Selective surface functionalization of polystyrene by inner-shell monochromatic irradiation and oxygen exposure
Daniel Eduardo
Weibel
*a,
Felipe
Kessler
a and
Gunar Vingre
da Silva Mota
b
aDepartamento de Físico-Química, IQ, UFRGS, Porto Alegre, RS, Brazil. E-mail: danielw@iq.ufrgs.br; Fax: + 55-5133087304; Tel: + 55-5133086204
bCurso de Física, Universidade Federal do Amapá, Macapá, AP, Brazil. E-mail: gun@unifap.br
First published on 8th February 2010
Abstract
The surfaces of polystyrene (PS) thin films were selectively functionalized by a combination of monochromatic synchrotron radiation with oxygen exposure. By selecting specific K-shell excitation energies, it was shown that highly efficient surface functionalization can be obtained. After the treatments, several oxygen functionalities were detected by XPS and NEXAFS spectroscopy. The functionalization strongly depended on the excitation energy. C 1s → σ*C–C transitions produced an extremely efficient oxidation of the uppermost monolayer of the PS films. The COO and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O elemental contributions in the C 1s envelope at the PS surface was higher than 70%. On the contrary, other excitation energies did not introduce COO functionalities efficiently. The obtained results were explained by taking into account the known Auger stimulated desorption mechanism. The present research will open new ways to functionalize a polymer surface by a combination of monochromatic inner-shell excitation together with exposure to a reactive gas atmosphere.
O elemental contributions in the C 1s envelope at the PS surface was higher than 70%. On the contrary, other excitation energies did not introduce COO functionalities efficiently. The obtained results were explained by taking into account the known Auger stimulated desorption mechanism. The present research will open new ways to functionalize a polymer surface by a combination of monochromatic inner-shell excitation together with exposure to a reactive gas atmosphere.
Introduction
Polymer interfaces are critical to the function of different materials, since the interfacial region either controls or plays a major role in determining several properties, such as flexural strength, durability, hydrophilicity or hydrophobicity and biocompatibility. Over the past few decades there has been an increasing demand for polymeric materials with controllable bulk and specific surface properties. For many applications, maintaining the bulk polymer properties while modifying the surface properties with respect to the pristine polymer is required. Several surface treatment techniques have been used in the past 30 years for polymer surface modification, such as: corona-discharge,1 plasma,2,3 plasma polymerization,4,5 graft co-polymerization,6 and ultraviolet (UV)/ozone treatment.7 Among these techniques, plasma treatments have became a common process for many industrial, medical and bioscience applications. However, traditional surface treatments based on discharge and plasma processes lack the ability to improve surface properties locally and selectively.With the field of nanotechnology in continuous expansion, new production methods combining micro-nano fabrication with surface selective functionalization will be needed. In recent years, processing of different materials by ultraviolet (UV) and vacuum-UV (VUV) radiation has received much attention largely due to the development of excimer lasers and VUV lamps based on dielectric barrier discharges.8,9 For instance, several polymers were modified by UV treatment in the presence of alkylsilanes, introducing Si–H groups onto the polymer surface, also providing new possibilities for further surface functionalization.10 Very important effects in the surface hydrophilicity changes of polysulfones and polyethersulfones have also been recently reported in films exposed to UV irradiation.11 N-rich organic surfaces for cell adhesion have been obtained by VUV functionalization and assisted polymerization of a hydrocarbon–ammonia mixture.8 López-Gelo et al. showed the differences between typical ozone oxidation and VUV photochemical treatments in the VUV modification of polystyrene (PS) surfaces in the presence of oxygen.12 Ozonization of the PS led to oxidative functionalization with a much smaller rate than with VUV irradiation. Oxidative fragmentation, by backbone dissociation, was only initiated by electronic excitation of the polymer leading to C–C bond breaking and a complex oxidation process. More interestingly, a few reports using “monochromatic” VUV radiation showed superior performance than its plasma counterpart.8,13 In particular, it was shown that VUV photo-polymerization can be obtained by irradiation of the inorganic monomer (C2H4) and ammonia mixtures at reduced pressures.13 The authors claimed that VUV photochemistry is superior to plasma chemistry in producing almost monofunctional organic thin films.
Considering all the light sources available at the moment, synchrotron radiation (SR) is by far the most useful if a wide range of excitation energy is needed, along with high intensity, monochromaticity and tunability. The high energy resolution of SR allows selective polymer bond breaking.14,15 Remarkably, a few reports have appeared in the literature on polymer surface functionalization by SR.16–18 In a series of interesting experiments on PTFE,17,18 the surface wettability was modified with unmonochromatic SR. Depending on the substrate temperature, the treated film could be hydrophilic, hydrophobic or even super hydrophilic. Nevertheless, including the above studies, there are no reports, to our knowledge, on selective surface functionalization using monochromatic SR.
Recently, we have carried out studies on the surface functionalization of polyurethane by low-pressure plasma treatment19,20 and UV-assisted surface modification.21 Those results showed the importance of trying to selectively control the functionality bonded to the polymer surface. Therefore, the aim of the present work was to investigate the hypothesis that by using highly monochromatic radiation, a selective functionalization of the polymer surface can be achieved. A simple polymer such as polystyrene (PS) was chosen because new functional groups containing oxygen, for example, would be easy detected by surface sensitive techniques. By using oxygen as the reactive atmosphere, we were able to find experimental conditions to selectively functionalize the polymer surface, introducing high concentrations of COO, CO and C–O groups at the uppermost monolayer of the films.
Experimental
Thin PS films (∼100 nm thickness) were prepared by the spin-coating technique from a ∼10−4 M chloroform solution to avoid charging problems.22 The films were cast on ∼10 × 10 mm polished stainless steel substrates. Film thicknesses were measured by a profilometer Ambios XP-2 with a 2.5 micron radius stylus. Oxygen, 99.99% from White Martins PRAXAIR INC, was used as received.SR experiments were carried out at the Brazilian Synchrotron Light Source (LNLS), Campinas, Brazil. The SGM (Spherical Grating Monochromator) beam line, for VUV and soft X-ray spectroscopy (250–1000 eV), which gives a spectral resolution (E/ΔE) better than 2000, was used as the monochromatic photon source. The experimental set up includes a XYZ sample manipulator housed in an ultra high vacuum (UHV) chamber with a base pressure of 10−7 Pa. The stainless steel substrates were directly attached to the sample holder by using conducting double sided tape and the SR beam was defocussed to ∼5 mm in diameter. No sample charging was observed throughout the experiments.
X-Ray photoelectron spectroscopy (XPS) data were obtained by using a hemispherical one-channel analyzer (Perkin Elmer 10-360) with a pass energy of 58.7 eV (Ee = 700 eV), whereas high-resolution (HR)-XPS C 1s spectra were recorded with a pass energy (Ee = 380 eV) of 23.5 eV. The full width at half maximum (FWHM) measured on a gold substrate (4 f7/2 transition) was 2.1 eV and 0.9 eV for survey and HR spectra respectively. The C 1s signal (position of the C–C C−1–H signals), 285 eV, was used for XPS energy calibration. The C 1s envelope was analyzed and peak-fitted after subtraction of the Shirley background using Gaussian-Lorenzian peak shapes obtained from the CasaXPS software package.
Near-edge X-ray absorption fine structure (NEXAFS) spectra were obtained by measuring the total electron yield (electron current at the sample) simultaneously with a photon flux monitor (Au grid). The final data were normalized by this flux spectrum to correct for fluctuations in beam intensity. NEXAFS spectra fitting was carried out using the BGAUSS multiline fitting program (BGAUSS-Version 3.05., 1994, T. Tyliszczak, Tolmar Instrument, Hamilton, Ontario, Canada) following the procedure already used for PS films.23,24 Spectra were analyzed by first subtracting the pre-edge linear baseline. Height, position and width of the Gaussian and error functions were all allowed to be varied (within limits) in the miniminization algorithm. The initial positions of the error functions were as follows:24 for the carbon edge the error function was 290.0 eV and for the oxygen edge the error functions were set at 535.0 and 545.0 eV.
When SR was used to functionalize the polymer surface, specific transitions were selected and during a fixed period of time of 15 min the polymer was irradiated under UHV conditions. After irradiation, pure oxygen at atmospheric pressure was introduced into the UHV chamber for 30 min to neutralize the remaining radicals on the PS surface. This treatment procedure was used for all the samples. Considering the collision frequency at a base pressure of 10−7 Pa and a sticking coefficient of unity, a monolayer is formed in ∼104 s. As a consequence, the radicals, photogenerated at the surface during SR irradiation, remained until oxygen was introduced into the UHV chamber. Finally, XPS and NEXAFS spectra were obtained before and after irradiation. The treated samples were analyzed after three hours of the process.
Results and discussion
Fig. 1 shows the NEXAFS carbon K-edge spectrum of pristine PS before irradiation. Three excitation energies were selected for the present study: C 1s → π*CC (285.2 eV and 287.2 eV) and C 1s → σ*C–C transitions (293.5 eV).25
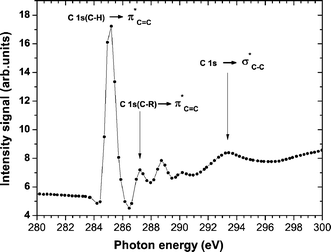 | ||
| Fig. 1 The carbon K-edge NEXAFS spectrum of untreated poly(styrene) (PS) showing the main transitions selected in the present work for selective surface functionalization. | ||
XPS was one of the surface sensitive techniques used to prove the surface chemistry changes. Survey XPS spectra of irradiated PS in UHV conditions did not have an O 1s signal. When oxygen gas was allowed to fill the UHV chamber after irradiation, the O 1s signal appeared in the XPS spectra indicating incorporation of oxygen on the surface (results not shown). The incorporation of oxygen was particularly efficient when the excitation energy was tuned to 293.5 eV, where it accounted for approximately 7.7% of the elemental contribution on the surface. This oxygen concentration is close to the plateau of 7% atomic oxygen concentration already found in the systematic oxidation of PS by UV-ozone treatment.24 Although the elemental oxygen concentration obtained in our work is similar to the above plasma results, the nature of the oxidation process is very different, as will be shown later in the paper.
High resolution (HR)-XPS spectra revealed a dramatic change in the surface chemistry of the PS films when the excitation energy was tuned to different transitions. Fig. 2 shows that while excitations from C 1s to π*CC mainly led to the introduction of C–O functionalities, excitation to a σ*C–C transition produced very efficient functionalization of the PS surface. The signal intensities corresponding to COO and CO groups were comparable to the aliphatic and aromatic contributions (see Fig. 2, bottom). Pristine XPS spectra of PS films were obtained by moving the sample up and down by ∼5 millimetres, indicating that the functionalization only took place in the irradiated area. The nature of the functionalization process, which was obtained in our approach by SR, is completely different from previous PS plasma treatment studies.26,27 In plasma experiments, the maximum total carbon-oxygen functionalities grafted on the surface, in so-called “saturated” conditions, was lower than 20%.27 The results obtained using selective SR/oxygen functionalization at 293.5 eV were higher than 50%, indicating that another functionalization mechanism had taken place. The difference is even larger for highly electronegative functionalities containing COO groups. With plasma treatment, the content of this electronegative contribution reached about 6%, while by using SR functionalization we obtained more than 40%.
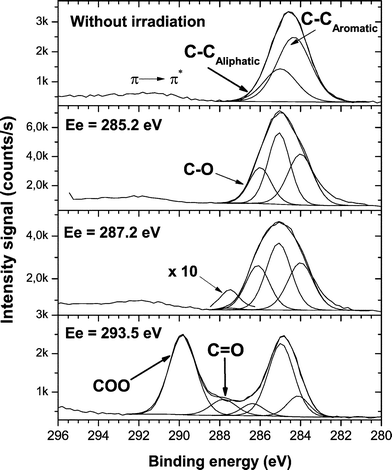 | ||
| Fig. 2 High-resolution XPS spectra of the C 1s envelope of PS without treatment and after SR functionalization at the selected excitation energies shown in Fig. 1. | ||
Fig. 2 shows a weak signal contribution, especially at an excitation energy of 285.2 eV, on the low energy side of the C 1s envelope. The determination of carbon sp2/sp3 ratios in similar polymers, such as polystyrene/polyethylene copolymers by XPS has been evaluated in the past.28 It was shown that the C 1s peak exhibited no useful variation in shape as the sp2 concentration was varied. Therefore, if new sp2 functionalities are formed by SR excitation it is difficult to assign them within a main C 1s peak. The films were ∼100 nm thick and the SR was defocused to ∼5 mm in diameter. Under these experimental conditions, charging problems were ruled out. Therefore, to rationalize the changes in the C 1s peak as a function of the excitation energy only the main aromatic and aliphatic contributions were taken into account.
Angle resolved XPS measurements revealed that the functionalization of the PS films occurred on uppermost monolayer of the polymer surface. Fig. 3 shows HR-XPS results obtained for electron emission angles of 45 and 75° (the EEA is the angle with respect to the surface normal). The COO plus CO contributions in the C 1s envelope increased from 54% to 73% when the EEA increased from 45 to 75° (Fig. 3). In addition, Fig. 4 shows the dependence of the different functionalities fitted in the C 1s envelope on the EEA. When the EEA was ∼85°, the contribution of the electronegative groups having oxygen is more than 70%. Those results showed that a selective functionalization of the PS surface occurred on the uppermost monolayer of the polymer surface.
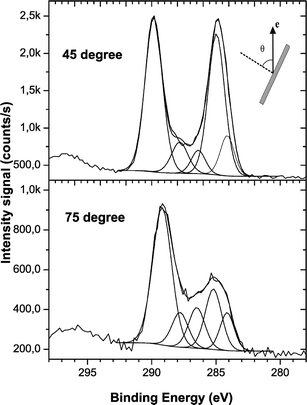 | ||
| Fig. 3 The evolution of the intensity of the components in the C1s envelope as a function of the electron emission angle (θ). θ is the angle with respect to the surface normal. | ||
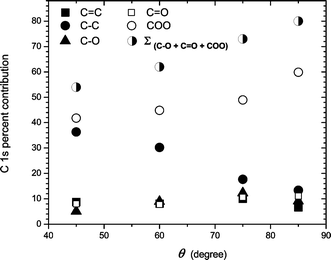 | ||
| Fig. 4 Angle-resolved XPS data for PS SR treated films at an excitation energy of 293.5 eV. Data obtained from C 1s HR-XPS spectra. | ||
The C 1s → σ*C–C transition can involve primary excitation to the aromatic or aliphatic part of the polymer. After the treatments, the functionalization mainly took place in the aromatic part of the PS (Fig. 2). The C–Caromatic contribution in the C 1s envelope decreased compared to the C–Caliphatic contribution. Those results indicated a highly selective bond breaking process, which cannot be obtained with classical surface treatment techniques.
Core-level excitation spectroscopy in its various forms is a very powerful probe of the short-range structure and bonding in molecules and solids. With respect to phenyl group-containing polymers, NEXAFS spectroscopy has shown its benefits over XPS in extracting phenyl ring sub-peak information.23,24,29 Because in NEXAFS spectroscopy the C 1s → π* phenyl ring resonance is one of the most prominent spectral features, its changes can be easily monitored during a surface modification treatment.
Fig. 5 shows the carbon K-edge NEXAFS spectra of PS before and after selective SR treatment at 293.5 eV. As can be seen in Fig. 5, important changes in the C 1s NEXAFS spectra were found after SR treatment. The C 1s → π*CC signal intensity strongly decreased after irradiation, indicating the lower contribution of this transition in the new modified polymer surface. The C 1s → σ*C–C primary excitation mainly affected the phenyl polymer structure. This result confirmed the XPS data that showed a decrease in the aromatic contribution of the C 1s peak (Fig. 2) at the three excitation energies studied. The A/B ratio of the peak labels in Fig. 5, top graph, decreased from 4.7 (untreated) to 2.0 (treated) when the excitation energy was tuned to 293.5 eV. In addition, it was necessary to add a new peak at ∼288.5 eV to get a satisfactory fitting in the treated spectrum. The new signal arose from carbon with a double bond to oxygen, π*CO, which appears to be located at energies between 286.5 and 289 eV24,30 depending on the polymer structure.
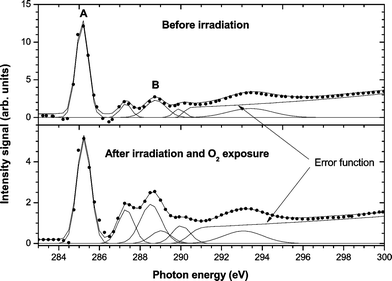 | ||
| Fig. 5 Carbon K-edge NEXAFS spectra of untreated and SR treated PS. | ||
The oxygen K-edge NEXAFS spectra clearly revealed the presence of oxygen functionalities in the SR treated polymer films. Fig. 6 shows that before irradiation no oxygen signal is detected. However, after treatment two signals corresponding to O 1s → π*CO and O 1s → σ* excitations24,25 can be seen in Fig. 6. Because PS does not have any oxygen contribution, the oxygen K-edge signal could be detected with high sensitivity over the background signals, demonstrating the presence of new functionalities on the PS surface.
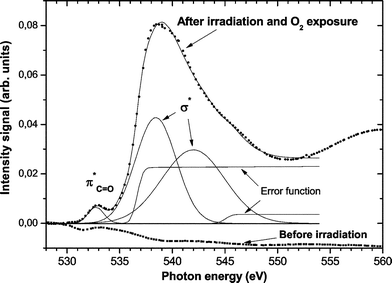 | ||
| Fig. 6 Oxygen K-edge NEXAFS spectra of untreated and SR treated PS. | ||
Our results can be interpreted taking into account previous photon stimulated positive-ions desorption studies of polymers.31–33 It was shown that core electron excitation to an antibonding σ* state, led to a much more effective bond scission at the site of excitation, compared to π*. The fragmentation and desorption of some polymer groups was taken as an indication of higher efficiency ion production by a σ* excitation. Excitation to σ* states has a higher probability of leaving radical species at the surface than π* excitations. These radicals can remain in UHV conditions and be efficiently neutralized with, for example, oxygen introduction in the last step of the process.
Conclusions
Selective surface functionalization by a combination of SR excitation with oxygen radical neutralization of PS thin films was investigated. The treatment methodology led to very important changes to the chemical composition of the surfaces of the PS substrates. After the treatments, high oxygen surface concentrations were found. By selecting a specific K-shell excitation (C 1s → σ*C–C), COO and CO groups were introduced on the PS surface with high efficiency. The COO and CO elemental contribution (C 1s envelope) in the PS surface was higher than 70%. This research will open new ways, that have not yet been investigated, to functionalize a polymer surface. A combination of monochromatic inner-shell excitation together with a radical neutralization process by reactive gas or monomer vapor addition, would allow specific functionalities to be introduced to a particular section of a polymer structure.
Acknowledgements
This work was partially supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil and LNLS – National Synchrotron Light Laboratory, Brazil. The authors would also like to acknowledge the technical assistance of the Accelerator Group, especially the VUV and Soft X-ray Spectroscopy Group.Notes and references
- A. R. Blythe, D. Briggs, C. R. Kendalll, D. G. Rance and V. J. I. Zichy, Polymer, 1978, 19, 1273–1278 CrossRef CAS.
- C. M. Chan, T. M. Ko and H. Hiraoka, Surf. Sci. Rep., 1996, 24, 1–54 CrossRef CAS.
- M. Dhayal, M. R. Alexander and J. W. Bradley, Appl. Surf. Sci., 2006, 252, 7957–7963 CrossRef CAS.
- S. A. Voronin, M. Zelzer, C. Fotea, M. R. Alexander and J. W. Bradley, J. Phys. Chem. B, 2007, 111, 3419–3429 CrossRef CAS.
- S. A. Voronin, M. R. Alexander and J. W. Bradley, Surf. Coat. Technol., 2006, 201, 768–775 CrossRef CAS.
- H. S. Choi, Y. S. Kim, Y. Zhang, S. Tang, S. W. Myung and B. C. Shin, Surf. Coat. Technol., 2004, 182, 55–64 CrossRef CAS.
- S. A. Mitchell, A. H. C. Poulsson, M. R. Davidson and R. H. Bradley, Colloids Surf., B, 2005, 46, 108–116 CrossRef CAS.
- F. Truica-Marasescu, S. Pham and M. R. Wertheimer, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 31–36 CrossRef CAS.
- J. P. Deng, L. F. Wang, L. Y. Liu and W. T. Yang, Prog. Polym. Sci., 2009, 34, 156–193 CrossRef CAS.
- J. Spanring, C. Buchgraber, M. F. Ebel, R. Svagera and W. Kern, Polymer, 2006, 47, 156–165 CrossRef CAS.
- E. Bormashenko, R. Pogreb, G. Whyman, Y. Bormashenko, R. Jager, T. Stein, A. Schechter and D. Aurbach, Langmuir, 2008, 24, 5977–5980 CrossRef CAS.
- J. López-Gejo, N. Manoj, S. Sumalekshmy, H. Glieman, T. Schimmel, M. Worner and A. M. Braun, Photochem. Photobiol. Sci., 2006, 5, 948–954 RSC.
- F. Truica-Marasescu and M. R. Wertheimer, Macromol. Chem. Phys., 2008, 209, 1043–1049 CrossRef CAS.
- Y. Baba, Low Temp. Phys., 2003, 29, 228–242 CrossRef CAS.
- M. L. M. Rocco, D. E. Weibel, L. S. Roman and L. Micaroni, Surf. Sci., 2004, 560, 45–52 CrossRef CAS.
- V. Skurat, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 27–34 CrossRef CAS.
- Y. Ukita, M. Kishihara, K. Kanda, S. Matsui, K. Mochiji and Y. Utsumi, Jpn. J. Appl. Phys., 2008, 47, 337–341 CrossRef CAS.
- Y. Kato, K. Kanda, Y. Haruyama and S. Matsui, Radiat. Phys. Chem., 2006, 75, 2049–2053 CrossRef CAS.
- D. E. Weibel, C. Vilani, A. C. Habert and C. A. Achete, J. Membr. Sci., 2007, 293, 124–132 CrossRef CAS.
- C. Vilani, D. E. Weibel, R. R. M. Zamora, A. C. Habert and C. A. Achete, Appl. Surf. Sci., 2007, 254, 131–134 CrossRef CAS.
- D. E. Weibel, A. F. Michels, F. Horowitz, R. D. S. Cavalheiro and G. V. d. S. Mota, Thin Solid Films, 2009, 517, 5489–5495 CrossRef CAS.
- S. G. Urquhart, A. P. Hitchcock, A. P. Smith, H. W. Ade, W. Lidy, E. G. Rightor and G. E. Mitchell, J. Electron Spectrosc. Relat. Phenom., 1999, 100, 119–135 CrossRef CAS.
- D. A. Outka and J. Stohr, J. Chem. Phys., 1988, 88, 3539–3554 CrossRef.
- R. J. Klein, D. A. Fischer and J. L. Lenhart, Langmuir, 2008, 24, 8187–8197 CrossRef CAS.
- I. Koprinarov, A. Lippitz, J. F. Friedrich, W. E. S. Unger and C. Woll, Polymer, 1998, 39, 3001–3009 CrossRef.
- R. M. France and R. D. Short, Langmuir, 1998, 14, 4827–4835 CrossRef CAS.
- R. M. France and R. D. Short, J. Chem. Soc., Faraday Trans., 1997, 93, 3173–3178 RSC.
- S. Turgeon and R. W. Paynter, Thin Solid Films, 2001, 394, 43–47 CrossRef.
- S. Hofmann, Appl. Surf. Sci., 2005, 241, 113–121 CrossRef CAS.
- O. Dhez, H. Ade and S. G. Urquhart, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 85–96 CrossRef CAS.
- K. Tanaka, M. C. K. Tinone, H. Ikeura, T. Sekiguchi and T. Sekitani, Rev. Sci. Instrum., 1995, 66, 1474–1476 CrossRef CAS.
- H. I. Sekiguchi, T. Sekiguchi and K. Tanaka, Phys. Rev. B: Condens. Matter, 1996, 53, 12655–12658 CrossRef CAS.
- N. Ueno and K. Tanaka, Jpn. J. Appl. Phys., 1997, 36, 7605–7610 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |