and there was a corresponding increase in Mn from 3300 to 5900, see Fig. 2. Unfortunately chain-end termination was again observed, as judged by the appearance of a low molecular tail in the GPC trace for PEO45–PAMA30–PAMA30 and the concomitant increase in Mw/Mn from 1.13 to 1.49. Thus it seems that PAMA chains prepared by ATRP under the stated conditions do not have sufficient living character to allow chain extension using other methacrylic monomers.
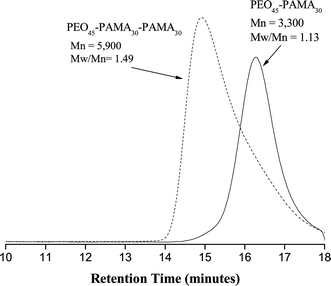 | ||
| Fig. 2 Aqueous GPC traces obtained for the chain extension of PAMA: PEO45–PAMA30–PAMA30 triblock copolymer (dashed line) and PEO45–PAMA30 diblock copolymer (black line) prepared by ATRP. | ||
In contrast, reasonably well-defined all-methacrylic diblock copolymers can be synthesised in good yield via sequential monomer addition provided that AMA is added as the second monomer (see entries 4–6 in Table 1).46
For example, the one-pot ATRP synthesis of PHPMA30–PAMA30 was conducted in 80 : 20 IPA–H2O for both blocks. High conversions were achieved (>95%) for the first- and second-stage polymerisation but the diblock copolymer polydispersity by DMF GPC (after modification of the AMA residues with methyl acrylate; see later) was a little higher than expected (Mw/Mn = 1.40). However, for the analogous syntheses of the PMPC–PAMA and PDPA–PAMA diblocks, it proved necessary to polymerise the MPC and DPA monomers in methanol and IPA, respectively. Using 80 : 20 IPA–H2O for the first block in the case of MPC led to solubility problems and hence some loss of control over the polymerisation. This is attributed to the well-known co-nonsolvency exhibited by PMPC in certain alcohol–water mixtures.47–49
Once these first PMPC or PDPA blocks had reached high conversion (>98%), AMA monomer was added as a 33% solution in 80 : 20 IPA–H2O. Final polydispersities were relatively narrow (Mw/Mn ≈ 1.11–1.28) and in all cases high conversions for the second block were achieved (>93%). An increase in molecular weight for the PAMA chain extension to give PDPA30–PAMA30 was confirmed by GPC, see Fig. 3.
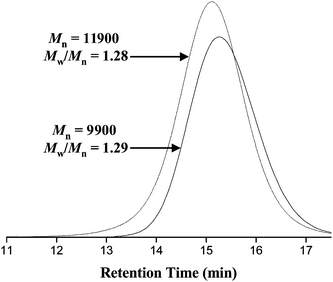 | ||
| Fig. 3 Aqueous GPC traces obtained for a PDPA30–PAMA30 diblock copolymer (dashed line; see entry 6 in Table 1) and the corresponding PDPA30 homopolymer precursor (solid line) prepared by ATRP using sequential monomer addition (with the DPA monomer polymerised first). | ||
Michael addition reactions
A series of Michael addition reactions were conducted using a near-monodisperse PAMA49 homopolymer (Mn = 1.24; Mw/Mn = 9500). The pendent primary amine groups on this homopolymer were modified with either acrylates or acrylamides. Reactions were conducted in aqueous solution at pH 9. This particular pH was chosen for two reasons. Firstly, the pKa of PAMA homopolymer is 7.6,20 hence above this value the AMA groups will be mainly in their neutral amine form, which is a prerequisite for successful Michael addition. Secondly, PAMA homopolymer is stable for up to 48 h at pH 9 (5.0 w/w% solution at 50 °C), but degrades in more alkaline media.18Michael addition between a primary amine and an acrylate is a well-known reaction in organic chemistry. Generally, polymer-analogous reactions tend to be slower than small molecule reactions and careful optimisation of the reaction conditions is usually required to ensure high conversions and to prevent side reactions.50 The first two acrylates studied were poly(ethylene glycol) methyl ether acrylate (PEGA), a well-known hydrophilic monomer, and 2-(dimethylamino)ethyl acrylate (DMAA), a pH-responsive monomer. Their chemical structures are shown in Scheme 2a.
 | ||
| Scheme 2 Modification of PAMA polymers by Michael addition with (a) acrylates (e.g. poly(ethylene glycol) methyl ether acrylate (PEGA) or 2-(dimethylamino)ethyl acrylate (DMAA)) or (b) acrylamides (e.g.N,N-dimethylacrylamide (DMAc), sodium acrylamido-2-methyl-1-propanesulfonate (AMPS), 3-acrylamidopropyltrimethylammonium chloride (ATAC), 4-acryloylmorpholine (AM)). | ||
Kinetic studies of the extent of acrylate addition were conducted with PEGA and DMAA. These two acrylates had very similar rates of reaction. In both cases essentially complete conversion was achieved within 5 h at 50 °C for 5.0 w/w% PAMA49 using an acrylate–amine molar ratio of 1.1 : 1, see Fig. 4. These reactions were conveniently monitored by the disappearance of vinyl groups at 5.5–6.5 ppm by 1H NMR in D2O/NaOD.
![Conversion vs. time plots for the reaction of PAMA49 with PEGA (triangles) and DMAA (squares) at relative molar ratios of [PEGA or DMAA] : [AMA] of 5 : 1 at 50 °C, pH 9 and 5.0 w/w% PAMA49 in D2O.](/image/article/2010/PY/b9py00320g/b9py00320g-f4.gif) | ||
| Fig. 4 Conversion vs. time plots for the reaction of PAMA49 with PEGA (triangles) and DMAA (squares) at relative molar ratios of [PEGA or DMAA] : [AMA] of 5 : 1 at 50 °C, pH 9 and 5.0 w/w% PAMA49 in D2O. | ||
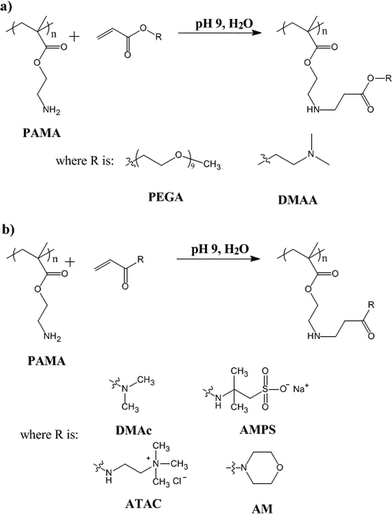
The same conditions mentioned above were used to synthesise PEGA-modified PAMA and DMAA-modified PAMA on a larger scale in H2O (5.0 w/w%, 50 °C, 24 h, 1.1 : 1 acrylate–amine molar ratio). After the reaction, any unreacted acrylate was removed via dialysis against water followed by lyophilisation. Comparing the 1H NMR spectra for PAMA and PEGA monomer with the PEGA-modified PAMA spectrum (Fig. 5) confirmed that Michael addition had occurred. Moreover, there was no sign of any vinyl signals (δ 5.5–7.0) from the monomer starting material after purification and new signals appeared (δ 2.6–3.4) from the two methylene protons, indicating formation of a β-aminoester. In addition, signals from the reacted PEGA were significantly broader compared to the monomer spectrum owing to their incorporation into polymer chains. The final apparent conversion of 103% for PEGA-modified PAMA49 (see Table 2) was calculated by comparison of the four methylene protons labelled ‘c’ (δ 4.1–4.6) and ‘f’ (δ 2.5–2.9). This result may simply reflect the (in)accuracy of our NMR measurements, but it is also possible that a small fraction of PAMA units has reacted with two acrylates to give tertiary amine residues. For DMAA-modified PAMA, 100% conversion was confirmed by comparison of signals from overlapping peaks ‘c’ and ‘g’ (δ 3.6–3.0) with peaks ‘d,’ ‘e,’ ‘f’ and ‘h’ (δ 2.3–2.7).
 | ||
| Fig. 5 Assigned 1H NMR spectra (D2O) for (a) PAMA49; (b) PEGA; (c) PEGA-modified PAMA49; (d) DMAA; (e) DMAA-modified PAMA49. | ||
One of the main problems with the analysis of cationic polymers is their unsuitability for most GPC protocols. PAMA homopolymers can only be analysed by aqueous GPC using an acidic buffer (pH 2–3). In other solvents such as THF or DMF, PAMA is insoluble. This means that the analysis of PAMA-based block copolymers is problematic when the comonomer(s) are unsuitable for acidic aqueous GPC (e.g. HPMA). In principle, protection of the PAMA block with PEGA should aid the solubility of the block copolymer. Surprisingly, PEGA–PAMA49 was only partially soluble in THF and DMF and was thus unsuitable for GPC analysis. In contrast, the PPO43–PAMA59 and PHPMA29–PAMA29 diblock copolymers were successfully derivatised directly in DMF by reaction with excess methyl acrylate. Triethylamine was added to the copolymer solution (10 g L−1) until complete dissolution occurred. This solution was stirred at 20 °C for 24 h and filtered prior to DMF GPC analysis. Monomodal GPC curves were obtained with Mn values ranging from 5000 to 15000 (vs. PMMA standards) and reasonably low polydispersities (Mw/Mn ∼ 1.3 to 1.4) in both cases. This indicates that derivatisation via Michael addition does not have any significant detrimental effect on the molecular weight distribution of the near-monodisperse precursor.
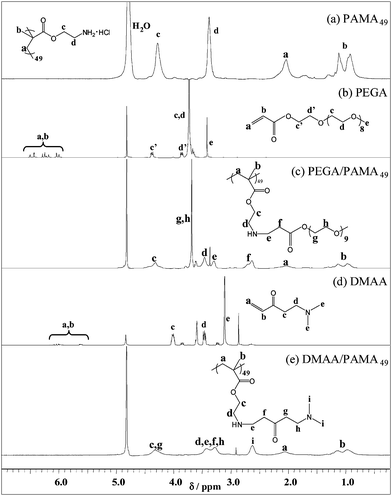
Four different acrylamides, DMAc, AMPS, ATAC and AM were selected for reaction with PAMA, see Scheme 2b. These particular monomers were selected for their known water solubility. First, the reaction between PAMA49 and DMAc was attempted at an acrylamide–amine molar ratio of 5 : 1 (50 °C, pH 9, 5.0 w/w% PAMA49). Even using this relatively large excess, it was found that the Michael addition of acrylamides with PAMA49 was much slower than that observed for acrylates. This has been attributed to delocalisation of the amide nitrogen lone pair, which renders acrylamides less reactive.51 Nevertheless, the Michael addition of DMAc proceeded to 96% conversion after 48 h, see Fig. 6. Under the same conditions, the other three acrylamides (AMPS, ATAC and AM) proved to be somewhat faster, requiring less than 10 h for >95% conversion.
 | ||
| Fig. 6 Conversion vs. time plots for the reaction of PAMA49 with DMAc, AMPS, ATAC and AM at a 5 : 1 acrylamide–amine molar ratio at 50 °C. Conditions: 5.0 w/w% PAMA49 in D2O/NaOD at pH 9. | ||
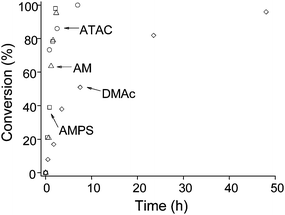
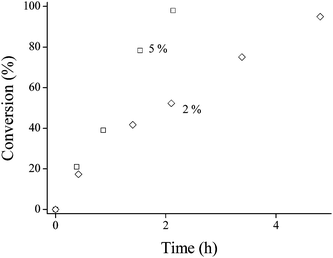
Reactions were also conducted at acrylamide–amine molar ratios of 3 : 1 and 1 : 1 for the reaction of AMPS with PAMA49 (50 °C, pH 9, 2.0 w/w%). These less forcing conditions resulted in slower reactions, as expected (see Fig. 7). The 1 : 1 reaction was significantly slower, requiring approximately one week to achieve ∼100% conversion, while the 3 : 1 and 5 : 1 reactions led to more than 95% conversion within 8.4 h and 4.8 h, respectively. Unsurprisingly, a higher PAMA49 concentration (5.0 w/w%) reduced the reaction time for 100% amine conversion to 2.1 h (50 °C, pH 9, using a 5 : 1 AMPS–amine molar ratio), see Fig. 8. Moreover, 1H NMR confirmed that this particular reaction continued to well beyond 100% conversion, producing essentially complete disubstitution of the AMA residues within 6 h, see Fig. 9.
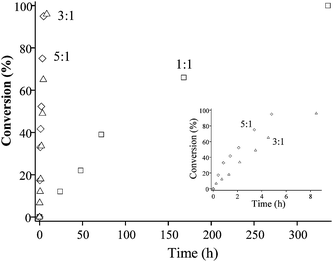 | ||
| Fig. 7 Conversion vs. time plots for the reaction of PAMA49 with AMPS at acrylamide–amine molar ratios of 5 : 1 (diamonds), 3 : 1 (triangles) and 1 : 1 (squares) at 50 °C. Conditions: 2.0 w/w% PAMA49 in D2O/NaOD at pH 9 (inset shows the same data for the 5 : 1 and 3 : 1 molar ratios over shorter time scales). | ||
 | ||
| Fig. 8 Conversion vs. time plots for the addition of AMPS to 2.0 w/w% and 5.0 w/w% PAMA49 in D2O/NaOD at pH 9 and 50 °C using an AMPS–amine relative molar ratio of 5 : 1. | ||
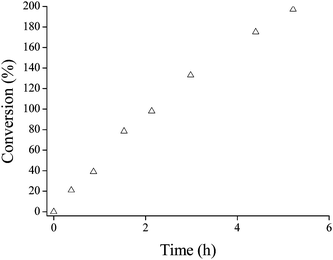 | ||
| Fig. 9 Conversion vs. time plot for the addition of AMPS to a 5.0 w/w% PAMA49 solution in D2O/NaOD at pH 9 and 50 °C using an AMPS–amine relative molar ratio of 5 : 1. | ||
1H NMR studies (conducted in D2O) provided a convenient method for monitoring and optimising the modification of PAMA49 with acrylamides. These conditions were successfully replicated in H2O on a larger scale so as to synthesise DMAc-, AMPS-, ATAC- and AM-modified PAMA49. Each acrylamide was added to a 5.0 w/w% solution of PAMA49 (acrylamide–amine molar ratio = 5 : 1) at pH 9 and stirred at 50 °C for 48 h. After Michael addition, any unreacted acrylamide was removed by dialysis against water, followed by lyophilisation. All four reactions led to high conversions with varying degrees of disubstitution (125–196%), see Table 3 and Fig. 10.
| Acrylamide type | Total conversion (%) | Percentage of monosubstituted AMA repeat units (%) | Percentage of disubstituted AMA repeat units (%) |
|---|---|---|---|
| DMAc | 190 | 10 | 90 |
| AMPS | 157 | 43 | 57 |
| ATAC | 125 | 75 | 25 |
| AM | 196 | 4 | 96 |
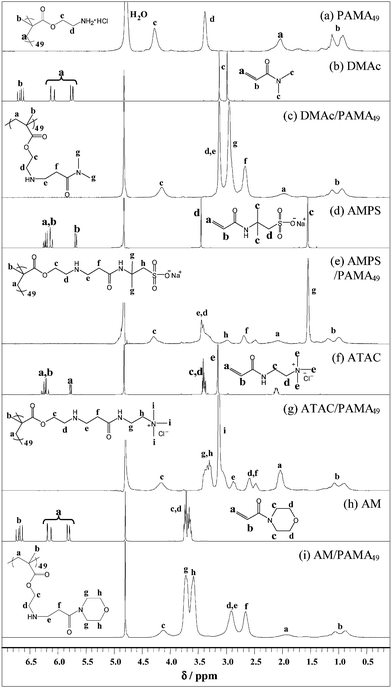 | ||
| Fig. 10 Assigned 1H NMR (D2O) spectra for (a) PAMA49; (b) DMAc; (c) DMAc-modified PAMA49; (d) AMPS; (e) AMPS-modified PAMA49; (f) ATAC; (g) ATAC-modified PAMA49; (h) AM; (i) AM-modified PAMA49 in D2O. | ||
It is worth emphasising that PAMA49 is stable with respect to hydrolytic degradation under the chosen reaction conditions. Our earlier study18 showed that negligible chemical degradation of PAMA49 (<1%) occurred after 48 h (50 °C, pH 9, 5.0 w/w% PAMA49). Moreover, there is no evidence for any PAMA degradation in any of the final 1H NMR spectra. This suggests that the reaction with acrylamides (or acrylates) is favoured over any degradation pathway and that the resulting derivatised polymer is much more stable than the PAMA precursor.
The reaction of PAMA49 with acrylamidoglycolic acid (AGA) and sodium vinyl sulfonate (SVS) was also attempted. AGA appeared to react slowly; after five days (50 °C, pH 9, 5.0 w/w% and an acrylamide–amine ratio of 5 : 1) approximately 67% conversion was achieved, as judged by 1H NMR. However, at the same time two new sharp peaks appeared in the spectrum and a control experiment conducted in the absence of any PAMA49 confirmed that AGA degradation occurred on the same time scale, under the same conditions. It is quite likely that this amino acid-type monomer would actually react with PAMA49 under different solvent/temperature conditions where monomer degradation either does not occur at all, or can be minimised. However, such optimisation is beyond the scope of the present study
SVS was selected in view of the facile Michael addition cross-linking chemistry of divinyl sulfone (DVS).52 However, this monofunctional analogue only reacted with PAMA49 very slowly at a SVS–amine molar ratio of 5 : 1 at 50 °C. Thus, for a 5.0 w/w% solution of PAMA49 the reaction reached only 47% conversion after 5 days. Moreover, leaving the reaction for two weeks only increased the conversion by a few percent. Because high conversions could not be achieved within a reasonable time scale, Michael addition derivatisation using this monomer was not pursued further.
AMPS was also utilised to modify selected PAMA-based diblock copolymers. Reaction with this reagent resulted in the synthesis of several novel anionic diblock copolymers with potentially interesting aqueous solution properties. The three diblock precursors utilised in these experiments were the pH-responsive PDPA30–PAMA28 and two thermo-responsive block copolymers, PHPMA29–PAMA29 and PPO43–PAMA59.
Michael addition was conducted using the same conditions as those employed for PAMA49 homopolymer. An AMPS–amine molar ratio of 5 : 1 was added to a 5.0 w/w% copolymer solution and stirred for 48 h at 50 °C. Under these conditions, an AMPS conversion of 62% was calculated for PDPA30–PAMA28 using 1H NMR by comparing the ‘c’ and ‘g’ signals at δ 4.0–4.5 (due to the PDPA and PAMA methylene protons) to the ‘p’ and ‘n’ signals at δ 2.5–3.0 (due to the methylene protons of the AMPS), see Table 4. The pH-induced micellisation behaviour of PDPA–PAMA has already been reported,20 with well-defined PDPA-core micelles being formed above pH 6.3. We speculate that the micellar nature of the PDPA–PAMA diblock precursor at pH 9 most likely has an adverse effect on the rate of Michael addition of the AMPS with the AMA residues, since such core-shell nanostructures are likely to resist the build-up of anionic charge in the micelle coronas.
| Diblock copolymer type | Time/h | Total conversion (%) | Percentage of monosubstituted AMA repeat units (%) | Percentage of disubstituted AMA repeat units (%) |
|---|---|---|---|---|
| PDPA30–PAMA28 | 48 | 62 | 62 | 0 |
| PHPMA29–PAMA29 | 48 | 157 | 43 | 57 |
| PPO43–PAMA59 | 48 | 93 | 93 | 0 |
In contrast, an AMPS conversion of 93% was calculated for PPO43–PAMA59 from the 1H NMR spectrum of the derivatised copolymer after purification by dialysis. This is a relatively high conversion for monosubstituted amines and may well reflect the much less compact, hydrated nature of the PPO-core micelles compared to the PDPA–PAMA micelles, since this reduced structural order is less likely to resist the build-up of anionic charge density. It is well-known that PPO is a thermo-responsive polymer that displays LCST behaviour with an estimated cloud point of 15 °C.11 Save et al. synthesised PPO43–PGMA70 (where PGMA = poly(glycerol monomethacrylate)) by ATRP and reported that this copolymer was molecularly dissolved at 5 °C while colloidal aggregates of 100–330 nm diameter were formed at 60 °C, as judged by DLS. In the present work, the PPO43–PAMA59 solution remained turbid throughout its reaction with the AMPS. It is likely that any reaction took place between the AMPS and the coronal PAMA59 chains of PPO43–core micelles, but further work is required to confirm this hypothesis.
The AMPS derivatisation of PHPMA29–PAMA29 was more encouraging, with a conversion of 157% being calculated from the 1H NMR spectrum. This suggests a mixture of mono- and disubstituted amines in this particular case. The diblock precursor solution was also turbid at the Michael addition reaction temperature of 50 °C due to the thermo-responsive nature of the PHPMA block.53 However, this appeared to have little effect on the rate of Michael addition; the overall conversion was comparable to that obtained for the PAMA49 homopolymer under the same conditions.
Conclusions
Well-defined PAMA-based diblock copolymers were successfully prepared via ATRP in alcohol–water mixtures at 50 °C using sequential monomer addition. However, AMA monomer must be polymerised second in order to ensure good blocking efficiencies, high overall conversions and reasonable control (i.e. low polydispersities). Attempted chain extension of PAMA homopolymer was unsuccessful, apparently due to both catalyst deactivation and also premature chain termination. The former problem presumably involves irreversible oxidation of Cu(I) to Cu(II) and can be at least partially overcome by addition of fresh catalyst just prior to the second charge of AMA monomer. However, the problem of premature chain termination could not be alleviated and this results in significant contamination of the final chain-extended polymer with the homopolymer precursor.PAMA homopolymer has been successfully reacted with a range of acrylates and acrylamides to obtain a library of new water-soluble polymers with the same relatively narrow molecular weight distribution. Acrylates proved to be more reactive than acrylamides, as expected. The former only require approximately stoichiometric molar ratios to form monosubstituted β-aminoesters. In contrast, the latter typically require excess reagents and more vigorous conditions, which often resulted in disubstituted primary amines.
Two reagents (sodium vinyl sulfonate and acrylamidoglycolic acid) proved to be much less successful, since they typically required extremely long reaction times and only gave incomplete conversions. Moreover, undesirable side reactions such as chemical degradation or in situ polymerisation were often observed. However, there may yet be some scope for further optimisation of the reaction conditions.
Michael addition also works well for the functionalisation of PAMA-based diblock copolymers. Appropriate derivatisation of selected PAMA-based block copolymers also allowed their GPC analysis in convenient eluents, whereas the precursors were sometimes not amenable to this technique. In addition, this facile chemistry has opened up routes to a wide range of functional block copolymers.
Acknowledgements
ESR thanks the University of Sheffield for an EPSRC PhD studentship. SPA is a recipient of a five-year Royal Society-Wolfson Research Merit Award. Unilever Research (Colworth) is thanked for CASE support of ESR and for permission to publish this work.References
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123 CrossRef CAS.
- C. L. McCormack and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2005, 38, 9075 CrossRef CAS.
- I. Y. Ma, E. J. Lobb, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. Salvage, Macromolecules, 2002, 35, 9306 CrossRef.
- M. Save, J. V. M. Weaver, S. P. Armes and P. McKenna, Macromolecules, 2002, 35, 1152 CrossRef CAS.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395 CrossRef CAS.
- Y. T. Li, S. P. Armes, X. P. Jin and S. P. Zhu, Macromolecules, 2003, 36, 8268 CrossRef CAS.
- X. Zhang, J. H. Xia and K. Matyjaszewski, Macromolecules, 1998, 31, 5167 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, S. J. Brooks and K. Matyjaszewski, Macromolecules, 2006, 39, 6817 CrossRef CAS.
- M. H. Dufresne and J. C. Leroux, Pharm. Res., 2004, 21, 160 CrossRef CAS.
- K. L. Thompson, E. S. Read and S. P. Armes, Polym. Degrad. Stab., 2008, 93, 1460 CrossRef CAS.
- Y. T. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81 CrossRef CAS.
- L. H. He, E. S. Read, S. P. Armes and D. J. Adams, Macromolecules, 2007, 40, 4429 CrossRef CAS.
- R. Narain and S. P. Armes, Macromolecules, 2003, 36, 4675 CrossRef CAS.
- Y. T. Li and S. P. Armes, Macromolecules, 2009, 42, 939 CrossRef CAS.
- J. Z. Du and S. P. Armes, Langmuir, 2008, 24, 13710 CrossRef CAS.
- A. H. Alidedeoglu, A. W. York, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5405 CrossRef CAS.
- X. W. Xu, A. E. Smith, S. E. Kirkland and C. L. McCormick, Macromolecules, 2008, 41, 8429 CrossRef CAS.
- R. Narain and S. P. Armes, Biomacromolecules, 2003, 4, 1746 CrossRef CAS.
- J. Ramos, A. Martin-Molina, M. P. Sanz-Izquierdo, A. Rus, L. Borque, R. Hidalgo-Alvarez, F. Galisteo-Gonzalez and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2404 CrossRef CAS.
- G. Sharma and M. Ballauff, Macromol. Rapid Commun., 2004, 25, 547 CrossRef CAS.
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487 CrossRef CAS.
- P. Ferruti, M. A. Marchisio and R. Barbucci, Polymer, 1985, 26, 1336 CrossRef CAS.
- P. Ferruti, E. Ranucci, F. Trotta, E. Gianasi, E. G. Evagorou, M. Wasil, G. Wilson and R. Duncan, Macromol. Chem. Phys., 1999, 200, 1644 CrossRef CAS.
- P. Ferruti, M. A. Marchisio and R. Duncan, Macromol. Rapid Commun., 2002, 23, 332 CrossRef CAS.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761 CrossRef CAS.
- C. S. Wu, Y. L. Liu and Y. S. Chiu, Polymer, 2002, 43, 1773 CrossRef CAS.
- M. Morpurgo, F. M. Veronese, D. Kachensky and J. M. Harris, Bioconjugate Chem., 1996, 7, 363 CrossRef CAS.
- V. S. Khire, T. Y. Lee and C. N. Bowman, Macromolecules, 2007, 40, 5669 CrossRef.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11 CrossRef CAS.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226 CrossRef CAS.
- S. A. Robb, B. H. Lee, R. McLemore and B. L. Vernon, Biomacromolecules, 2007, 8, 2294 CrossRef CAS.
- R. Roy and C. A. Laferriere, Chem. Commun., 1990, 1709 RSC.
- D. L. Elbert and J. A. Hubbell, Biomacromolecules, 2001, 2, 430 CrossRef CAS.
- R. Duncan, S. Gac-Breton, R. Keane, R. Musila, Y. N. Sat, R. Satchi and F. Searle, J. Controlled Release, 2001, 74, 135 CrossRef CAS.
- S. Y. Liu and S. P. Armes, J. Am. Chem. Soc., 2001, 123, 9910 CrossRef CAS.
- K. Jankova, X. Y. Chen, J. Kops and W. Batsberg, Macromolecules, 1998, 31, 538 CrossRef CAS.
- In this context, it is interesting to note that He et al. found that well-defined PAMA–PDPA diblock copolymers could only be successfully synthesised via a ‘one-pot’ reaction provided that AMA was polymerised first.20 In this earlier study, PDPA–PAMA could only be synthesised if PDPA homopolymer was first isolated and purified (i.e. the so-called macro-RAFT CTA approach) prior to chain extension with AMA, although this restriction may be mainly due to the differing solubility requirements of the two monomers and the corresponding polymers under the chosen RAFT conditions.
- Y. Matsuda, M. Kobayashi, M. Annaka, K. Ishihara and A. Takahara, Polym. J., 2008, 40, 479 CrossRef CAS.
- Y. Kiritoshi and K. Ishihara, J. Biomater. Sci., Polym. Ed., 2002, 13, 213 CrossRef CAS.
- Y. Kiritoshi and K. Ishihara, Sci. Technol. Adv. Mater., 2003, 4, 93 CrossRef CAS.
- X. Bories-Azeau, S. P. Armes and H. J. W. van den Haak, Macromolecules, 2004, 37, 2348 CrossRef CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, UK, 2001 Search PubMed.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021 RSC.
- J. Madsen, S. P. Armes, K. Bertal, H. Lomas, S. MacNeil and A. L. Lewis, Biomacromolecules, 2008, 9, 2265 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |