DOI:
10.1039/B9PY00227H
(Paper)
Polym. Chem., 2010,
1, 303-311
Received
29th August 2009
, Accepted 22nd October 2009
First published on 21st December 2009
Abstract
The local conformation of poly(methyl methacrylate) (PMMA) chains at the nitrogen (N2) and water interfaces was studied by infrared-visible sum-frequency generation (SFG) spectroscopy. Although SFG spectra in the C–H region for PMMA at the N2 interface have been hitherto reported, the peak assignments are not in accord with one another. Thus, we first made accurate assignments of SFG peaks using films, which had been well annealed at a temperature above the glass transition temperature for a long time, of three different deuterated PMMAs as well as normal protonated PMMA. At the N2 interface, hydrophobic functional groups such as α methyl, ester methyl and methylene groups were present. While the α methyl group was oriented along the direction parallel to the interface, ester methyl and methylene groups were oriented normal to the interface. Quantitative discussion concerning the orientation of the functional groups of PMMA at the N2 interface was aided by a model calculation. Once the PMMA film contacted water, the carbonyl groups of the PMMA side chains were oriented to the water phase to form hydrogen bonds with water molecules, resulting in the migration of ester methyl into the internal region of the film. Concurrently, the methylene groups became randomly oriented at the water interface and/or in part migrated into the internal region. Interestingly, the α methyl groups still existed at the water interface oriented along the parallel direction. The outermost region of PMMA in water can consist of hydrophilic and hydrophobic domains with sub-nanometre scale. Water molecules H-bond to themselves near the hydrophobic domains, leading to the formation of an ice-like structure of water molecules. However, water molecules adjacent to the hydrophilic domains H-bond with carbonyl groups.
Introduction
The interfaces of polymers with “nonsolvents” play an important role in their functional properties such as wettability, friction with lubricants, cell adhesion, biocompability, etc.1,2 To design and construct highly functionalized polymers for applications that exploit these characteristics, the aggregation states of the polymers at the liquid interfaces must be understood as the first benchmark. However, this is experimentally difficult because such interfaces are buried.
Aggregation states of polymers at the air-facing surface in solid films can be conveniently analyzed by established surface sensitive techniques.3,4 However, such information may not be good enough to deduce the structure at the outermost surface after the film is contacted with a liquid. This is because the structure of the outermost region of the film is generally reorganized under the presence of the liquid to minimize the free energy at the interface.5,6 To overcome this problem, the technique of freeze-drying has been widely used.7,8 This method should be effective for studying relatively large-scale structures, such as those which occur in a phase-separated system. However, this may not be the case for aggregation states with a scale smaller than a chain dimension, such as local conformations, because they can be easily restructured during the drying process, even at liquid nitrogen temperatures. Thus, the local structure of polymers at the liquid interface should be examined in situ without taking the sample out of contact with the liquid.
Recently, sum frequency generation (SFG) vibrational spectroscopy has been used to study molecular structures at surfaces and interfaces.9–15 To obtain SFG signals, which originate in a second-order nonlinear optical process, centrosymmetry in the system must be broken. This condition can be satisfied only at interfaces, leading to an SFG signal that is highly interface specific. The surface and interfacial sensitivities of SFG are much better than those of other techniques such as attenuated total reflectance Fourier transform infrared spectroscopy16 and surface enhanced Raman spectroscopy.17 The other intriguing advantage of SFG vibrational spectroscopy is the ability to gain insight into the orientation of functional groups by using different polarization combinations of input and output beams. Thus, SFG enables us to study in situ local conformation of polymer chains at various interfaces.
Poly(methyl methacrylate) (PMMA) is widely used in many biomedical materials such as bone cement,18 intraocular lenses,19 artificial kidneys,20 and so on. In these applications, PMMA is generally in contact with water, which is a nonsolvent for this polymer. So far, we have examined the density profile of a perdeutrated PMMA (d8-PMMA) at the interface with water along the direction normal to the interface by specular neutron reflectivity (NR).21 The interface of the d8-PMMA with water was much more diffuse than that of the pristine surface in air. This implies that the d8-PMMA had partly dissolved into the water and was swollen by water molecules. Therefore, as the next step of the investigation, we need to address how the aggregation states of PMMA at the outermost region of the film changes after contact with water.
Poly(methacrylate)s are some of the polymers most studied by SFG, and spectra have been reported by several groups. 12,14,22–28 Unfortunately, peak assignments are not consistent among these reports. Chen et al. studied aggregation states of poly(n-alkyl methacrylate)s with different side chain lengths at the air and water interfaces by SFG.12,14,22,27,29,30 While a structural change was clearly observed for poly(n-alkyl methacrylate)s between the air and water interfaces, this was not the case for PMMA itself.14,22,27 At a glance, this result looks to be inconsistent with our findings which indicate that the outermost region of a PMMA film is partially dissolved and is swollen by water molecules.
In general, polymer chains in a glassy solid film are frozen in a non-equilibrium state. Thus, to study the structure as well as the physical properties of polymers, the film should be well annealed to reach a quasi-equilibrium state under conditions where polymer chains can be fully relaxed. However, it seems to us that so far, care in establishing such a quasi-equilibrium state has been lacking in surface and interfacial characterization of polymers by SFG.
In this study, we use SFG spectroscopy to study the local conformation of PMMA chains at the interface with gaseous nitrogen, which was chosen as a controlled, water-free, model for air, and at water interfaces. We first assign the SFG spectra, focusing on the C–H stretching region, for spin-coated films of various PMMAs in which different portions were labeled by deuterium. The effect of annealing on the local conformation of PMMA at the nitrogen interface is next examined. Then, any conformational changes in PMMA at the interface in a quasi-equilibrium film in contact with water are discussed. Finally, the structure of the water in contact with the PMMA is discussed.
Experimental
Materials and film preparation
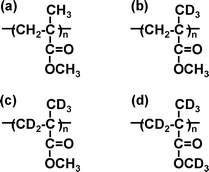
Samples of monodisperse PMMA, in which the syndiotactic triad is dominant, with a number-average molecular weight (Mn) of 212.9k were used. To assist in the assignment of SFG peaks, the partially deuterated PMMAs shown in Fig. 1 were also used namely, d3-PMMA, d5-PMMA, and d8-PMMA. The characteristics of the PMMA samples are shown in Table 1. A hemi-cylindrical prism made of silica (IR Grade) was used as the substrate. Prior to use, the prism was cleaned with a piranha solution composed of sulfuric acid and hydrogen peroxide (7/3 w/w) at 383 K for 1 h to remove any organic residues and rinsed several times with Milli-Q®water. In addition, a hemi-cylindrical CaF2 prism was used as the substrate for the samples examined in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O region of the spectrum where silica absorbs. The prism was first cleaned with chloroform and then was treated with an air plasma. PMMA samples were directly spin-coated onto the prisms from their 4 wt% toluene solutions and then annealed under vacuum at 433 K for 24 h, unless stated otherwise. Since the bulk glass transition temperature (Tgb) of the PMMA used here was found by differential scanning calorimetry to be 400 K, the annealing procedure should have made the polymer chains fully relaxed. The thickness of the films measured by ellipsometry was approximately 200 nm.
O region of the spectrum where silica absorbs. The prism was first cleaned with chloroform and then was treated with an air plasma. PMMA samples were directly spin-coated onto the prisms from their 4 wt% toluene solutions and then annealed under vacuum at 433 K for 24 h, unless stated otherwise. Since the bulk glass transition temperature (Tgb) of the PMMA used here was found by differential scanning calorimetry to be 400 K, the annealing procedure should have made the polymer chains fully relaxed. The thickness of the films measured by ellipsometry was approximately 200 nm.
Table 1 Characteristics of PMMAs
The SFG spectra were collected with visible and tunable IR beams traveling through the prism and overlapping at the PMMA/N2 or PMMA/water interfaces, as depicted in Fig. 2. The visible beam with a wavelength of 532 nm was generated by frequency-doubling the fundamental output pulses from a picosecond Nd:YAG laser (PL2143, EKSPLA). The IR beam, which was tunable over the wavenumber range from 1000 to 4300 cm−1, was generated from an EKSPLA optical parametric generation/amplification and difference frequency generation (OPG/OPA/DFG) system based on LBO and AgGaS2 crystals. The intensity of the SFG signals was normalized to those of the original IR and visible beams. The measurements were carried out at room temperature using two types of polarization combinations, namely ssp and ppp (SF output, visible input, and infrared input).
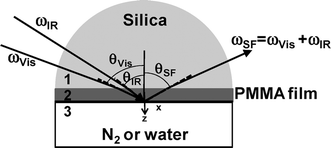 |
| | Fig. 2 A schematic representation of the optical geometry used in our SFG spectroscopy experiments. | |
Theory
The SFG intensity (ISFG) is proportional to the square of the absolute value of the effective sum frequency susceptibility tensor of the surface (χeff(2)), which is itself related to the second-order nonlinear susceptibility tensor χ(2) in the laboratory coordinate system, as well as the incident laser intensities of the two input beams (Ii(ωVis) and Ii(ωIR)). ISFG in the reflective direction can be formulated as11| | 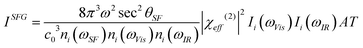 | (1) |
where ni(ω) is the refractive index of medium i at frequency ω, θSF is the reflection angle of the sum-frequency field, and c0 is the speed of light in the vacuum. In eqn (1), A and T are the beam overlap cross section at the interface and the pulse width, respectively. In the case of an azimuthally isotropic interface, there are only four independent non-vanishing components of χ(2). With the laboratory coordinates chosen such that z is along the interface normal and x is in the incidence plane, they are χxxz = χyyz, χxzx = χyzy, χzxx = χzyy, and χzzz. In this study, SFG spectra were collected with the ssp and ppp polarization combinations. The χeff(2) for the two polarization combinations can be expressed using χ(2).11| | | χeff,ssp(2) = Lyy(ωSF)Lyy(ωVis)Lzz(ωIR)sinθIRχyyz | (2a) |
| | 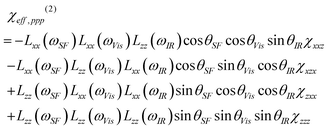 | (2b) |
where Lii(i = x, y, z) are the Fresnel coefficients. The angle between the surface normal and the beam is expressed as θ, as shown in Fig. 2. The subscripts Vis and IR denote the input beams. To calculate the Fresnel factors for the PMMA/N2 and PMMA/H2O interfaces, the following equations are used,31| | 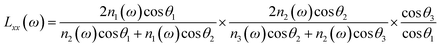 | (3a) |
| | 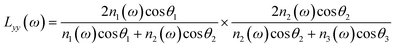 | (3b) |
| | 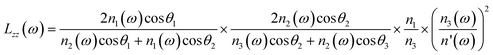 | (3c) |
where n′ is the refractive index of the PMMA/N2 or PMMA/water interfacial layer. The n′ values of 1.18 and 1.41 were used for the PMMA/N223 and PMMA/water32 interfaces, respectively. In eqn (3), θi is the incidence angle in the medium i. When the IR frequency is near vibrational resonance, χeff(2) can be formulated as| | 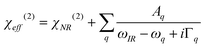 | (4) |
where χNR(2) arises from the non-resonant background contribution. Here, Aq, ωq, and Γq are the strength, resonant frequency, and damping coefficient of the qth vibrational mode, respectively. Each peak on the SFGspectra was fitted by eqn (4) using Aq, ωq, and Γq as fitting parameters to find the peak intensity, ISFG.
Results and discussion
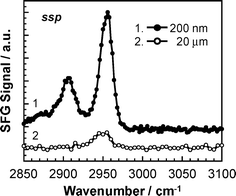
The SFG signal is generated at a location where centrosymmetry is broken. This means that in our system, the signal might be generated from either, or both, of the polymer/substrate or polymer/air (or water) interfaces. To examine which interface makes the dominant contribution to the SFG signal in our set-up, we made measurements on thin and thick PMMA films in a nitrogen (N2) atmosphere. In the case of a thick film , the IR beam is mostly absorbed in the internal regions of the film on the way to the PMMA/N2 interface, and hence the contribution of that interface to the SFG signal will be reduced. On the other hand, the signal from the polymer/substrate interface should be unaffected by the thickness of the film. Fig. 3 shows typical SFG spectra of thin and thick PMMA films in the wavenumber range from 2850 to 3100 cm−1. The SFG signal was much weaker for the thick film . This makes it clear that the SFG signal obtained under the current optical geometry is mostly coming from the polymer/N2 interface.
The signal intensity, ISFG, is proportional to the square of χeff(2), which can be given by eqn (2). The square of χeff(2) for the PMMA/N2 interface for ssp polarization combination was calculated to be a factor of 8 larger than that for the substrate interface. The difference became even larger for the PMMA/water interface, increasing to a factor of 13. Therefore, it seems most likely that the SFG signal obtained in our current set-up is mainly coming from the PMMA/N2 or PMMA/water interface. Therefore, in the subsequent experiments, we used films that were approximately 200 nm thick to avoid IR absorption on the way to the PMMA/N2 or PMMA/water interface. This thickness is safely out of the range in which the aggregation structure and physical properties of polymers in thin films differ from the corresponding bulk ones due to an ultrathinning effect.
II. Peak assignments of the SFG spectra
A number of reports dealing with SFG spectra for PMMA films have been published.12,14,22–28 However, the peak assignments of the spectra differ between the various reports. Therefore, in this work, we used films of partially deuterated PMMA to assist in the peak assignment. Fig. 4 shows SFG spectra over the wavenumber region from 2850 to 3050 cm−1 for the films of various PMMAs for the ssp and ppp polarization combinations. Since this wavenumber region corresponds to the C–H stretching region, no peaks were observed for d8-PMMA. In the case of d5-PMMA, C–H bonds are only present in ester methyl groups. While an intense peak was also observed for this compound at 2955 cm−1 for the ssp combination, an additional weak peak was found at 2990 cm−1 for the ppp combination. Thus, the SFG peaks at 2955 and 2990 cm−1 can be assigned to the C–H symmetric and antisymmetric stretching vibrations of the ester methyl groups.
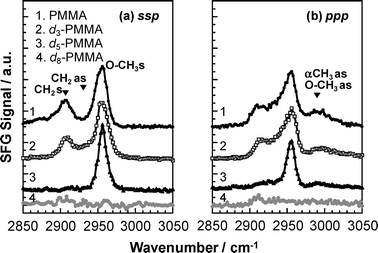 |
| | Fig. 4
SFG spectra for various PMMA films under a N2 atmosphere. (a) ssp and (b) ppp polarization combinations. | |
In the case of d3-PMMA, C–H bonds are present both in ester methyl and methylene groups. For this compound, an additional peak was observed at 2908 cm−1 for the d3-PMMA film with the ssp and ppp combinations. Thus, there is no doubt that the peak at 2908 cm−1 can be assigned to the C–H symmetric stretching vibrations of the methylene groups. An overlapping peak around 2930 cm−1 arising from the antisymmetric stretching of methylene groups might be present in both the ssp and pppspectra. The ppp peak intensity at 2990 cm−1 for d5-PMMA was similar to that for d3-PMMA. This is quite reasonable because the peak arises from the C–H antisymmetric stretching vibrations of the ester methyl groups, which are present in both d5-PMMA and d3-PMMA. Only PMMA possesses α methyl groups. The sspspectra of PMMA and d3-PMMA seem to be identical. On the other hand, the pppspectrum of PMMA is slightly different from that of d3-PMMA in that the spectral intensity at 2990 cm−1 for PMMA was more intense. Taking into account that the only structural difference between PMMA and d3-PMMA is the presence of α methyl groups in the former, the above result implies that the peak observed at 2990 cm−1 in the pppspectrum involves the C–H antisymmetric stretching of the α methyl groups. These assignments were in good agreement with those found from Fourier-transform infrared spectroscopy using bulk films of various PMMAs.
SFG
ssp
spectra for the PMMA/N2 interface obtained here, curve 1 in Fig. 4, does not have the same shape as those in the previous reports, especially around the CH2 stretching region at 2908 cm−1.12,14,22–28 In the previous reports, the PMMA films were annealed at a temperature below the glass transition temperature. This may result in the PMMA chains in the films not being fully relaxed and thus adopting unstable conformations dependent on the history of the spin-coating process.33 Hence, we examined the effect of annealing on the local conformation of chains at the surface.
Fig. 5 shows the SFG spectra for PMMA, annealed at 298, 333, 353 and 433 K for 24 h, at the N2 interface for ssp and ppp polarization combinations. It should here be noted that the bulk Tg of the sample was 400 K. The peak around 2908 cm−1 assigned to the C–H symmetric stretching vibration for the methylene groups grew with increasing annealing temperature, particularly at the final temperature that is some 33 K above the bulk Tg. This makes it clear that once the segmental motion of PMMA chains is free, hydrophobic methylene groups in the main chain partially orient in the plane of the N2 interface to minimize the free energy. The details of the orientation will be discussed in the next section.
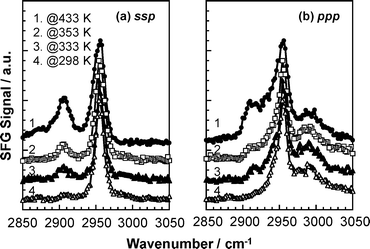 |
| | Fig. 5
Annealing temperature dependence of SFG spectra for PMMA at a N2 interface with (a) ssp and (b) ppp polarization combinations. | |
Careful analysis of the ssp and pppspectra shows that the width of the peak near 2955 cm−1 broadens with increasing temperature, especially at the lower wavenumber side. When the methylene groups orient in the plane of the N2 interface, the corresponding C–H antisymmetric stretching vibration, at 2930 cm−1, should be also observed. Thus, the peak broadening with temperature reflects the overlap of the C–H symmetric vibration of the ester methyl groups and the C–H antisymmetric stretching vibration of the methylene groups. Interestingly, this peak starts to grow, even at temperatures lower than the bulk Tg, suggesting that chain mobility at the N2 interface is enhanced in comparison with that in the internal bulk phase. This observation is also in good agreement with a previous finding that the Tg is lower at the surface than in the bulk.34,35 Hereafter, all films used were well annealed at 433 K for 24 h so that we can assume that a local conformation is in a quasi-equilibrium in our discussions.
IV. Local conformation of chains at the surface
(a) Ester methyl groups.
First, we focus on the SFG spectra for the d5-PMMA film, curves 3 in the right and left panels of Fig. 4, in order to extract the orientation of the ester methyl groups at the N2 interface. For the ssp combination, only the symmetric stretching vibration was observed, qualitatively indicating that the ester methyl groups orient almost totally along the direction perpendicular to the surface. On the other hand, the peak for the antisymmetric stretching vibration of the ester methyl groups was seen at 2990 cm−1only for the ppp combination. We estimated the tilt angle (θ) of the ester methyl groups following a method proposed by Hirose et al.,36–40 namely, using the intensity ratio of the SFG peaks of the symmetric (Is) to antisymmetric stretching modes (Ias). The tilt angle is the angle between the c and z axes. The z axis is already defined and is along the interface normal in the laboratory coordinate system, and is hence always fixed. On the other hand, since the c axis is in the molecular coordinate system, its direction depends on the molecular symmetry. Thus, the definition of this axis will vary for different functional groups.
The macroscopic sum frequency susceptibility tensor χijk(2) is related to the microscopic hyperpolarizability tensor elements βi′j′k′(2) in the molecular coordinate system defined by a, b and c axes. Both α methyl and ester methyl groups can be treated as having C3v symmetry.36,37 In this case, there are two non-vanishing independent elements in the symmetric stretch hyperpolarizability tensor: βccc and βaac = βbbc.36 In the molecular-fixed coordinates for a methyl group, the c axis coincides with the C3 axis, and the a axis lies in the plane of symmetry σv of the methyl group. For the C–H symmetric stretching vibrations, the components of χ(2) for an isotropic surface are given by11,37
| | 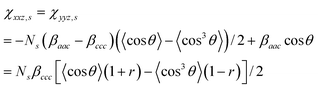 | (5a) |
| | 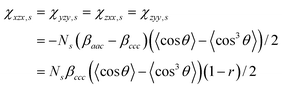 | (5b) |
| | 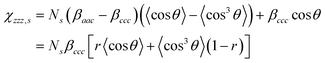 | (5c) |
where
Ns is the surface density of molecules and
r =
βaac/
βccc. The
r value for the
methyl groups of
PMMA was given as 1.8 in
ref. 23. The brackets < > denote the average over polar angles. For the antisymmetric stretch, the components of χ
(2) for an isotropic surface are expressed as follows.
37,40| | | χxxz,as = χyyz,as = −Nsβcaa(〈cosθ〉 − 〈cos3θ〉) | (6a) |
| | | χxzx,as = χyzy,as = χzxx,as = χzyy,as = Nsβcaa〈cos3θ〉 | (6b) |
| | | χzzz,as = 2Nsβcaa(〈cosθ〉−〈cos3θ〉) | (6c) |
Generally, functional groups existed at the surface possess an orientation angle with a certain fluctuation. Here, the angle fluctuation was modeled by a Gaussian distribution.41,42
| | | 〈cosnθ〉 = ∫π0cosnθ × f(θ)sinθdθ | (7a) |
| | | f(θ) = Cexp[−(θ − θ0)2/2σ2] | (7b) |
where
θ0 is the average over tilt angles.
Using these equations we can determine the orientation of the ester methyl groups at the N2 interface. Fig. 6 shows simulated values of Ias/Is as functions of the average tilt angle θ0 and the angle distribution σ for the ssp and ppp polarization combinations. The simulation used eqn (1)–(3) and (5)–(7) assuming βccc ≈ βcaa43 and a value of n′ of 1.18.23 To find Ias/Is from experiment, the SFG spectra for d5-PMMA were separated into two components by curve-fitting using eqn (4). The ratios for the ssp and ppp combinations were, respectively, 0 and 0.22. Comparing these values with the simulation results shown in Fig. 6, it seems most likely that θ0 for the ester methyl groups is approximately 20° with zero angular distribution or that θ0 is smaller than 15° with an angular distribution less than 15°. In either case, this result indicates that the ester methyl groups at the N2 interface are well ordered and orient along the direction normal to the interface.
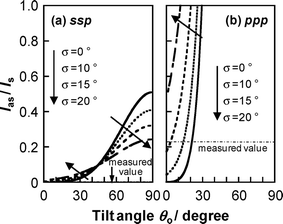 |
| | Fig. 6 Ratio of the SFG intensity of the C–H antisymmetric and symmetric stretching modes for ester methyl groups of PMMA under a N2 atmosphere as functions of the average tilt angle θ0 and angular distribution σ. Left and right panels show the plots for (a) ssp and (b) ppp polarization combinations. | |
(b)
Methylene groups
.
The methylene groups are treated as having C2v symmetry.36 In the case of C2v, there are three non-vanishing independent elements in the symmetric stretch hyperpolarizability tensor: βaac, βbbc, and βccc. In the molecular-fixed coordinates for methylene, the c axis is defined as the direction along the bisector line of the two C–H bonds, the a axis is in the H–C–H plane, and b is out of plane. The components of χ (2) for an isotropic surface are given by37| | 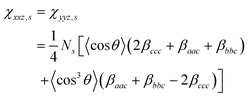 | (8a) |
| | 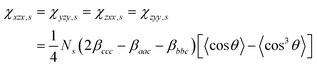 | (8b) |
| | | χzzz,s = ½[(βaac + βbbc)〈cosθ〉 − (βaac + βbbc − 2βccc)〈cos3θ〉] | (8c) |
The antisymmetric stretch hyperpolarizability tensor has only one independent element: βaca = βcaa. Thus, in this case, the components of χ(2) for an isotropic surface are given by
| | | χxxz,as = χyyz,as = −½Nsβcaa(〈cosθ〉 − 〈cos3θ〉) | (9a) |
| | | χxzx,as = χyzy,as = χzxx,as = χzyy,as = −½Nsβcaa〈cos3θ〉 | (9b) |
| | | χzzz,as = Nsβcaa(〈cosθ〉−〈cos3θ〉) | (9c) |
The relationship of each β for the methylene stretching modes has been already reported as41,44
| βaac ≈ 0.82β0, βbbc ≈ 0.16β0, βccc ≈ 0.49β0 |
where
β0 is a constant defined for a single C–H bond.
The SFG spectra for the PMMA film were again separated into two components, the symmetric and antisymmetric modes for the methylene groups, by curve-fitting using eqn (4). The intensity ratio of the antisymmetric mode to the symmetric one was 0.06 for the ssp polarization combination and 4.48 for the ppp combination. Fig. 7 shows multiple simulated solutions for Ias/Is as functions of θ0 and σ. Comparing the experimental Ias/Is values with those from the simulation, it seems most likely that the methylene groups are tilted at about θ0 = 20-40° with zero angular distribution or, assuming on a Gaussian distribution, the angular distribution is less than 15°. These results indicate that the main chain of PMMA orients along the direction parallel to the N2 interface.
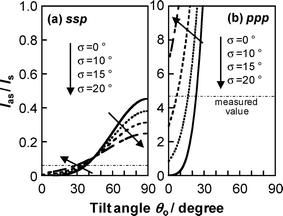 |
| | Fig. 7 Ratio of the SFG intensity of the C–H antisymmetric and symmetric stretching modes for methylene groups of PMMA under a N2 atmosphere as functions of the average tilt angle θ0 and angle distribution σ. Left and right panels show the plots for (a) ssp and (b) ppp polarization combinations. | |
(c) α methyl groups.
Comparing the d3-PMMAspectra to those of PMMA, the C–H antisymmetric stretching vibration of the α methyl groups was observed only for the ppp combination. Thus, it seems reasonable to infer that the α methyl groups are oriented almost parallel to the interface.
To summarize this section, the hydrophobic methyl and methylene groups were preferentially segregated at the surface, as expected. To achieve this, the main part of the PMMA chain orients in the interfacial plane. Such a conformation corresponds to that expected for a space-filling Corey, Pauling and Kultun molecular model, and is shown in Fig. 8.
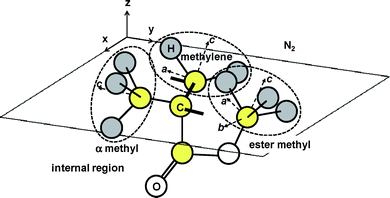 |
| | Fig. 8 A possible local conformation of PMMA at the N2 interface. | |
V. Local conformation of chains at the water interface
(a)
Methylene groups
.
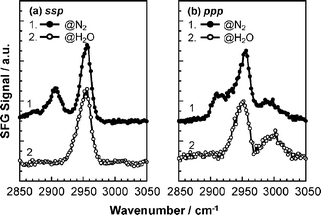
Fig. 9 shows SFG spectra for PMMA films under N2 and in water. The spectra under N2 are imported from Fig. 4. Interestingly, the 2908 cm−1methylene C–H symmetric stretch disappeared in the water. This was a common feature of both ssp and ppp combinations. Thus, it seems most likely that the methylene groups that were oriented at the N2 interface became randomly oriented at the water interface. This can be easily understood if we assume that the side chains of PMMA are segregated to the water phase because of an attractive interaction between carbonyl groups and water molecules.
(b)
Carbonyl groups
.
To confirm whether the above assumption related to the orientation of the carbonyl groups is realistic, SFG spectra for PMMA in the CO region were acquired before and after the contact with water using the ssp polarization combination. Fig. 10 shows the result. At the N2 interface, a clear peak was observed at 1736 cm−1 and could be assigned to free carbonyl groups.45 A smaller accompanying peak was observed at 1706 cm−1. At this time we cannot provide an unambiguous assignment for this peak. Possible origins are a harmonic or combination rocking vibration of the methylene groups,46,47 which are enhanced by Fermi resonances. Another candidate for the assignment of the minor peak at 1706 cm−1 is that some of the carbonyl groups are already bonded to water molecules. Although the measurement was carefully made under a N2 atmosphere, inevitably it is impossible to completely remove all of water molecules from the sample cell. We prefer this last assignment for the 1706 cm−1 peak based on the results from a water interface discussed below.
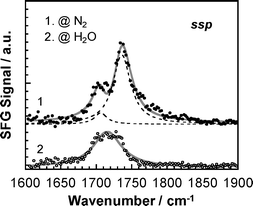 |
| | Fig. 10
SFG spectra for PMMA at N2 and water interfaces in the CO region for the ssp polarization combination. | |
The CO SFG spectrum dramatically changed shape after contact with water. The peak became broader and shifted to 1714 cm−1. Interfacial hydrogen bonding between the carbonyl groups and water molecules has been discussed in several publications, and the extent of the wavenumber shift was reported to be 10–20 cm−1.14,30,48 The peak may be composed of two overlapping peaks from free and hydrogen bonded carbonyl groups. However, it was impossible to resolve it unambiguously into two peaks. Since the extent of the red shift observed here was 22 cm−1, it seems reasonable to claim that the most of carbonyl groups present at the interface formed hydrogen bonds with water molecules.49
(c) Ester methyl groups.
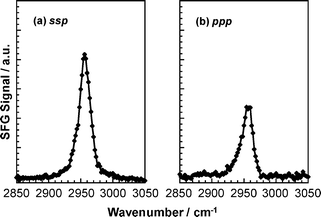
To gain further insight into the orientation of the PMMA side chains, a SFG measurement for the d5-PMMA film was performed in deuterated water. This experiment probes the orientation of the ester methyl group directly. Fig. 11 shows that the C–H symmetric stretching vibration of the ester methyl groups was observed in both polarization combinations, whereas the antisymmetric vibration was not observed in either. This means that the ester methyl groups orient at the water interface along the direction perpendicular to the interface. However, the discussion of Fig. 10 showed that the carbonyl groups were segregated at the water interface because of an attractive interaction with water molecules. To account for both these observations, the ester methyl groups must orient toward to the internal bulk region along the direction normal to the interface. Taking into account the hydrophobicity of the ester methyl groups, such a conformation seems to be reasonable.
(d) α methyl groups.
Fig. 11 also shows that, in the case of the d5-PMMA film in deuterated water, no SFG peaks were observed around 2990 cm−1 for both the ssp and ppp combinations, as shown in Fig. 11. Similarly, a clear peak was not observed either for the PMMA film in water at the corresponding wavenumber with the ssp combination, as shown by curve 2 in Fig. 9(a). On the other hand, a clear peak appeared for the PMMA film in water near 2990 cm−1 for the ppp combination, as shown by curve 2 in Fig. 9(b). Based on these results, it is conceivable that the α methyl groups orient parallel to the interface even in the presence of water. Fig. 12 shows a schematic illustration of a likely local conformation of PMMA at the water interface deduced from the SFG data.
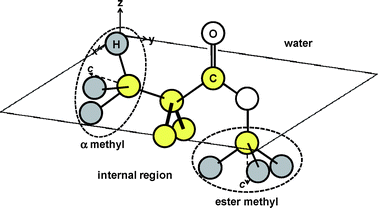 |
| | Fig. 12 A possible local conformation of PMMA at the water interface. | |
VI. Water structure at the PMMA interface
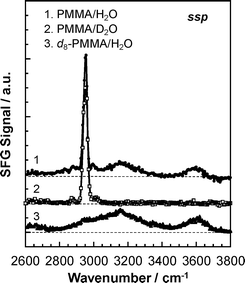
We finally come to the aggregation states of water molecules at the PMMA interface. Fig. 13 shows SFG spectra for PMMA with water, PMMA with deuterated water and d8-PMMA with water. In the case of PMMA/water, broad peaks were observed around 3150 and 3600 cm−1 in addition to the intense peak corresponding to the C–H symmetric stretching vibration for the ester methyl groups at 2955 cm−1. While the broad peaks at 3150 and 3600 cm−1 disappeared for the PMMA film in deuterated water, they again appeared for the d8-PMMA in water. Therefore, those peaks are assignable to the O–H stretching of water molecules. Comparing our results with the wavenumbers appearing in published reports,13,50,51 the peaks at 3150 and 3600 cm−1 were specifically assigned to ice-like water and free O–H, respectively.
At the water interface, the outermost region of PMMA might consist of hydrophilic and hydrophobic domains at a sub-nanometre scale. If this is the case, it can be envisaged that water molecules H-bond among themselves near the hydrophobic domains composed of methylene and α methyl groups. This leads to the formation of an ice-like structure of water molecules. The peak at 3600 cm−1 is a signature of the stretching of only one free O–H of a water molecule, not of two. This is simply because the O–H antisymmetric stretch, which should appear at a wavenumber higher than 3700 cm−1 for a free molecule was not observed. Water molecules are supposed to establish hydrogen bonds with their nearest neighbours in a tetrahedral geometry.50 However, at the interface, water molecules hydrogen bonded to the carbonyl groups are located in a confined geometry. Such a situation may not allow the water molecules to form a second hydrogen bond, resulting in the presence of free O–H groups.
Conclusions
The interfacial local conformation of PMMA was studied by SFG vibrational spectroscopy. We assigned SFG peaks using three different deuterated PMMA as well as normal protonated PMMA. In addition, we examined the effect of annealing on the aggregation states of PMMA at the N2 interface. This was of pivotal importance in explaining the disagreement with published reports, and allowed unambiguous peak assignments in the C–H region for the SFGspectrum of PMMA. At the N2 interface, the main chain of PMMA oriented along the direction parallel to the surface. The α methyl and the ester methyl groups were present at the interface along the directions parallel and normal to the interface, respectively. In contact with water, the chains in the outermost region of the PMMA reorganized to turn the carbonyl groups towards the water interface to form hydrogen bonds, while the hydrophobic ester methyl groups retreated to the internal bulk phase. To attain such a conformation, the main chains of PMMA must randomize. However, the α methyl groups remained in plane at the interface even in the presence of water. The outer most region of PMMA in water probably possesses both hydrophilic and hydrophobic domains at a sub-nanometre scale. On the hydrophobic domains, water molecules form an ice-like network structure without interacting with the PMMA. In contrast, on the hydrophilic regions, they form hydrogen bonds with carbonyl groups.
Acknowledgements
This work was in part supported by an Industrial Technology Research Grant Program in 2006 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan, and by Grant-in-Aids for Young Scientists A (No. 21685013), Science Research in a Priority Area “Soft Matter Physics” and Innovative Areas “Molecular Soft-Interface Science” from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Notes and references
-
F. Carbassi, M. Morra and E. Occhiello, Polymer Surfaces: From Physics to Technology, John Wiley and Sons, Chichester, 1994 Search PubMed
 .
.
-
Surface and Interfaces for Biomaterials, ed. P. Vadgama, CRC, Boca Raton, FL, 2005 Search PubMed .
-
Surface and Interfacial Aspects of Biomedical Polymers, ed. Andrade, J. D., Plenum Press, New York, 1985, vol. 1 Search PubMed .
- T. Kajiyama, K. Tanaka, S. R. Ge and A. Takahara, Prog. Surf. Sci., 1996, 52, 1 CrossRef CAS .
- E. Ruckenstein and S. V. Gourisankar, J. Colloid Interface Sci., 1986, 109, 557 CrossRef CAS .
- J. K. Pike, T. Ho and K. J. Wynne, Chem. Mater., 1996, 8, 856 CrossRef CAS .
- A. Takahara, K. Takahashi and T. Kajiyama, J. Biomater. Sci., Polym. Ed., 1993, 5, 183 CAS .
- K. B. Lewis and B. D. Ratner, J. Colloid Interface Sci., 1993, 159, 77 CrossRef CAS .
- X. D. Zhu, H. Suhr and Y. R. Shen, Phys. Rev. B: Condens. Matter, 1987, 35, 3047 CrossRef CAS .
- Y. R. Shen, Nature, 1989, 337, 519 CrossRef CAS .
- X. Zhuang, P. B. Miranda, D. Kim and Y. R. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12632 CrossRef CAS .
- Z. Chen, Y. R. Shen and G. A. Somorjai, Annu. Rev. Phys. Chem., 2002, 53, 437 CrossRef CAS .
- G. L. Richmond, Chem. Rev., 2002, 102, 2693 CrossRef CAS .
- Z. Chen, Polym. Int., 2007, 56, 577 CrossRef CAS .
- X. L. Lu, N. Shephard, J. L. Han, G. Xue and Z. Chen, Macromolecules, 2008, 41, 8770 CrossRef CAS .
- P. A. Wilks, Spectrochim. Acta, 1962, 18, 884 .
- Y. J. Chen, W. P. Chen and E. Burstein, Phys. Rev. Lett., 1976, 36, 1207 CrossRef CAS .
- S. Shinzato, M. Kobayashi, W. F. Mousa, M. Kamimura, M. Neo, Y. Kitamura, T. Kokubo and T. Nakamura, J. Biomed. Mater. Res., 2000, 51, 258 CrossRef CAS .
- P. G. Ursell, D. J. Spalton, M. V. Pande, E. J. Hollick, S. Barman, J. Boyce and K. Tilling, J. Cataract Refract. Surg., 1998, 24, 352 CAS .
- H. H. Lin, Y. L. Liu, J. H. Liu, C. Y. Chou, Y. F. Yang, H. L. Kuo and C. C. Huang, Artif. Organs, 2008, 32, 468 CrossRef CAS .
- K. Tanaka, Y. Fujii, H. Atarashi, K. Akabori, M. Hino and T. Nagamura, Langmuir, 2008, 24, 296 CrossRef CAS .
- J. Wang, S. E. Woodcock, S. M. Buck, C. Chen and Z. Chen, J. Am. Chem. Soc., 2001, 123, 9470 CrossRef CAS .
- J. Wang, C. Chen, S. M. Buck and Z. Chen, J. Phys. Chem. B, 2001, 105, 12118 CrossRef CAS .
- T. Miyamae and H. Nozoye, Surf. Sci., 2003, 532–535, 1045 CrossRef CAS .
- A. Rao, H. Rangwalla, V. Varshney and A. Dhinojwala, Langmuir, 2004, 20, 7183 CrossRef CAS .
- S. J. Kweskin, K. Komvopoulous and G. A. Somorjai, Langmuir, 2005, 21, 3647 CrossRef CAS .
- M. L. Clarke, C. Chen, J. Wang and Z. Chen, Langmuir, 2006, 22, 8800 CrossRef CAS .
- X. F. Wang, H. G. Ni, D. W. Xue, X. P. Wang, R. R. Feng and H. F. Wang, J. Colloid Interface Sci., 2008, 321, 373 CrossRef CAS .
- J. Wang, Z. Paszti, M. A. Even and Z. Chen, J. Am. Chem. Soc., 2002, 124, 7016 CrossRef CAS .
- C. Chen, M. L. Clarke, J. Wang and Z. Chen, Phys. Chem. Chem. Phys., 2005, 7, 2357 RSC .
- H. Noguchi, H. Minowa, T. Tominaga, J. P. Gong, Y. Osada and K. Uosaki, Phys. Chem. Chem. Phys., 2008, 10, 4987 RSC .
- The n′ value for the PMMA/water interface was difficult to determine. Hence, it was assumed to be an average value of refractive index for PMMA and water, 1.49 and 1.33 at the wavelength.
- R. L. Jones, S. K. Kumar, D. L. Ho, R. M. Briber and T. P. Russell, Nature, 1999, 400, 146 CrossRef CAS .
- Y. Fujii, K. Akabori, K. Tanaka and T. Nagamura, Polym. J., 2007, 39, 928 CrossRef CAS .
- Q. F. Li, R. Hua, I. J. Cheah and K. C. Chou, J. Phys. Chem. B, 2008, 112, 694 CrossRef CAS .
- C. Hirose, N. Akamatsu and K. Domen, J. Chem. Phys., 1992, 96, 997 CrossRef CAS .
- C. Hirose, N. Akamatsu and K. Domen, Appl. Spectrosc., 1992, 46, 1051 .
- C. Hirose, H. Yamamoto, N. Akamatsu and K. Domen, J. Phys. Chem., 1993, 97, 10064 CrossRef CAS .
- N. Akamatsu, K. Domen and C. Hirose, J. Phys. Chem., 1993, 97, 10070 CrossRef CAS .
- N. Watanabe, H. Yamamoto, A. Wada, K. Domen, C. Hirose, T. Ohtake and N. Mino, Spectrochim. Acta, Part A, 1994, 50, 1529 CrossRef .
- X. Wei, X. Zhuang, S.-C. Hong, X. Zhuang, T. Goto and Y. R. Shen, Phys. Rev. Lett., 1999, 82, 4256 CrossRef CAS .
- G. J. Simpson and K. L. Rowlen, J. Am. Chem. Soc., 1999, 121, 2635 CrossRef CAS .
- S. Ye, S. Morita, G. Li, H. Noda, M. Tanaka, K. Uosaki and M. Osawa, Macromolecules, 2003, 36, 5694 CrossRef CAS .
- X. Wei, S.-C. Hong, X. Zhuang, T. Goto and Y. R. Shen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 5160 CrossRef CAS .
- C. A. Fleischer, A. R. Morales and J. T. Koberstein, Macromolecules, 1994, 27, 379 CrossRef CAS .
- S. Dirlikov and J. L. Koenig, Appl. Spectrosc., 1979, 33, 551 CrossRef CAS .
- B. Schneider, J. Štokr, P. Schmidt, M. Mihailov, S. Dirlikov and N. Peeva, Polymer, 1979, 20, 705 CrossRef .
- G. Li, S. Ye, S. Morita, T. Nishida and M. Osawa, J. Am. Chem. Soc., 2004, 126, 12198 CrossRef CAS .
- L. S. Wan, X. J. Huang and Z. K. Xu, J. Phys. Chem. B, 2007, 111, 922 CrossRef CAS .
- Q. Du, R. Superfine, E. Freysz and Y. R. Shen, Phys. Rev. Lett., 1993, 70, 2313 CrossRef CAS .
- Q. Du, E. Freysz and Y. R. Shen, Science, 1994, 264, 826 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.