Furans as offspring of sugars and polysaccharides and progenitors of a family of remarkable polymers: a review of recent progress
Alessandro
Gandini
*
CICECO and Chemistry Department, University of Aveiro, 3810-193, Portugal. E-mail: agandini@ua.pt; Fax: +351 234 370084; Tel: +351 234 370 711
First published on 16th December 2009
Abstract
The recent literature on polymers incorporating furanheterocycles or moieties derived from them is reviewed to highlight an important area of polymers from renewable resources. Emphasis is placed on novel applications of furfuryl alcohol, conjugated polymers, polyesters and the application of the Diels–Alder reaction to prepare thermoreversible macromolecular architectures.
Background
Interest in polymers from renewable resources is growing vigorously1 as part of the general concern for sustainability, which obviously also includes the more pressing problem of alternative energy sources. The chemistry involved in the synthesis of these biomass -based macromolecules is much more frequently associated with the modification of natural polymers (cellulose, starch, chitin, etc.) or oligomers (lignin, tannins, vegetable oils, etc.) rather than with the more classical fashion involving monomers and their polymerization. Within the latter strategy, the materials obtained from monomers like terpenes, lactic acid and sugars bear intrinsically the structure imprint of their precursors and hence a specific set of related properties. The situation is qualitatively different with polymers prepared with furan monomers or through furan chemistry, because the approach here resembles the petrochemical counterpart in the sense that it provides entry to a whole host of monomers and hence a variety of macromolecular structures with different properties. In this peculiar version of a biorefinery, two first-generation furan derivatives, furfural (F) and 5-hydroxymethylfurfural (HMF), readily prepared from ubiquitous C5 and C6 carbohydrate resources, respectively, represent the precursors to that rich array of monomer structures, suitable for any type of polymerization process.2Schemes 1–3 illustrate this point through a choice of monomers which have been synthesised and polymerised.2
 | ||
| Scheme 2 A selection of monomers derived from 5-hydroxymethylfurfural (HMF). | ||

In addition to the straightforward research activities consisting of preparing these monomers, studying their behaviour in polymerisation and copolymerisation systems and assessing the properties and possible applications of the ensuing materials, another domain of investigation calls upon some furan-specific chemical features, which enable original polymers to be prepared. These include end-functionalised macromolecules, block and graft copolymers2 and, more originally, the exploitation of the marked dienic character of the furanheterocycle through the application of the Diels–Alder (DA) reaction, as shown in Scheme 4, to prepare thermoreversible polymer architectures and hence, e.g., readily recyclable and mendable materials.3
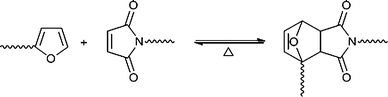 | ||
| Scheme 4 The DA equilibrium between growing species bearing respectively furan and maleimide end groups. | ||
The purpose of the present review is to highlight the remarkable progress that the field of furanpolymers has undergone since the late 1990s, when the last comprehensive appraisal was published.2c
Furfural and 5-hydroxymethylfurfural
Despite the well-established acid-based technologies which produce some 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of F yearly, mostly from the pentoses of corncobs and sugarcane bagasse, recent interest in novel systems associated with the chemical exploitation of biomass ,4 has revived studies on its preparation. These include the use of zeolites,5 and of conventional Brønsted acids in an ionic liquid.6 The study of F as a monomer or comonomer in the elaboration of often ill-defined “furan resins” has never been a particularly stimulating issue,2 and no relevant contribution has enriched this topic in the past decade.
000 tons of F yearly, mostly from the pentoses of corncobs and sugarcane bagasse, recent interest in novel systems associated with the chemical exploitation of biomass ,4 has revived studies on its preparation. These include the use of zeolites,5 and of conventional Brønsted acids in an ionic liquid.6 The study of F as a monomer or comonomer in the elaboration of often ill-defined “furan resins” has never been a particularly stimulating issue,2 and no relevant contribution has enriched this topic in the past decade.
Within the same context, the sudden surge of investigations related to the synthesis of HMF from biomass -derived mono- and poly-saccharides is one of the characteristic indicators of the growing impact of research on renewable resources.1,7 These investigations include mostly the use of D-fructose, but also inulin and glucose, as the precursors, and a broad selection of novel catalysts, reaction media, and/or process conditions.4b,6,8 In the space of a few years this issue has thus been advanced so considerably, that it brought HMF to the verge of becoming an industrial commodity. Given, however, that it is difficult and impractical to store HMF because of its proneness to degradation even under relatively mild surroundings, its in situ transformation into a stable derivative seems most appropriate.
The chemistry of HMF is well documented9 and two obvious pathways are likely to be privileged in the search of that viable derivative, viz. the oxidation to the corresponding dialdehyde or diacid. Indeed, together with the recent interest in preparing HMF through novel and more efficient procedures, studies have began appearing on its oxidation to 2,5-diformylfuran10 and 2,5-furandicarboxylic acid (FDA) or its esters,8o,11 which are all highly stable molecules and, of course, very valuable potential monomers. Interestingly, the reduction of HMF to the corresponding diol2,5-bis(hydromethyl)furan has not stimulated any tangible study, probably because its alternative preparation from furfuryl alcohol is more viable.
Furfuryl alcohol and its polymers
The vast majority of F is converted into furfuryl alcohol (FA), a well-established industrial commodity, which has found growing applications as a source of a variety of materials,2 with notable recent progress.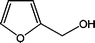
The acid-catalysed polycondensation of FA is a complex system whose intricacies were unravelled for the most part in a systematic investigation that called upon the use of various model compounds.12 The mechanistic complexity arises from the fact that two inevitable side reactions alter profoundly the “normal” course of the linear growth, generating unsaturated sequences, which undergo intermolecular Diels–Alder (DA) reactions leading to black cross-linked products, as in the simplified depiction of Scheme 5.
 | ||
| Scheme 5 The two mechanisms responsible for conjugated sequences (colour formation) and subsequent cross-linking in the acid-catalysed polymerisation of FA.12 | ||
A chemo-rheological analysis of this polymerization13 confirmed both the clear-cut evidence and the tentative mechanisms previously put forward12 and provided an interesting kinetic insight into the competition between linear growth and the growing role of interchain DA couplings, responsible for the early attainment of a diffusion-controlled regime. Similar conclusions were drawn in a very detailed ATR-IR study of this polycondensation.14FTIR, Raman and UV-vis spectroscopies were also adopted to follow FApolymerization in the H–Y confined domains of a protonic Y zeolite.15 The reduced reaction rate opened the way to the identification of some intermediate structures, including active carbenium ion species. This study was extended to the carbonisation process suffered by the cross-linked polyFA upon heating, as further discussed below.
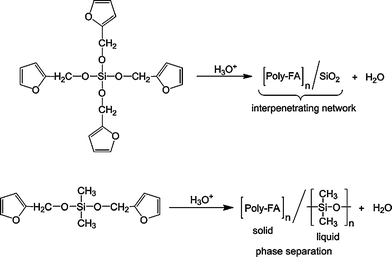
Turning to materials, FA has attracted considerable attention in recent investigations aimed at preparing and characterising carbonaceous and other materials, through its polymerisation and subsequent pyrolisis. Examples include mesoporous16 and microporous17 carbon, silica nanocomposites,18 carbon nanocomposites,19 glass-like carbon,20 ZnO-carbon composites,21 carbon films22 and foams ,23 as well as mesoporous crystalline TiO2.24 Another area where polyFA is gaining momentum is the synthesis of organic-inorganic hybrids,25 including nanoscopic morphologies26 and biobased nanomaterials.27 An interesting aspect of some of these systems is the formation of furfuryl-alkoxide intermediates, e.g. from siloxanes incorporating one or several furfuryl moieties through alkoxide exchanges, followed by the acid-promoted polymerisation of the furfuryl motifs and/or the sol–gel process of the hydrolysed siloxanes, as shown in Scheme 6.
Another field in which FA has been successfully applied is wood preservation and modification. Its impregnation of wood and subsequent polymerisation promoted by acidic catalysts has been optimised to provide remarkable improvements in such properties as dimensional stability, mechanical and chemical strengthening, excellent resistance to microbial decay and insect attack, as well as ecological soundness, which have led to its commercialisation.28 It is unfortunate that very little work on the chemistry associated with this impregnation accompanied the technical development. Recent attempts to unravel the basic query of whether the polymerising FA structures react with any of the wood components has been limited to the study of lignin model compounds29 with unconvincing conclusions. This state of affairs is quite common in the realm of the wide range of studies related to the many applications of polyFA,2 where empirical approaches dominate to the detriment of deeper chemical investigation, which would certainly provide better materials.
A recent addition to the numerous applications of polyFA has to do with its role as a mechanical reinforcement for highly porous polymeric matrices, introduced by the vapour-phase adsorption of FA into these materials and its subsequent in situpolymerisation.30
The realpolyFA, namely the ideal structure associated with a linear colourless thermoplastic polymer, however, regrettably remains elusive and its synthesis therefore represents a beautiful challenge.

Conjugated polymers
Although polyfuran, like polythiophene and polypyrrole, has been the subject of numerous publications, unlike its two homologues, the structural evidence pointing to a regular sequence of 2,5-substituted heterocycles has never been irrefutably provided, whatever the synthetic procedure adopted.2 This is still today a critical missing link, most probably because, during the polymerization, the pronounced dienic character of the furan ring induces different modes of insertion and hence irregular macromolecular structures difficult to minimise. The development of materials associated with such a potentially useful conjugated polymer has therefore been hampered. Attention has thus moved onto the synthesis of copolymers with other heterocycles like thiophene,31 whose alternating oligomers had previously been characterised,32 and porphirines,33 or the polymerisation of monomers incorporating furan moieties together with aniline34 and phenylene motifs.35 All these well-defined materials displayed interesting opto-electronic properties.
Poly(2,5-furylene vinylene) (PFV) represents undoubtedly the best achievement in the context of conjugated furanmacromolecules. Whereas its synthesis by the classical approach through 2,5-bis(tetrahydrothiopeniomethyl)furan dichloride36 is rather cumbersome, the much simpler base-catalysed polycondensation of 5-methylfurfural provides a straightforward route to both PFV and its oligomers , additionally bearing a terminal aldehyde function.2d,37 The structure depicted here bears that terminal aldehyde moiety, which proved particularly useful in a variety of chemical modifications, including chain extensions, grafting and preparation of Schiff bases.2d,37
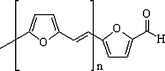
While the polymer displays good conductivity, solubility in common solvents and resistance to atmospheric oxidation and can be plasticized by attaching polyether chains to its reactive endgroup, the individual oligomers , viz. dimer to pentamer, display photo- and electro-luminescence covering essentially the entire visible spectrum. The peculiar photochemical behaviour of the dimer was moreover exploited to prepare photocrosslinkable materials from poly(vinyl alcohol)38 and chitosan.39 It follows that 5-methylfurfural, which is obtained industrially as a secondary product in the manufacture of F, is a very useful precursor to a series of conjugated molecules and macromolecules with high tech applications.37
A very recent interesting addition to the realm of furan-based conjugated polymers describes the use of a bisfuranyl monomer incorporating a 1,6-methano[10]annulene moiety to prepare conducting materials.40
Polyesters
The literature on furan polyester is quite rich,2 starting in earnest with the ground-breaking work carried out in the seventies in Moore's laboratory.2c For the sake of clarity it seems useful to separate two families of macromolecular structures, namely those derived from FDA (hence based on HMF) and those incorporating a bis-furan acid residue of the kind shown in Scheme 3 (hence based on F).The former type of polyesters remained practically ignored after Moore's work and was only revived recently in our laboratory. The first investigation dealt with a hitherto virtually neglected polymer, despite its obvious relevance, viz. poly(2,5-ethylene furandicarboxylate) (PEF), which is the heterocycle homologue of the most important commercial polyester, poly(ethylene terephthalate) (PET). Both the classical polycondensation between FDA dichloride and ethylene glycol and the polytransesterification of the monomer prepared by the reaction of FDA with an excess of ethylene glycol (Scheme 7) were successful, but the latter yielded higher molecular weights (DPn > 200) and a remarkably elevated crystallinity for the pristine polymers.41
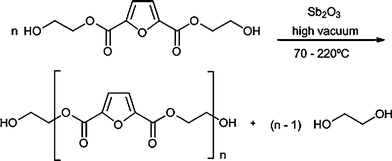 | ||
| Scheme 7 Polytransesterification mechanism leading to high-DP PEF. | ||
A thorough characterisation of this novel polyester showed its remarkable interest as a potential alternative to PET based on renewable resources (ethylene glycol can also be classified in this category as a glycerol, sorbitol or cellulose derivative, following recent catalytic conversions), since its properties simulated rather closely those of the classical petroleum-based counterpart.
The study has now been extended to other poly(2,5-furan dicarboxylate)s using aliphatic diols like propylene glycol, sugar diols like isosorbide, benzylic structures like 1,4-bishydroxymethyl benzene, and bisphenols like hydroquinone.42,43 This ongoing project has set a new stage, in which polyesters with a very wide range of properties and hence of possible applications, can be readily prepared using one monomer, if not both, from renewable resources. The recent upsurge of interest in improving the process of HMF production8 is particularly beneficial to the future of these polyesters and of other polymers based on FDA and 2,5-furancarboxydialdehyde.
The second family of furan-based polyesters had been intensively studied over the last two decades of the 20th century, as previously reviewed.2 A large variety of combinations between differently bridged difuran diacids and a comprehensive series of diols (see Scheme 8) allowed sound criteria of structure/properties relationships and consequently of the domains of applications to be established for the ensuing materials.44 This research strategy, which calls upon F as the precursor to the diacids, is being pursued in the direction of amorphous furanic-aromatic random copolyesters involving the terephthalate and difuran diacid units both esterified with ethylene glycol.45
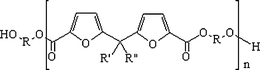 | ||
| Scheme 8 General structure of a wide family of furanpolyesters.2,44 | ||
Diels–Alder systems
Studies on the application of the reversible DA reaction between furan (diene) and maleimide (dienophile) (Scheme 4) for the synthesis of novel macromolecular materials have bloomed during the past decade, even though the field is some forty years old.2e,3a It is important to emphasise that the furan/maleimideadduct is in fact a mixture of its threo and erithro forms, whose proportion varies as a function of the reaction parameters. This aspect is highly relevant to most organic syntheses which call upon the DA reaction, but not to the present context, since neither polymer growth nor degradation are affected by the actual adduct stereochemistry.The different approaches applied to this general strategy can be summarised in terms of (i) linear polymerisations, (ii) reactions leading to networks, (iii) formation of dendrimers or hyperbranched polymers.3 In all instances, the underlying motivation is of course the possibility of reverting to the starting reagents in a clean-cut fashion through the thermal reversibility of the construct. One is dealing therefore with a remarkably useful type of click chemistry, because of its straightforward reversibility coupled with the fact the both DA and retro-DA reactions are not marred by side events. The temperatures associated with the equilibrium in Scheme 4 depend on the desired rates of either forward or backward reactions, but, typically, 60–65 °C insure a good forward rate and a negligible retro-DA contribution, whereas 100–110 °C are quite adequate to revert the situation to a fast and essentially complete adduct decomposition.
Before reviewing the recent polymer contributions, it seems appropriate to refer to a recent study on monofunctional model compounds, which, on the one hand, examined the kinetics of the DA reaction and, on the other, tested a combined spectroscopic approach to follow both the forward and the reverse course of these reactions.46
The association of UV and NMR spectroscopy proved particularly useful in this context and was therefore extended to the study of polymerization–depolymerisation systems. The first such investigation deals with linear DApolycondensations of difuran and bismaleimide monomers, like those shown in Scheme 9.42,46,47
 | ||
| Scheme 9 Reversible DApolycondensation between complementary bifunctional monomers. | ||
These polymerisations proceeded without any interference from side reactions and their DA/retro-DA reversibility was assessed over several cycles. The next topic concerns non-linear polycondensations involving monomers (one or both) with functionality higher than two, as in the system shown in Scheme 10.42,47
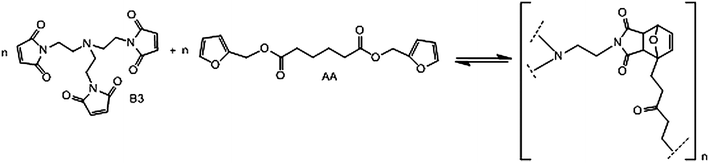 | ||
| Scheme 10 Reversible DA non-linear polycondensation between a tris-maleimide and a difuran monomer. | ||
The molar ratio between the monomers was varied in order to examine both non-gelling and crosslinking situations. Again, both types of systems displayed a clean-cut behaviour and good recyclability.
This broad approach also includes AB monomers, i.e. molecules bearing a furanheterocycle at one end and a maleimide moiety at the other.42,47 In order to avoid premature polymerization, the maleimide function (or indeed the furan ring) can be protected in the form of a DAadduct , which is readily deprotected by heating the compound just before studying its polymerisation, as shown in Scheme 11.
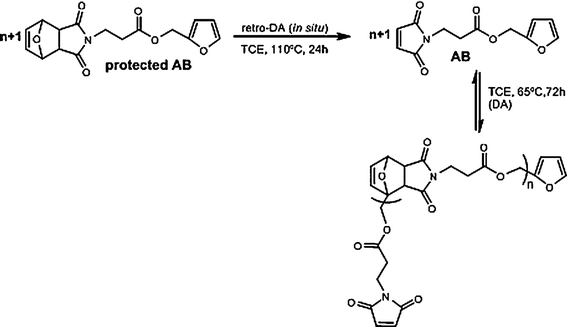 | ||
| Scheme 11 Deprotection and reversible DApolymerisation of an AB monomer.47 | ||
Finally, this ongoing work also includes the synthesis and polymerisation of ABn and AnB monomers (n > 1) and the characterisation of the ensuing hyperbranched macromolecular materials, as well as other macromolecular architectures, namely graft- and comb-shaped copolymers.
Apart from the recent contribution from the Aveiro laboratory summarised above, numerous other studies have been published in the past several years on the reversible coupling of furan and maleimideheterocycles aimed at preparing thermally reversible networks. The strategies vary somewhat, but the overall scenario is essentially the same, viz. to build a cross-linked material, which can be readily reversed to the starting monomers or polymers. In other words, the original ideas developed for this general purpose3 are maintained and only specific issues are in fact modified.
Thus, copolymers bearing furfuryl methacrylate units were crosslinked with a bis-maleimide;48 shape-memory materials prepared thanks to this reversible DA reaction;49 and thermally reversible cross-linked polyamide,50epoxy,51–53 hydrogels54 and biobased polymers,55 including self-healing structures,56 described. It is worth mentioning that these studies focus essentially on the syntheses of monomers and polymers and on the retro-DA applied to the latter, with little or no emphasis on kinetic aspects and materials properties. In the same vein, a star-shaped polymer was reversibly dismembered through the furan/maleimideDA reaction.57 Likewise block dendrimers were joined/disjoined through the same mechanism.58 In a different vein, the retro-DA reaction of the N-phenylmaleimide-FAadduct was studied in different polymer matrices kept in a viscous state in order to assess the role of diffusion limitations on its decoupling.59Multi-walled carbon nanotubes (MWCNTs) have been decorated with both furan and maleimide moieties and the subsequent inter-MWCNTDA reactions studied.60
Miscellaneous systems
Few contributions have appeared in recent years within the realm of chain polymerisations involving furan monomers and none has provided new insight.The synthesis of 5-hydroxymethyl-2-vinylfuran from HMF61 and its free radical polymerisation62 have indicated that this structure is not particularly suited as a monomer.
The reaction of urea with 2,5-diformylfuran was claimed to give a linear polymer , but its limited characterisation and low thermal stability suggest that more work is needed to assess the interest of the system and of the ensuing product.63
Novel furan polyhydrazides and polyoxadiazoles have been reported.64 Their good thermal stability, particularly that of the latter structures, is the salient feature of these materials.
Biodegradability
To the best of my knowledge, furanpolymers have never been the subject of biodegradability studies. The reasons for this serious gap are most probably related to the fact that such materials are not identified as “renewable” like, e.g., cellulose, lignin, and starch and hence not associated with a natural life cycle. The issue therefore deserves a close scrutiny in terms of a structure–property relationship capable of establishing under what configurations (if any) the presence of the furanheterocycle imparts biodegradable features to the polymers that incorporate it.Conclusions
The field of furanpolymers in which major advances have been achieved during the beginning of the new millennium are, on the one hand, novel (intelligent) materials based on the DA reaction or on conjugated structures, and, on the other, a vibrant renewal of interest in polyesters and in the exploitation of furfuryl alcohol in new original directions. The fact that numerous laboratories worldwide are actively engaged in optimising the process of conversion of C6 sugars and/or polysaccharides into hydroxymethylfuraldehyde, must be viewed as a critical cornerstone, because it will open the road to viable ways to prepare a whole host of polycondensates from renewable resources capable of replacing fossil-based conventional and high-tech materials.References
- (a) Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008 Search PubMed; (b) A. Gandini, Macromolecules, 2008, 41, 9491 CrossRef CAS.
- (a) A. Gandini, Adv. Polym. Sci., 1977, 25, 47 CAS; (b) A. Gandini, ACS Symp. Ser., 1990, 433, 195 CAS; (c) A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203 CrossRef CAS; (d) C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS; (e) A. Gandini and M. N. Belgacem, Furan Derivatives and Furan Chemistry at the Service of Macromolecular Materials, in reference 1a, ch. 6 Search PubMed.
- (a) A. Gandini and M. N. Belgacem, ACS Symp. Ser., 2007, 954, 280; (b) S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41 RSC.
- (a) F. W. Lichtenthaler and S. Peters, C. R. Chim., 2004, 7, 65 CrossRef CAS; (b) J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC; (c) M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924 CrossRef CAS; M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 423 CrossRef CAS.
- (a) S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144 CrossRef CAS; (b) R. O'Neill, M. N. Ahmad, L. Vanoye and F. Aiouache, Ind. Eng. Chem. Res., 2009, 48, 4300 CrossRef CAS.
- C. Sievers, I. Musin, T. Marzialetti, M. B. Valenzuels Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2, 665 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS.
- (a) C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2004, 275, 111 CrossRef CAS; (b) M. Bicker, D. Kaiser, L. Ott and H. Vogel, J. Supercrit. Fluids, 2005, 36, 118 CrossRef CAS; (c) Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS; (d) F. S. Asghari and H. Yoshida, Carbohydr. Res., 2006, 341, 2379 CrossRef CAS; F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163 CrossRef CAS; (e) H. Zhao, J. E. Holladay, H. Brown and C. Zhang, Science, 2007, 316, 1597 CrossRef CAS; (f) A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021 CrossRef CAS; (g) S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. LI, J. Song and Y. Xie, Green Chem., 2008, 10, 1280 RSC; (h) G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345 CrossRef CAS; (i) X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Green Chem., 2008, 10, 799 RSC; X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Ind. Eng. Chem. Res., 2008, 47, 9234 CrossRef CAS; (j) S. Hu, Z. Zhang, Y. Zhou, J. Song, H. Fan and B. Han, Green Chem., 2009, 11, 873 RSC; (k) Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297 CrossRef CAS; (l) J. Y. G. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731 CrossRef; (m) C. Li, Z. Zhang, Z. Bao and K. Zhao, Tetrahedron Lett., 2009, 50, 5403 CrossRef CAS; (n) X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Green Chem., 2009, 11, 1327 RSC; (o) Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672 CrossRef CAS; (p) M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859 CrossRef CAS; (q) S. P. Verevkin, V. N. Emel'yanenko, E. N. Stepurko, R. V. Ralys and D. H. Zaitsau, Ind. Eng. Chem. Res., in press, DOI:10.1021/ie901012g; (r) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS; (s) A. Boisen, T. B. Christensen, W. Fu, Y. Y. Gorbanev, T. S. Hansen, J. S. Jensen, S. K. Klitgaard, S. Pedersen, A. Riisager, T. Ståhlberg and J. M. Woodley, Chem. Eng. Res. Des., 2009, 87, 1318 CrossRef CAS; (t) X. Qi, M. Watanabe, T. A. Aida and R. L. Smith Jr., ChemSusChem, 2009, 2, 944 CrossRef CAS.
- J. Lewkowski, ARKIVOC, 2001,(1), 17.
- (a) G. A. Halliday, R. J. Young Jr. and V. V. Grushin, Org. Lett., 2003, 5, 2003 CrossRef CAS; (b) C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197 CrossRef CAS; (c) A. S. Amarasekara, D. Green and E. McMillan, Catal. Commun., 2008, 9, 286 CrossRef CAS; (d) O. Casanova Navarro, A. Corma Canós and S. Iborra Chornet, Top. Catal., 2009, 52, 304 CrossRef.
- (a) E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75 CrossRef CAS; (b) O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839 CrossRef CAS.
- N. Guigo, A. Mija, L. Vincent and N. Sbirrazzuoli, Phys. Chem. Chem. Phys., 2007, 9, 5359 RSC.
- S. Barsberg and L. G. Thygesen, Vib. Spectrosc., 2009, 49, 52 CrossRef CAS.
- S. Bertarione, F. Bonino, F. Cesano, A. Damin, D. Scarano and A. Zecchina, J. Phys. Chem. B, 2008, 112, 2580 CrossRef CAS.
- D. Kawashima, T. Aihara, Y. Kobayashi, T. Kyotani and A. Tomita, Chem. Mater., 2000, 12, 3397 CrossRef CAS.
- J. Yao, H. Wang, J. Liu, K.-Y. Chan, L. Zhang and N. Xu, Carbon, 2005, 43, 1709 CrossRef CAS.
- A. J. G. Zarbin, R. Bertholdo and M. A. F. C. Oliveira, Carbon, 2002, 40, 2413 CrossRef CAS.
- (a) H. Wang and J. Yao, Ind. Eng. Chem. Res., 2006, 45, 6393 CrossRef CAS; (b) B. Yi, R. Rajagopalan, H. C. Foley, U. J. Kim, X. Liu and P. C. Ecklund, J. Am. Chem. Soc., 2006, 128, 11307 CrossRef CAS.
- T. Hirasaki, T. Meguro, T. Wakihara, J. Tatami and K. Komeya, J. Mater. Sci., 2007, 42, 7604 CrossRef CAS.
- F. Cesano, D. Scarano, S. Bertarione, F. Bonino, A. Damin, S. Bordiga, C. Prestipino, C. Lamberti and A. Zecchina, J. Photochem. Photobiol., A, 2008, 196, 143 CrossRef CAS.
- S. Bertarione, F. Bonino, F. Cesano, S. Jain, M. Zanetti, D. Scarano and A. Zecchina, J. Phys. Chem. B, 2009, 113, 10571 CrossRef CAS.
- (a) G. Tondi, A. Pizzi, H. Pasch, A. Celzard and K. Rode, Eur. Polym. J., 2008, 44, 2938 CrossRef CAS; (b) G. Tondi, A. Pizzi, H. Pasch and A. Celzard, Polym. Degrad. Stab., 2008, 93, 968 CrossRef CAS; (c) A. Pizzi, G. Tondi, H. Pasch and A. Celzard, J. Appl. Polym. Sci., 2008, 110, 1451 CrossRef CAS.
- J. Yao and H. Wang, Ind. Eng. Chem. Res., 2007, 46, 6264 CrossRef CAS.
- Y. Zhai, B. Tu and D. Zhao, J. Mater. Chem., 2009, 19, 131 RSC.
- S. Grund, P. Kempe, G. Baumann, A. Seifert and S. Spange, Angew. Chem., Int. Ed., 2007, 46, 628 CrossRef CAS; S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111 CrossRef CAS.
- L. Pranger and R. Tannenbaum, Macromolecules, 2008, 41, 8682 CrossRef CAS.
- S. Lande, M. Westin and M. Schneider, Mol. Cryst. Liq. Cryst., 2008, 484, 367.
- L. Nordstierna, S. Lande, M. Westin, O. Karlsson and I. Furó, Holzforschung, 2008, 62, 709 CrossRef CAS.
- O. Lépine, M. Birot and H. Deleuze, Macromol. Mater. Eng., 2009, 294, 599 CrossRef CAS.
- F. Alakhras and R. Holze, Synth. Met., 2007, 157, 109 CrossRef CAS.
- Y. Miyata, T. Nishinaga and K. Komatsu, J. Org. Chem., 2005, 70, 1147 CrossRef CAS.
- T. Umeyama, T. Takamatsu, N. Tezuka, Y. Matano, Y. Araki, T. Wada, O. Yoshikawa, T. Sagawa, S. Yoshikawa and H. Imahori, J. Phys. Chem. C, 2009, 113, 10798 CrossRef CAS.
- L. C. Baldwin, A. P. Chafin, J. R. Deschamps, S. A. Hawkins, M. E. Wright, D. L. Witker and N. Prokopuk, J. Mater. Sci., 2008, 43, 4182 CrossRef CAS.
- H. Yoneyama, K. Kawabata, A. Tsujimoto and H. Goto, Electrochem. Commun., 2008, 10, 965 CrossRef CAS.
- B. R. Cho, T. H. Kim, K. H. Son, Y. K. Kim, Y. K. Lee and S.-J. Jeon, Macromolecules, 2000, 33, 8167 CrossRef CAS.
- A. Gandini, C. Coutterez, C. Goussé, R. Genheim and S. Waig Fang, ACS Symp. Ser., 2001, 784, 98 and references therein.
- S. Waig Fang, H. J. Timpe and A. Gandini, Polymer, 2002, 43, 3505 CrossRef.
- A. Gandini, S. Ariri and J.-F. Le Nest, Polymer, 2003, 44, 7565 CrossRef CAS.
- P. A. Pearl and J. D. Tovar, Macromolecules, in press, DOI:10.1021/ma9006494.
- A. Gandini, A. J. D. Silvestre, C. Pascoal Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295 CrossRef CAS.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656 RSC.
- A. Gandini, M. Gomes and A. J. D. Silvestre, submitted.
- A. Chaabouni, S. Gharbi, M. Abid, S. Boufi, R. El Gharbi and A. Gandini, J. Soc. Chim. Tunis., 1999, 4, 547 Search PubMed.
- M. Abid, W. Kamoun, R. El Gharbi and A. Fradet, Macromol. Mater. Eng., 2008, 293, 39 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Eur. Polym. J., 2008, 44, 4029 CrossRef CAS.
- A. Gandini, D. Coelho and A. J. D. Silvestre, submitted.
- A. A. Kavitha and N. K. Singha, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4441 CrossRef CAS.
- M. Yamashiro, K. Inoue and M. Iji, Polym. J., 2008, 40, 657 CrossRef CAS.
- Y.-L. Liu, C.-Y. Hsieh and Y.-W. Chen, Polymer, 2006, 47, 2581 CrossRef.
- Y.-L. Liu and C.-Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905 CrossRef CAS.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces., in press, DOI:10.1021/am900104w.
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289 RSC.
- H.-L. Wei, Z. Yang, L.-M. Zheng and Y.-M. Shen, Polymer, 2009, 50, 2836 CrossRef CAS.
- K. Ishida and N. Yoshie, Macromol. Biosci., 2008, 8, 916 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906 CrossRef CAS.
- N. Aumsuwan and M. Urban, Polymer, 2009, 50, 33 CrossRef CAS.
- M. M. Kose, G. Yesilbag and A. Sanyal, Org. Lett., 2008, 10, 2353 CrossRef CAS.
- C. Jegat and N. Mignard, Polym. Bull., 2008, 60, 799 CrossRef CAS.
- C.-M. Chang and Y.-L. Liu, Carbon, 2009, 47, 3041 CrossRef CAS.
- A. Mehner, A. L. Montero, R. Martinez and S. Spange, Molecules, 2007, 12, 634 Search PubMed.
- N. Yoshida, N. Kasuya, N. Haga and K. Fukuda, Polym. J., 2008, 40, 1164 CrossRef CAS.
- A. Afli, S. Abid and R. El Gharbi, e-Polymer, 2008 Search PubMed paper 064.
- A. Amarasekara, D. Greenand and L. D. Williams, Eur. Polym. J., 2009, 45, 595 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |