DOI:
10.1039/B9PY00273A
(Paper)
Polym. Chem., 2010,
1, 634-644
Facile conversion of RAFT polymers into hydroxyl functional polymers: a detailed investigation of variable monomer and RAFT agent combinations†
Received
30th September 2009
, Accepted 7th November 2009
First published on
1st March 2010
Introduction
Reversible addition fragmentation chain transfer polymerization (RAFT) is currently one of the most established techniques applied for controlling radical polymerizations.1–5 RAFT has proven itself to be a flexible method, featuring a broad range of polymerizable monomer systems as well as a high solvent compatibility.2–6 Current examples of the use of RAFT include pathways towards thermo-responsive protein conjugates,7 the synthesis of surface initiated stimuli-responsive diblock co-polymers,8 applications in polymer therapeutics and drug delivery9 as well as the synthesis of surface functional nanomaterials.10
The living nature and control over the molecular weight is achieved by a reversible chain transfer step of the propagating polymer radical onto a dormant polymer chain. Usually a dithioester or di- or trithiocarbonate group is employed as the controlling agent. The RAFT process thus yields polymers bearing the leaving- or R-group of the controlling agent as the initiating chain end and the thiocarbonyl thio- or Z-group as the terminating functional group.6
Although the RAFT process has high industrial potential, the presence of thioester or trithiocarbonate groups in the synthesized polymer is a significant drawback of the technique. These functional groups also lead to a generally coloured material and further problems with potentially toxic or ill-smelling end-group degradation products. Even though the dithioester group is a highly reactive functional group, there is a shortage of chemical transformations in the literature that are of synthetic industrial interest.6,11
These characteristics of the RAFT process are disfavoured in commercial applications and have lead to research into developing an efficient and low cost reaction to eliminate the Z-group or transform it into a more desirable functional group, which enables one to perform further reaction steps with the functionalized polymer. Recently, Perrier and colleagues have shown an elegant approach for the end-group modification of RAFT polymers. The advantage of this procedure is that is allows for the recovering of the chain transfer agent. Their employed procedure requires the polymer to be reacted with a functional azo-initiator after isolation of the polymer.11 In addition, dithioesters and trithiocarbonates may undergo aminolysis12,13 and hydrolysis14 to yield thiol functional polymer. When methacrylates are employed, they can undergo intramolecular cyclization to form thiolactone end-groups. Thiol groups may also be reversibly oxidized to form disulfide-linkages.13,15 3-arm star polymers can be formed, which was recently reported, by a dimethylphenylphosphine catalyzed thiol-ene reaction after aminolysis of the RAFT group.16 In a recent procedure, dithioester capped polymers were aminolyzed in the presence of S-3-butynyl methane thiosulfonate, enabling the use of the generated acetylene end-group in Cu(I) catalyzed cycloaddition reactions.17
Pyrolytic elimination of dithioesters and trithiocarbonates at temperatures above 200 °C yields the vinyl functional polymer.18–20 Other transformation reactions include oxidation by singlet oxygen, hydroperoxides or peroxyacids to form sulfines, rendering the carbonyl sulfur prone to electrophilic attack.21 Photolysis or radiolysis of the RAFT group has also been investigated, including contributions from our group.22–24 However, these degradative transformations have found no perceptible application in synthetic polymer chemistry.
We have recently devised a novel strategy of coupling RAFT polymers with other functional polymers without the need for R-group functionalization. Making use of the ability of the activated thiocarbonyl group to selectively undergo (in some cases Lewis acid catalyzed) hetero Diels–Alder reactions with dienes. This reaction is an atom economical way to obtain well controlled complex structures from a great range of radical or non-radical derived polymer systems under very mild reaction conditions and with extremely rapid reaction times.25–27
In addition, our group has previously reported on the degradation of dithiobenzoate capped poly(methyl methacrylate) in peroxide-containing tetrahydrofuran.28 Surprisingly, it was found that polymer degradation did not proceed towards the formation of sulfines and thioesters, but instead the single product of this degradation reaction appeared to be hydroperoxide functional polymer. It was further shown that for dithiobenzoate capped pMMA and pyridine-2-carbodithioate poly(isobornyl acrylate) (piBoA) the conversion into hydroperoxide- or hydroxyl- functional polymers can easily be achieved in a quantitative fashion.29 The conversion reaction was performed in a simple one-pot procedure, employing an azo-initiator and tetrahydrofuran as solvent and reagent.
In the current contribution we systematically investigate, via ESI-MS, ESI-MS-CID, NMR and SEC, the applicability of this novel synthetic strategy by employing various monomers (tert-butyl acrylate, methyl acrylate, butyl acrylate, styrene, isobornyl acrylate) and RAFT agents (cyanoisopropyl dithiobenzoate, cumylphenyldithioacetate, dibenzyltrithiocarbonate).
Experimental
Materials
Methyl acrylate (99%, Acros Organics), butyl acrylate (99%, Acros Organics), tert-butyl acrylate (99%, Acros Organics), and isobornyl acrylate (technical grade, Sigma Aldrich) were freed from inhibitor by percolating through a column of basic alumina prior to use. The RAFT agents cyanoisopropyl dithiobenzoate (CPDB),30 cumyl phenyldithioacetate31 and dibenzyl trithiocarbonate32,33 were synthesized according to literature and their purity was confirmed by 1H-NMR spectroscopy. 2,2′-Azobis(isobutyronitrile) (98%, Sigma Aldrich) was recrystallized twice from ethanol prior to use. Sodium iodide (puriss. p.a., Fluka), tetrahydrofuran (multisolvent, 250 ppm BHT, Scharlau), triphenylphosphine (Merck) and methanol (chromasolv, Sigma Aldrich) were used as received.
Solutions of cyanoisopropyl dithiobenzoate (CPDB) and 2,2′-azobis(isobutyronitrile) in the monomer (50 mL) were degassed by purging with nitrogen for 15 min. The solutions were heated to 60 °C for variable time intervals, after which the reactions were stopped by cooling in liquid nitrogen. The residual monomers were removed under vacuum and the polymers precipitated in cold methanol. Polymerization times and exact concentrations are collated in Table 1. Molecular weight averages were determined by SEC after precipitation.
Table 1 Reaction conditions for the CPDB mediated polymerizations of various monomers at 60 °C as well as the obtained molecular weights and polydispersities, where c0CPDB is the concentration of the employed RAFT agent, c0AIBN is the initial concentration of AIBN
| Monomer |
c
0
CPDB/mmol L−1 |
c
0
AIBN/mmol L−1 |
t/min |
M
n/g mol−1 |
PDI
|
| MMA |
48.70 |
5.11 |
150 |
3500 |
1.18 |
| MA |
7.85 |
3.41 |
150 |
3100 |
1.09 |
| BA |
7.89 |
3.61 |
120 |
2300 |
1.18 |
|
iBoA |
21.08 |
2.19 |
300 |
3100 |
1.20 |
The same experimental procedure as with CPDB was employed. Polymerization times and exact concentrations are shown in Table 2. Molecular weight averages were determined by SEC after precipitation.
Table 2 Reaction conditions for the CPDA mediated polymerizations of various monomers at 60 °C as well as the obtained molecular weights and polydispersities, where c0CPDA is the concentration of the employed RAFT agent, c0AIBN is the initial concentration of AIBN
| Monomer |
c
0
CPDA/mmol L−1 |
c
0
AIBN/mmol L−1 |
t/min |
M
n/g mol−1 |
PDI
|
| MA |
8.84 |
3.65 |
40 |
3100 |
1.30 |
| BA |
8.80 |
3.74 |
30 |
6300 |
1.23 |
An identical experimental procedure as in the case of the CPDB mediated polymerization was employed. The polymerization times and exact concentrations are collated in Table 3. Molecular weight averages were determined by SEC after precipitation.
Table 3 Reaction conditions for the DBTC mediated polymerizations of various monomers at 60 °C as well as the obtained molecular weights and polydispersities, where c0DBTC is the concentration of the employed RAFT agent, c0AIBN is the initial concentration of AIBN
| Monomer |
c
0
DBTC /mmol L−1 |
c
0
AIBN /mmol L−1 |
t/min |
M
n /g mol−1 |
PDI
|
| Sty |
10.27 |
4.36 |
960 |
20000 |
1.33 |
| MA |
29.52 |
3.65 |
35 |
3500 |
1.32 |
| MA |
17.06 |
5.05 |
45 |
34100 |
1.07 |
| BA |
12.00 |
3.50 |
60 |
1800 |
1.41 |
| BA |
6.10 |
1.83 |
60 |
56800 |
1.12 |
|
tBA |
29.48 |
3.71 |
60 |
1000 |
1.34 |
|
iBoA |
13.77 |
7.31 |
20 |
3600 |
1.30 |
Analytical (small scale) end-group conversion
A solution of 2,2′-azobis(isobutyronitrile) (10 mmol L−1) in THF was heated to 60 °C for 60 min in the presence of ambient air. A solution of the RAFT-polymer (5–10 mmol L−1based on Mn) in 2 mL of the pre-treated THF was prepared in a 4 mL glass vial under ambient atmosphere. The vial was heated to 60 °C. After a discoloration of the solution indicated full conversion, the temperature was reduced to 40 °C and 3 eq. triphenylphosphine were added. The reader is cautioned that although triphenylphosphine acts as a quenching agent, formation of small amounts of potentially explosive THF-peroxides during the reaction is possible and liquid solvent waste should be tested and treated accordingly.
Larger scale end-group conversion
A solution of 2,2′-azobis(isobutyronitrile) (10 mmol L−1) in THF was heated to 60 °C for 60 min under ambient air. A solution of 500 mg RAFT-polymer in the pre-treated THF (10 mmol L−1 based on Mn) was prepared in a 50 mL round flask under ambient atmosphere. The flask was heated to 60 °C under vigorous stirring. After a discoloration of the solution indicated full conversion, the temperature was reduced to 40 °C and 3 equiv. triphenylphosphine were added. After 15 min the polymer was precipitated in cold methanol. Please refer to the note of caution in the previous paragraph; it applies similarly to this procedure.
SEC-ESI-MS
Spectra were recorded on an LXQ mass spectrometer (ThermoFisher Scientific, San Jose, CA, USA) equipped with an atmospheric pressure ionization source operating in the nebulizer assisted electrospray mode. The instrument was calibrated in the m/z range 195–1822 using a standard containing caffeine, Met-Arg-Phe-Ala acetate (MRFA) and a mixture of fluorinated phosphazenes (Ultramark 1621) (all from Aldrich). A constant spray voltage of 4.5 kV and a dimensionless sweep gas flow rate of 2 and a dimensionless sheath gas flow-rate of 12 were applied. The capillary voltage, the tube lens offset voltage and the capillary temperature were set to 60 V, 110 V and 275 °C respectively. The LXQ was coupled to a Series 1200 HPLC-system (Agilent, Santa Clara, CA, USA) consisting of a solvent degasser (G1322A), a binary pump (G1312A), a high-performance autosampler (G1367B), followed by a thermostatted column compartment (G1316A). Separation was performed on two mixed bed size exclusion chromatography columns (Polymer Laboratories, Mesopore 250 × 4.6 mm, particle dia. 3 μm) with pre-column (Mesopore 50 × 4.6 mm) operating at 30 °C. THF at a flow rate of 0.30 mL min−1 was used as the eluent. The mass spectrometer was coupled to the column in parallel to an RI-detector (G1362A with SS420x A/D) in a setup described previously.34 0.27 mL min−1 of the eluent were directed through the RI-detector and 30 μL min−1 infused into the electrospray source after post-column addition of a 100 μM solution of sodium iodide in methanol at 20 μL min−1 by a micro-flow HPLC syringe pump (Teledyne ISCO, Model 100DM). 20 μL of a polymer solution with a concentration of approximately 3 mg mL−1 were injected onto the HPLC system. Measurements can also be conducted via direct infusion ESI-MS. However, preseparation via SEC provides an improved ionization due to the absence of low molecular weight impurities and the slice by slice ionization of the investigated polymers.
Molecular weight analysis via SEC
For the determination of molecular weight distributions (MWD), an SEC system (Polymer Laboratories PL-GPC 50 Plus) comprising an auto injector, a guard column (PLgel Mixed C, 50 × 7.5 mm) followed by three linear columns (PLgel Mixed C, 300 × 7.5 mm, 5 μm bead-size) and a differential refractive index detector was employed. THF was used as the eluent at 40 °C with a flow rate of 1 mL min−1. The SEC system was calibrated using narrow polystyrene standards ranging from 160 to 6 × 106 g mol−1 (Polymer Standard Service). The resulting molecular weight distributions were reassessed by universal calibration using Mark–Houwink parameters for piBoA (K = 5.00 × 10−5 dL g−1, α = 0.745),35 pMA (K = 19.5 × 10−5 dL g−1, α = 0.66),36 pBA (K = 12.2 × 10−5dL g−1, α = 0.70),37 pMMA (K = 7.56 × 10−5dL g−1, α = 0.731)38 and for polystyrene (K = 14.1 × 10−5 dLg−1 and α = 0.70).39 As no Mark–Houwink–Kuhn–Sakurada parameters are available in the case of ptBA, pSty equivalent molecular weights are reported instead.
1H-NMR measurements
1H NMR spectra were recorded in CDCl3 at room temperature on a Bruker Advance 400 NMR spectrometer operating at 400 MHz.
Results and discussion
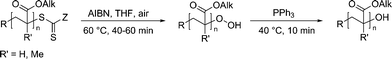
Scheme 1 depicts the general procedure for the conversion of dithioester functional (meth)acrylate polymers: A dilute solution of the RAFT polymer and a radical initiator (2,2′-azobisisobutyronitril, AIBN) in THF is stirred at elevated temperatures (T = 60 °C) in the presence of atmospheric oxygen to yield hydroperoxide functional polymer. Subsequent reduction by triphenylphosphine leads to a quantitative conversion into the hydroxide.
Mechanism of the end-group conversion
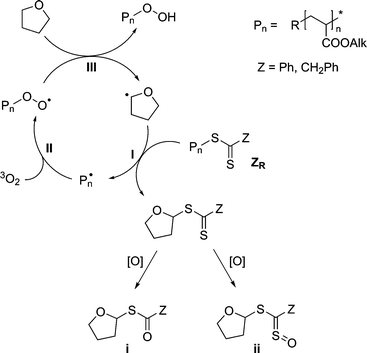
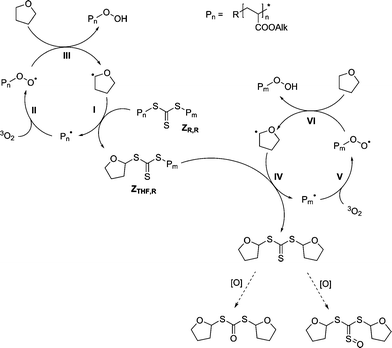
Based on earlier experiments28 and on the results from SEC-ESI-MS analysis of the degraded polymer solutions, a tentative mechanism for the end-group conversion has already been proposed (see Scheme 2).28,29 The mechanism consists of a radical oxidation cycle similar to the radical autoxidation of ethers,40,41 yet comprises an intermediate chain transfer step.
Initially the dithioester functional vinyl polymer ZR is cleaved by a tetrahydrofuranyl radical, yielding the macroradical Pn˙ and the tetrahydrofuranyl dithioester in a degenerative chain transfer step [Scheme 2 (I)]. The formed dithioester is relatively inert against radical attack, because of the low leaving group ability of the tetrahydrofuranyl substituent. It may be further oxidized to the sulfine (ii) or the thioester (i) by molecular oxygen or by hydroperoxide [Scheme 2 (IV)]. As shown in ref. 29, these species were identified during the SEC-ESI-MS analysis of the raw polymer solution after end-group conversion. The polymer radical undergoes irreversible addition to oxygen dissolved in the solution with close to diffusion controlled rates [Scheme 2 (II)].42 Finally, the hydroperoxide functional polymer is expelled by radical chain transfer to THF (III), starting a new oxidation cycle. Initiation of the radical oxidation process is most likely achieved by addition of oxygen onto a cyanoisobutyryl radical and subsequent chain transfer to THF, analogous to steps II and III of the reaction mechanism but with the cyanoisobutyryl radical instead of the macroradical. The proposed conversion mechanism is most likely to work with macroradicals Pn˙ that have a good leaving group ability. The leaving group ability depends on the stability of the formed macroradical Pn˙. The better the macroradical Pn˙ is stabilized, the better is its leaving group ability. Thus, the stability of the macroradical is based on the class of monomer, and therefore only monomer classes that yield stable polymer radicals are most likely to work in the conversion mechanism. Monomers which adhere to this requirement include functional acrylates, methacrylates and styrene. Highly reactive monomers such as vinylacetate are not capable to undergo the conversion into hydroxyl functional polymers using the here applied method because the degenerative chain transfer step yielding the destabilized macroradical Pn˙ [Scheme 2 (I)] is not favourable. Employing a reagent/solvent forming a less stable radical than THF may provide a solution to overcome this limitation.
End-group conversion of dithiobenzoate functional poly(alkyl acrylate)s and poly(alkyl methacrylate)s
The quantitative conversion of dithiobenzoate functional poly(methyl methacrylate) pMMA into hydroxyl functional pMMA was previously demonstrated.29 Here we examine the ability of various poly(alkyl acrylate)s prepared via cyanoisopropyl dithiobenzoate (CPDB) mediated RAFT polymerization to undergo conversion into hydroxyl functional poly(alkyl acrylate)s.
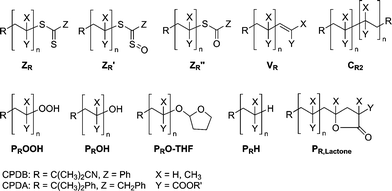
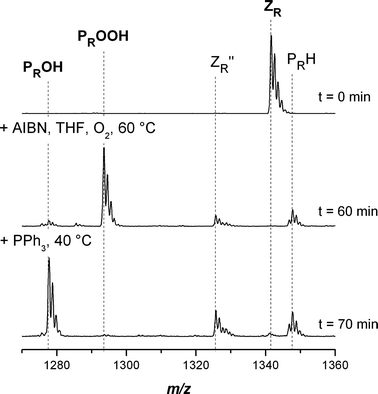
Fig. 1 displays the SEC-ESI-MS spectra of dithiobenzoate functional poly(methyl acrylate) (pMA) before (top) and after conversion into the hydroxyl functional pMA (bottom). Inspection of the figure indicates that the conversion towards the hydroxyl functional pMA involves the formation of some side products. At the start of the reaction (t = 0), the only species present in the spectrum is the dithiobenzoate capped pMA ZR (see Scheme 3 for structural formulas). After 60 min reaction time the dithiobenzoate capped pMA ZR has disappeared. The main product is the hydroxyl functional pMA PROOH, yet some thioester functional polymer ZR′′ can also be identified (for a full overview of the SEC-ESI-MS spectra, see ESI Fig. S1†). It is already known that hydroperoxides are able to oxidize dithiobenzoate functional polymers to sulfine and thioester capped polymers.21,43 A similar result is also obtained in the case of alternative polymers prepared from acrylic monomers, i.e. poly(isobornyl acrylate) (piBoA) and poly(butyl acrylate) (pBA) (see ESI, Fig. S2 and S3). The theoretical m/z ratios of the involved species are collated in Table 4. Within experimental error, excellent agreement between the measured m/z ratios and the theoretical m/z ratios of the expected structures is observed. From the above observations it can be concluded that although CPDB functionalized poly(acrylate)s are adequate precursors for the synthesis of hydroxyl functional poly(acrylate)s, some thioester functional poly(acrylate) ZR′′ side product will always be present during subsequent reaction steps (e.g. ring opening polymerization) that maybe conducted with the transformed polymer. These evolving side products are a drawback compared to the conversion of CPDB functionalized pMMA. Poly(acrylate)s are more susceptible to oxidation compared to pMMA, as was previously shown.29 An explanation can be found in the radical leaving group abilities of the macroradicals associated with different RAFT polymers. As the macroradical Pn˙ derived from pMA is less stable than the macroradical Pn˙ of the pMMA, radical reactions occur slower and hence non-radical reactions play a more important role for poly(acrylate)s.
Table 4 Theoretical and measured m/z ratios of the main species involved in the end-group conversion of pMA carrying a dithiobenzoate end-group into hydroxyl functional pMA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| ZR |
1362.50 |
1362.58 |
0.08 |
| ZR′′ |
1346.52 |
1346.67 |
0.15 |
| PROOH |
1328.55 |
1328.58 |
0.03 |
| PROH |
1312.55 |
1312.75 |
0.20 |
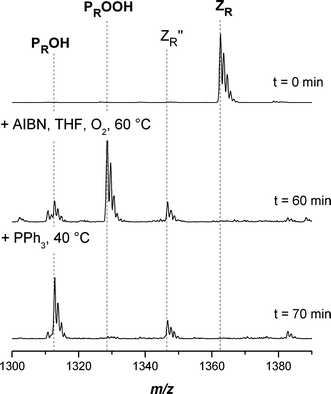 |
| | Fig. 1 Electrospray ionization mass spectra of the end-group transformation of poly(methyl acrylate) carrying a dithiobenzoate end-group into hydroxyl functional pMA in the charge state z = 1. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 60 min. Full conversion was reached after 70 min. The PPh3 acts as the reducing agent from –OOH to –OH. | |
End-group conversion of phenyldithioacetate functional poly(alkyl acrylate)s
As the conversion of dithiobenzoate functional pMA, piBoA and pBA into hydroxyl functional polymer leads to formation of the thioester ZR′′ as a side-product, phenyldithioacetate was employed as an alternative RAFT agent for the preparation of dithioacetate capped pMA and pBA to study the influence of the Z-group on the conversion process. Fig. 2 displays the SEC-ESI-MS spectra of phenyldithioacetate functional pMA before (top) and after conversion to the hydroxyl functional pMA (bottom). Similarly to the observation when using dithiobenzoate functionalized pMA, it can be observed that the conversion is not fully quantitative towards the hydroxyl functional pMA, which also applies to the conversion of pBA. The ESI-MS spectra for the conversion of phenyldithioacetate functional pBA and the theoretical and measured m/z ratios can be found in the ESI (see Fig. S4 and Table T3).† The thioester functional poly(acrylate) ZR′′ (see Scheme 3 for structural formulas) is generated in noticeable amounts. In addition, species corresponding in mass to the hydrogen terminated pMA PRH can be observed. From the chain transfer reaction of a macroradical Pn˙ with a THF molecule, a hydrogen terminated pMA PRH and a THF radical both evolve, which is plausible in light of the reactive nature of acryloyl radical species. Table 5 collates the experimental m/z ratios, which are in very good agreement with the theoretical m/z ratios of the involved species.
Table 5 Theoretical and measured m/z ratios of the main species involved in the end-group conversion of pMA carrying a phenyldithioacetate end-group into hydroxyl functional pMA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| PRH |
1347.67 |
1347.60 |
0.07 |
| ZR |
1341.52 |
1341.58 |
0.06 |
| ZR′′ |
1325.54 |
1325.67 |
0.13 |
| PROOH |
1293.55 |
1293.58 |
0.03 |
| PROH |
1277.55 |
1277.75 |
0.20 |
End-group conversion of symmetrical trithiocarbonate functional poly(alkyl acrylate)s
In an attempt to optimize the conversion procedure, a symmetrical trithiocarbonate was therefore investigated as an alternative end-group for the quantitative conversion of acrylate based RAFT-polymers. For this purpose dibenzyltrithiocarbonate (DBTC) was employed as RAFT agent. Using such a symmetrical trithiocarbonate, the polymer chain is inserted on both sides of the RAFT agent, placing the trithiocarbonate in the middle of the polymer chain. Therefore the mechanism of transformation into hydroxyl functional polymers is slightly different to that proposed for the case when dithiobenzoates and phenyldithioacetates are employed as RAFT agents. As shown in Scheme 4, in the first step the tetrahydrofuranyl radical cleaves the trithiocarbonate capped polymer ZR,R to leave a macroradical Pn˙ and the tetrahydrofuranyltrithio functional polymer ZTHF,R. The tetrahydrofuranyltrithio functional polymer ZTHF,R subsequently reacts with a further tetrahydrofuranyl radical, yielding another macroradical Pn˙ and a ditetrahydrofuranyltrithiocarbonate molecule. The macroradicals Pn˙ undergo irreversible addition to oxygen dissolved in the solution, analogue to the case for the dithiobenzoate and phenyldithioacetate conversion mechanism [Scheme 2 (II)].42 Eventually, the hydroperoxide functional polymer is expelled by radical chain transfer to THF (III), starting a new oxidation cycle.
Upon inspection of Scheme 4, it can be observed that the Mn of the polymer after the conversion should be half the Mn of the polymer before conversion into a hydroxyl functional polymer. To assess this theoretical expectation, SEC traces before and after the conversion sequence were recorded for variable polymers carrying a trithiocarbonate function in the mid-chain position.
In Fig. 3 the SEC traces of the trithiocarbonate functional and the degraded polymers are depicted. To map the progress of the reaction, samples of the reaction mixture were taken at preset time intervals. It can be observed that Mn decreases with time, congruent with the proposed conversion mechanism. Table 6 depicts the different polymer classes, molecular weights of the polymers before and after the conversion and also the ratios of the number average molecular weights MnRAFT/MnOH,exp. The ratio MnRAFT/MnOH,exp is close to two for pMA and pBA. This observation underpins the proposed conversion mechanism in Scheme 4. The ratios MnRAFT/MnOH,exp for pSty and piBoA are not as close to two as for pMA and pBA. The derivation may be explained by side reactions, such as recombination of macroradicals Pn˙ or oxidation of the trithiocarbonate capped polymer ZR,R to the dithiocarbonate capped polymer ZR,R′′ (see Scheme 5 for structural formulas).
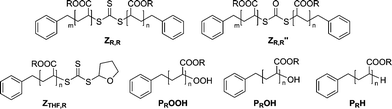 |
| | Scheme 5 Possible species involved in the transformation of various poly(acrylates) synthesized with DBTC as RAFT agent into hydroxyl functional polymers. | |
Table 6 Number average molecular weights, Mn, and polydispersities, PDI, for variable polymers with a trithiocarbonate moiety in the middle of the chain before and after conversion into hydroxy functional polymers
| |
pMA |
pBA |
pSty |
piBoA |
|
M
n
RAFT /g mol−1 |
34000 |
56800 |
19900 |
23300 |
|
M
n
OH,theo/g mol−1 |
17000 |
28400 |
9950 |
11650 |
|
M
n
OH,exp/g mol−1 |
17600 |
28900 |
11000 |
13300 |
|
M
n
RAFT/MnOH,exp |
1.93 |
1.96 |
1.81 |
1.75 |
|
PDI
RAFT
|
1.07 |
1.12 |
1.30 |
1.47 |
|
PDI
OH
|
1.11 |
1.20 |
1.31 |
1.80 |
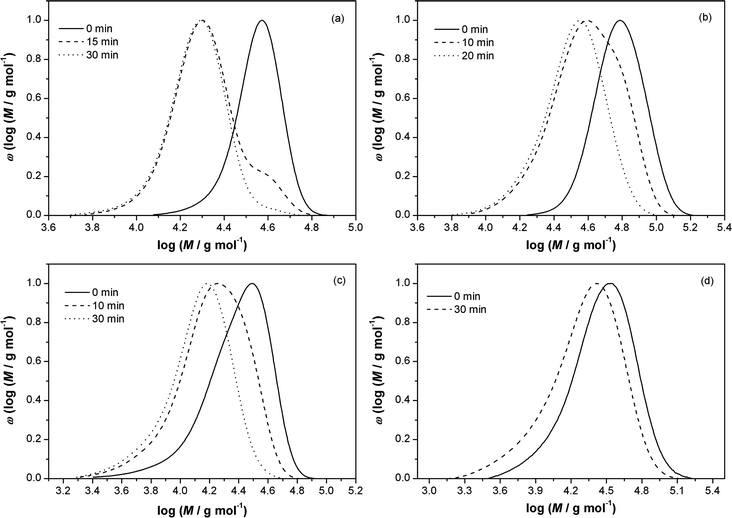 |
| | Fig. 3 SEC traces of variable polymers with a trithiocarbonate moiety in the middle of the chain after reaction with AIBN/THF as a function of time. (a) pMA, (b) pBA, (c) pSty, (d) piBoA. Samples were taken at preset time intervals and precipitated in methanol. | |
For further investigations into the mechanism an SEC-ESI-MS analysis was performed on the polymers. Employing SEC/ESI-MS, polymers of a molecular weight of up to 15 kDa (up to 4 kDa in the charge state z = 1) can be investigated with quadrupole ion trap mass spectrometry.34,44 To achieve high resolution spectra, lower molecular weight polymers were synthesized to study their conversion into hydroxyl capped entities. Ionization of non-polar polymers such as polystyrene is achieved only with some difficulty by soft ionization methods such as MALDI or ESI-MS.45 Although there is an existing method available for the ionization of dithioester capped polystyrene, only very low ion counts were obtained disallowing a meaningful spectral data interpretation.
Fig. 4 displays the SEC/ESI-MS spectra of the symmetrical trithiocarbonate functional pMA before and after conversion into the hydroxyl functional pMA recorded at variable reaction times. It can be seen that after the conversion reaction, there are nearly quantitative amounts of the hydroxyl functional pMA PROH present. There are only traces of hydrogen terminated PRH present in the spectrum.
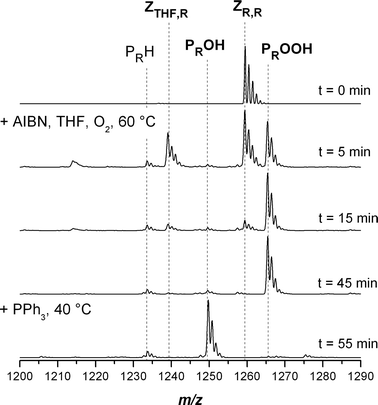 |
| | Fig. 4 Electrospray ionization mass spectra of the transformation of poly(methyl acrylate) with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional pMA in the charge state z = 1. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 45 min. Full conversion was reached after 55 min. | |
The state of the reaction can conveniently be monitored by SEC/ESI-MS. After five minutes reaction time the intermediate tetrahydrofuranyl functional polymer ZTHF,R can be observed. Ten minutes later, ZTHF,R is only present in trace amounts. The intermediate appearance of ZTHF,R further supports the proposed mechanism depicted in Scheme 4.
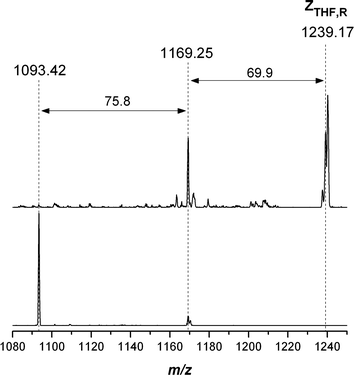
Table 7 displays the experimental m/z ratios, which are in good agreement with the theoretical m/z ratios of the involved species. However, the difference between the theoretical and measured m/z of the species ZTHF,R was somewhat enhanced compared to the other species. A great advantage of quadrupole ion trap instrumentation is the ability to perform multi-stage dissociation experiments, yielding valuable information about the chemical structure of unknown species in the mass spectrum. We therefore performed collision induced dissociation experiments on the putative species with m/zmeas = 1239.1 to obtain proof of the intermediate product structure. As can be observed in Fig. 5, a neutral loss of m/zmeas = 69.9 corresponding to a loss of dihydrofuran (m/ztheo = 70.0) in the first dissociation step (Scheme 6), followed by a loss of m/zmeas = 75.8 corresponding to CS2 (m/ztheo = 76.0) in the second dissociation step proves the species to be the THF trithioester-capped polymer ZTHF,R.
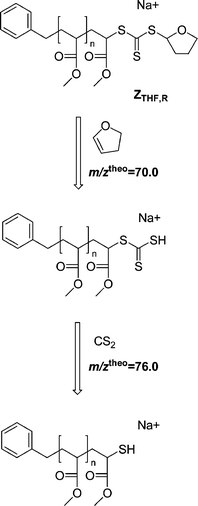 |
| | Scheme 6 Dissociation pathway and theoretical masses of the neutral losses during the collision induced dissociation of the intermediate THF-trithiocarbonate carrying poly(methyl acrylate) ZTHF,R. MS2 of ZTHF,R leads to loss of dihydrofuran (m/ztheo = 70.0, m/zmeas = 69.9), MS3 of the product species leads to loss of a CS2 molecule (m/ztheo = 76.0, m/zmeas = 75.8) confirming the structure of the intermediate. | |
Table 7 Theoretical and measured m/z ratios of the main species involved in the conversion of pMA with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional pMA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| ZR,R |
1259.42 |
1259.50 |
0.08 |
| ZTHF,R |
1239.42 |
1239.08 |
0.34 |
| PROOH |
1265.52 |
1265.42 |
0.10 |
| PROH |
1249.53 |
1249.67 |
0.14 |
| PRH |
1233.53 |
1233.67 |
0.14 |
In Fig. 6 the 1H-NMR spectrum before the transformation reaction (t = 0 min) of the symmetrical trithiocarbonate functional pMA and at full conversion (t = 55 min) is depicted. Disappearance of peak (a) at 4.7 ppm, corresponding to the proton in geminal position to the trithioester sulfur indicates the complete replacement of the trithiocarbonate group.
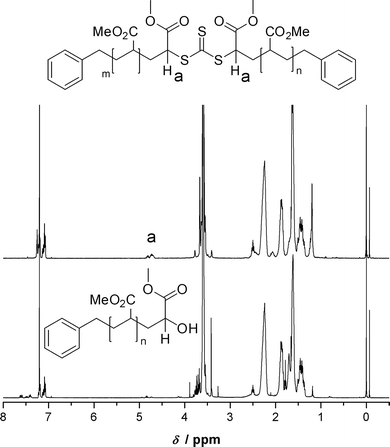 |
| | Fig. 6 (Top) 1H-NMR spectrum of poly(methyl acrylate) with a trithiocarbonate moiety in the middle of the chain. (Bottom) Spectrum of the same polymer after 70 min reaction time at the end of the transformation reaction. Disappearance of peak (a) corresponding to the proton Ha in geminal position to the trithioester sulfur indicates complete conversion of the RAFT endgroup. | |
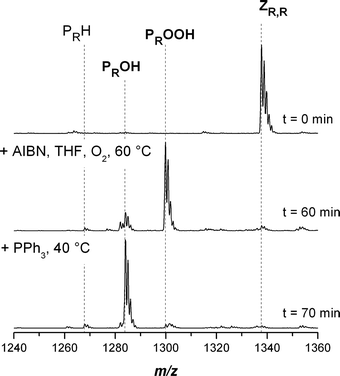
Fig. 7 displays the SEC-ESI-MS spectra of symmetrical trithiocarbonate functional pBA before (top) and after conversion to the hydroxyl functional pBA (bottom). It can be seen that the conversion is nearly quantitative towards the hydroxyl functional pBA PROH and only traces of hydrogen terminated PRH are present in the spectrum. Table 8 displays the experimental m/z ratios, which are in very good agreement with the theoretical m/z ratios of the involved species.
Table 8 Theoretical and measured m/z ratios of the main species involved in the conversion of pBA with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional pBA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| ZR,R |
1337.69 |
1337.75 |
0.06 |
| PROOH |
1299.83 |
1299.80 |
0.03 |
| PROH |
1283.80 |
1284.00 |
0.20 |
| PRH |
1267.81 |
1268.00 |
0.19 |
 |
| | Fig. 7 Electrospray ionization mass spectra of the transformation of poly(butyl acrylate) with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional pBA in the charge state z = 1. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 60 min. Full conversion was reached after 70 min. | |
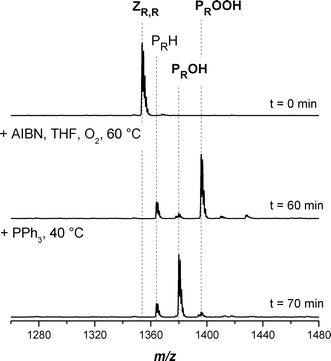
Fig. 8 depicts the SEC-ESI-MS spectra of symmetrical trithiocarbonate functional piBoA before (top) and after conversion to the hydroxyl functional piBoA (bottom). One can conclude from Fig. 8 that there is also an almost quantitative conversion towards the hydroxyl functional piBoA. The symmetrical trithiocarbonate functionalized piBoA after conversion has a slightly higher content of hydrogen terminated PRH than the pMA and pBA. This could be due to saturation of the macroradical Pn˙ with a THF molecule. Table 9 displays the experimental m/z ratios, which are in good agreement with the theoretical m/z ratios of the involved species.
Table 9 Theoretical and measured m/z ratios of the main species involved in the conversion of piBoA with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional piBoA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| ZR,R |
1353.75 |
1353.75 |
0.00 |
| PRH |
1363.93 |
1363.92 |
0.01 |
| PROOH |
1187.78 |
1187.75 |
0.03 |
| PROH |
1171.78 |
1171.75 |
0.03 |
 |
| | Fig. 8 Electrospray ionization mass spectra of the transformation of poly(isobornyl acrylate) with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional piBoA in the charge state z = 1. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 60 min. Full conversion was reached after 70 min. | |
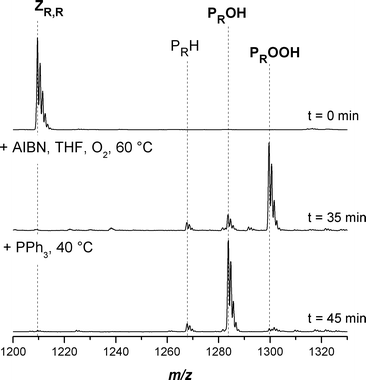
Water soluble polymers, such as poly(acrylic acid) pAA, are interesting due to their potential use in e.g. the construction of nano-containers.46 Thus, it seemed important to investigate the suitability of pAA precursor polymers such ptBA for the conversion process, as ptBA can readily be transformed via hydrolysis into pAA under mild conditions. Concerning the symmetrical trithiocarbonate functional ptBA, it can be seen in Fig. 9 that the conversion is almost quantitative and there is only a small amount of hydrogen terminated PRH evolving during the reaction. Table 10 displays the experimental m/z ratios, which are in excellent agreement with the theoretical m/z ratios of the concerned species. The conversion process does not affect the tert-butyl group.
 |
| | Fig. 9 Electrospray ionization mass spectra of the transformation of poly(tert-butyl acrylate) with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional ptBA in the charge state z = 1. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 35 min. Full conversion was reached after 45 min. | |
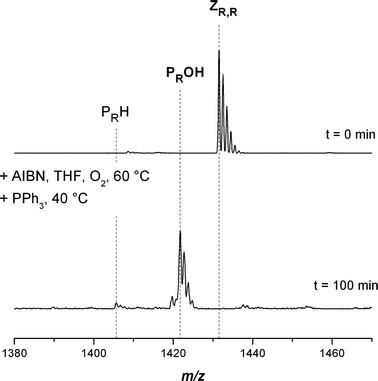
To enable the application of the hydroxyl functional polymer in further reaction steps the conversion procedure was carried out on a larger scale using around ten times more material (500 mg) of the trithiocarbonate capped pMA. The polymer was isolated by precipitation and subsequently investigated by SEC-ESI-MS. It can be elucidated from Fig. 10 that the conversion reaction is also almost quantitative towards the hydroxyl functional pMA, as shown before in Fig. 4. It was observed that the only difference between performing the reaction on a small and larger scale was the longer reaction time (by a factor of approx. 1.5) required for the large scale process. The dissolved oxygen in the reaction mixture is depleted more rapidly than it is replaced by oxygen from the atmosphere. The results additionally confirm the introduced method as a useful strategy for converting trithiocarbonate functional polymers into hydroxyl functional polymers.
 |
| | Fig. 10 Electrospray ionization mass spectra of the preparative transformation of poly(methyl acrylate) with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional pMA in the charge state z = 1. DBTC was used as RAFT agent. The reagents AIBN/THF and PPh3 were added sequentially at t = 0 and 90 min. Full conversion was reached after 100 min. The transformation was carried out with 500 mg pMA. The resulting polymer was precipitated in methanol. | |
Table 10 Theoretical and measured m/z ratios of the main species involved in the conversion of ptBA with a trithiocarbonate moiety in the middle of the chain into hydroxyl functional ptBA
| Structure |
|
[M + Na]+ |
|
|
m/ztheo |
m/zexp |
Δm/z |
| ZR,R |
1209.60 |
1209.50 |
0.10 |
| PRH |
1267.81 |
1267.75 |
0.06 |
| PROH |
1283.80 |
1283.75 |
0.05 |
| PROOH |
1299.80 |
1299.67 |
0.13 |
Conclusions
It has been demonstrated that transformation reactions of trithiocarbonate functional acrylate polymers into hydroperoxide and hydroxyl functional polymers proceeds with a very high efficiency. The amount of side products is negligible and the conversion is almost quantitative. A mechanism for the transformation is proposed which is backed by mass spectrometric evidence. A further advantage of symmetrical trithiocarbonates lies in the fact that they can be synthesized in one pot reactions and are mostly easily purified.32,33 We additionally demonstrate that the presented one-pot conversion method is applicable to a number of different monomer systems and can also be carried out on a larger scale. The transformation reaction can also be performed with dithiobenzoate and phenyldithioacetate capped acrylate polymers, however there is a small amount of side products evolving. The above described one-pot procedure is simple and versatile, as only low cost solvents and chemicals are utilized. Having access to a stabile and versatile –OH functional group allows for the first time the direct use of RAFT polymers in certain end-group conjugation and block-co-polymer formation reactions: ring-opening polymerization (ROP) with ethylene oxide and lactones or, after tosylation, cationic ROP of oxazolines for the formation of well defined block co-polymers are imaginable. We are currently evaluating the various synthetic possibilities in our laboratories.
Acknowledgements
C.B.-K. acknowledges financial support from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative for leading German universities as well as the German Research Council (DFG) and the Ministry of Science and Arts of the state of Baden-Württemberg. C.S. acknowledges additional financial support from the Federal Institute for Materials Research and Testing (BAM). M.D. thanks the Fraunhofer Institute of Chemical Technology for financial support.
References and Notes
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28(5), 539–559 CrossRef CAS.
-
C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, 1st edn, 2008 Search PubMed.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(17), 5715–5723 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27(9), 653–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58(6), 379–410 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130(34), 11288–11289 CrossRef CAS.
- M. D. Rowe, B. A. G. Hammer and S. G. Boyes, Macromolecules, 2008, 41(12), 4147–4157 CrossRef CAS.
- Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef CAS.
- S. R. S. Ting, A. M. Gregory and M. H. Stenzel, Biomacromolecules, 2009, 10(2), 342–352 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38(6), 2033–2036 CrossRef CAS.
- A. Previero, A. Gourdol, J. Derancourt and M.-A. Coletti-Previero, FEBS Lett., 1975, 51(1–2), 68–72 CrossRef CAS.
- Y.-Z. You, D. S. Manickam, Q.-H. Zhou and D. Oupický, Biomacromolecules, 2007, 8, 2038–2044 CrossRef CAS.
- N. Malic and R. A. Evans, Aust. J. Chem., 2006, 59, 763–771 CrossRef CAS.
- Y. K. Goh, A. K. Whittaker and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(17), 4150–4153 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(12), 3118–3130 CrossRef CAS.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- B. Chong, G. Moad, E. Rizzardo, M. A. Skidmore and S. H. Thang, Macromolecules, 2007, 40, 9262–9271 CrossRef.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38(13), 5371–5374 CrossRef CAS.
- P. Metzner, Pure Appl. Chem., 1996, 68(4), 863–868 CAS.
- T. M. Lovestead, G. Hart-Smith, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 4142–4153 CrossRef CAS.
- L. Lu, H. Zhanh, N. Yang and Y. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41(12), 4120–4126 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008,(17), 2052–2054 RSC.
- A. J. Inglis, S. Sinnwell, M. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- T. Gruendling, R. Pickford, M. Guilhaus and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(22), 7447–7461 CrossRef CAS.
- T. Gruendling, M. Dietrich and C. Barner-Kowollik, Aust. J. Chem., 2009, 62, 806–812 CrossRef CAS.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300 CrossRef CAS.
- C. Barner-Kowollik, J. F. Quinn, T. L. Uyen Nguyen, J. P. A. Heuts and T. P. Davis, Macromolecules, 2001, 34, 7849 CrossRef CAS.
- B. Aoyagi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3702 CrossRef.
- A. M. Bivigou-Koumba, J. Kristen, A. Laschewsky, P. Mueller-Buschbaum and C. M. Papadakis, Macromol. Chem. Phys., 2009, 210, 565 CrossRef CAS.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80(18), 6915–6927 CrossRef CAS.
- B. Dervaux, T. Junkers, M. Schneider-Baumann, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci. A Polym. Chem., 2009, 47(23), 6641–6654 CrossRef CAS.
- M. Buback, C. H. Kurz and C. Schmaltz, Macromol. Chem. Phys., 1998, 199, 1721 CrossRef CAS.
- S. Beuermann, D. A. Paquet and J. H. McMinn, Macromolecules, 1996, 29, 4206 CrossRef CAS.
- Y.-J. Chen, J. Li, N. Hadjichristidis and J. W. Mays, Polym. Bull., 1993, 30, 575 CrossRef CAS.
- C. Strazielle, H. Benoit and O. Vogl, Eur. Polym. J., 1978, 14, 331 CrossRef CAS.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1969, 47(20), 3809–3815 CAS.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1970, 48(6), 873–880 CrossRef CAS.
- B. Maillard, K. U. Ingold and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 5095–5099 CrossRef CAS.
- P. Vana, L. Albertin, L. Barner, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4032–4037 CrossRef CAS.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Macromolecules, 2009, 42(17), 6366–6374 CrossRef CAS.
- T. Gruendling, G. Hart-Smith, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41(6), 1966–1971 CrossRef CAS.
- L. Serra, J. Doménech and N. A. Peppas, Eur. J. Pharm. Biopharm., 2009, 71(3), 519–528 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.