Structured hydrophilic domains on silicone elastomers†
Vinodh
Rajendra
,
Yang
Chen
and
Michael A.
Brook
*
Department of Chemistry, McMaster University, 1280 Main St. W., Hamilton, ON, Canada L8S 4M1. E-mail: mabrook@mcmaster.ca; Fax: +1 905 522 2509; Tel: +1 905 525 9140
First published on 21st December 2009
Abstract
The controlled generation of hydrophilic structures within hydrophobic polymers can be challenging. Very few examples of such structures have been described for silicones. We now report that such structures can be encoded in the air-contacting layer of a silicone elastomer by the formation of silica domains from tetraethoxysilane, optionally in the presence of poly(ethylene glycol) (PEG), using a surface active aminopropylsilicone catalyst and moisture cure. The control of the relative modulus at the upper versus lower layers and the degree and type of hydrophilic structuring requires control over the efficiency of delivery of water to the core of the pre-elastomer, which is facilitated by the surface active catalyst and may additionally be manipulated by the addition of PEG.
Introduction
Silicone elastomers are widely used as biomaterials, amongst other reasons, because of their facile fabrication in a variety of complex shapes, their high oxygen permeability, which is required in ophthalmic applications such as contact lenses,1and the very low degree to which the human body responds to these synthetic materials.2 However, silicones are exceptionally hydrophobic, with surface energies at or below 23 nN m−1.3,4 While this can be beneficial in treatments to reduce the intensity of scar tissue,5 more frequently, consequent lipid uptake6 and protein adsorption7 at biomaterials interfaces can compromise otherwise useful silicone-based devices.Silicone elastomers have also been broadly used as components in drug delivery systems. With rare exception,8–10only hydrophobic drugs that readily diffuse through silicones have been delivered, either internally or topically.11The delivery of hydrophilic drugs from silicones, by contrast, requires internal structuring of the silicone such that water can penetrate, and the hydrophilic drug can then escape the body of the silicone elastomer. Such structuring has previously been achieved by the use of proteins,8 or the use of surfactants generated by the drug itself.9
The objective of the current study was to develop synthetic strategies that would permit the structuring of silicones in two different ways. First, it should be possible to control the hydrophilicity and roughness of the interface that would ultimately contact a biological environment and, second, the silicone elastomer proximal to the external interface should be internally structured so that the modulus can be controlled and that hydrophilic domains can be incorporated, which would ultimately mediate the release profile of hydrophilic drugs. Both elements should affect in a positive way the biocompatibility of silicone elastomers.
The strategy adopted makes use of previous observations9 that poly(ethylene glycol) (PEG), a polymer widely recognized for its biocompatibility,12 can be incorporated in silicones to facilitate water ingress and subsequent rates of bioactive release. Although there have been several studies in which internal structuring of two different domains was accomplished using covalent linkages,13,14 we chose to use PEG that was not chemically tethered to the silicone. Instead, structuring was provided by the presence of silica synthesized in situ during polymerization. We describe below the preparation of silicone elastomers with structurally controlled surface chemistry, morphology and, additionally, internal structuring of the silicone in the vicinity of the air interface, by use of a combination of cure kinetics and surface-active agents.
Experimental section
Materials
Fluorescein (Aldrich), ninhydrin (Pierce), tetraethoxysilane (TEOS, 99.999%, Aldrich), hydroxy-terminated PDMS (1800–2200 cSt, ∼36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Aldrich) aminopropyl-terminated PDMS (10–15 cSt, ∼875 g mol−1, Gelest), poly(ethylene glycol) (PEG, 1000 g mol−1, Aldrich), and the rake surfactant seen in 1 (silicone backbone, ethylene oxide/propylene oxide sidechains, DC 3225C (Fig. 1), Dow Corning) were used as received.
000 g mol−1, Aldrich) aminopropyl-terminated PDMS (10–15 cSt, ∼875 g mol−1, Gelest), poly(ethylene glycol) (PEG, 1000 g mol−1, Aldrich), and the rake surfactant seen in 1 (silicone backbone, ethylene oxide/propylene oxide sidechains, DC 3225C (Fig. 1), Dow Corning) were used as received.
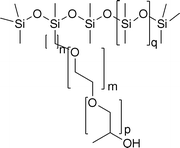 | ||
| Fig. 1 Structure of DC 3225c. | ||
Characterization
Detection and characterization of PDMS, aminopropyl-terminated PDMS and PEG content of elastomers were established using 1H NMR performed on a Bruker AV-200 spectrometer operating at 200.13 MHz. In the case of extracts from the elastomer disks, trimethoxybenzene (TMB) was used as an internal standard and was prepared by first making a stock solution containing approximately 0.03 g of TMB in 1 mL of CDCl3.Elastomer preparation
Silicone elastomers were prepared by mixing various quantities of hydroxy-terminated PDMS, PEG, aminopropyl-terminated PDMS, and TEOS (Table 1). Efficient mixing required a precise order of adding ingredients. PDMS was measured using a 5 ml plastic syringe and poured into a 20 ml glass vial. PEG was pre-weighed and then placed into the glass vial where it was weighed once again, TEOS was added using an Eppendorf 1000 μl pipette and then the mixture was gently heated to a temperature of ∼60 °C for approximately 1–2 min. This step was continued until the PEG liquefied. After cooling for 30 s, the aminosilicone catalyst was added to the mixture and then manually mixed for approximately 5 min, leading to a cloudy liquid. Some of the formulations additionally made use of the surfactant DC 3225c, in which case it was added using a 1000 μl Eppendorf pipette immediately after the mixture had cooled to room temperature. The mixture was then poured into a plastic Petri dish (35 mm diameter) lined with a Teflon film and allowed to cure at room temperature for 3–4 days. Some of the elastomers were cured under constant humidity using an ESL-2CA constant humidity chamber (ESPEC) at room temperature at 90% relative humidity, over the course of 3–4 days. In some experiments, disposable polystyrene cuvettes (1 × 1 × 4.5 cm, Aldrich) were used instead of Petri dishes. Once cured, the elastomers were cut out as circular disks using a 0.6 cm diameter coring tool for further use.| Compound | Controls (entries 1–5)a | Varying TEOS (entries 6–10) | Varying catalyst (entries 11–13) | Controls at 90% humidity (entries 14–18) | Varying TEOS at 90% humidity (entries 19–23) | 1% DC 3225C (entry 24) | 5% DC 3225C (entries 25–27) |
|---|---|---|---|---|---|---|---|
| a Refers to specific formulations described in more detail on Table 2. | |||||||
| PDMS | 2.125–1.625 (85–65%) | 2.000–1.500 (80–60%) | 1.900–1.750 (76–70%) | 2.125–1.625 (85–65%) | 2.000–1.500 (80–60%) | 1.725 (69%) | 1.775–1.700 (71–68%) |
| TEOS | 0.125–0.625 (5–25%) | 0.125–0.625 (5–25%) | 0.375 (15%) | 0.125–0.625 (5–25%) | 0.125–0.625 (5–25%) | 0.375 (15%) | 0.375 (15%) |
| Aminopropyl-terminated PDMS | 0.250 (10%) | 0.250 (10%) | 0.100–0.250 (4–10%) | 0.250 (10%) | 0.250 (10%) | 0.250 (10%) | 0.100–0.175 (4–7%) |
| PEG | 0 | 0.125 (5%) | 0.125 (5%) | 0 | 0.125 (5%) | 0.125 (5%) | 0.125 (5%) |
| DC 3225C | 0 | 0 | 0 | 0 | 0 | 0.025 (1%) | 0.125 (5%) |
Preparation of ninhydrin solution and testing for the location of amines in the elastomer
Circular elastomer disks were prepared as shown in Fig. 2, and placed within 20 ml glass vials to which approximately 5 ml of a 1wt% ninhydrin solution in reagent grade methanol were added. Samples were left for 30 min in the solution and thereafter the samples were checked by eye for a positive response (purple color change).15 | ||
| Fig. 2 Preparation of elastomers for fluorescence microscopy. | ||
Soxhlet extraction
Elastomer disks were placed in cellulose extraction thimbles and then Soxhlet extracted with reagent grade hexanes overnight. Elastomers were then dried in a vacuum oven at room temperature overnight. The differences in weight were noted and 1H NMR spectra were taken of the extracted solution after evaporation of the hexanes.Characterization of elastomer extracts
Exceptionally, the elastomers made with 5% TEOS exhibited a phase-separated liquid on the surface. The characterization of the surface liquid involved coring a circular disk 0.6 cm in diameter and then washing it with 1 mL of CDCl3, with which the 1H NMR was taken (see ESI† ).Surface imaging
Circular disks were cut out as described below and characterized by X-ray photoelectron spectrophotometry (XPS, University of Toronto) using a Thermo Scientific Theta Probe XPS spectrometer (ThermoFisher, E. Grinstead, UK) in standard mode. The angles were taken relative to the surface normal. A monochromatic Al KαX-ray source was used, with a spot area of 400 μm. Charge compensation was provided utilizing the combined e−/Ar+ flood gun. The position of the energy scale was adjusted to place the main C 1s feature (C–C) in the high resolution spectrum at 284.4 eV, the literature value for PDMS.16Data processing was performed using the software (Avantage) provided with the instrument. Fig. 4 shows an approximately linear decrease in the atomic% of nitrogen measured on elastomer surfaces at take-off angles of 30°, 50° and 70° as a function of TEOS concentration. For one sample, XPS was performed using the Kα probe at 20°, 30°, and 90°.Scanning electron microscope (SEM) images of elastomers were obtained using a JEOL 7000F SEM at an accelerating voltage of 5 kV. Energy dispersive X-ray (EDX) analysis was performed on the JEOL 7000F SEM at 10 kV. When obtaining cross sections, elastomers were initially frozen in liquid nitrogen and then fractured before mounting.
Surface roughness – profilometry
Circular elastomer disks, with a diameter of 0.6 cm, were gold coated with an Edwards Sputter Coater S150B (using a rate of ∼15 nm min−1) for 30 s to give a 7.5 nm thick coating. Surface roughness was measured with a WYKO NT1100 optical profiler equipped with the Vision32 software that imaged the surface texture of the silicone elastomers. Imaging was performed at 5× and 20× magnification with the low magnification PSI setting turned on at the profilometer, but run in VSI mode in the software, as greater image quality was achieved.Preparation of fluorescein-containing elastomers and use of the optical microscope
Optical microscopy was used to image the surface of the elastomers and to determine the type of internal structuring of the silicone. Surfaces and hydrophilic PEG/silica domains were visualized using a 1.2 mM solution of fluorescein (0.008 g, 0.024 mmol) in distilled water (20 mL). The circular elastomer disks of interest were cut as shown in Fig. 2 and then were placed within 20 ml glass vials. Approximately 5 ml of fluorescein solution were added to the vials using a glass pipette, which were stored in the dark for approximately 3–4 days and then analyzed using fluorescence spectroscopy. For imaging, the samples were first quickly rinsed with distilled water and then placed onto glass slides underneath an Olympus BX51 microscope fitted with a Q-imaging, Retiga EXi camera. Fluorescence and light microscopy images were taken of the elastomers and recorded on the Image Pro-Plus software by Media Cybernetics. Wavelengths of the elastomer surfaces were performed using the Image Pro-Plus software.Results
Silicone elastomers were prepared using a room temperature vulcanization (RTV) system that involved TEOS (Si(OEt)4) as the crosslinker and hydroxy-terminated dimethylsilicone (HO(Me2SiO)nH) 36000 Mw as the bridging linear polymer (Fig. 3). No filler was added. However, excess TEOS was added to the recipe, which led to the generation of silicain situ, a process described by Mark and others.17 Low molecular weight aminopropyl-terminated PDMS (referred to as ‘catalyst’ below) was utilized as the hydrolysis/condensation catalyst. Note that aminecatalysts are far inferior, with respect to rate of cure, to the more commonly used tin- and titanium-based catalysts.18 However, we wished to avoid the use of metals in these elastomers that may be targeted for biomaterials’ applications. The final requirement for cure is water, which is important both for cure, and for the generation of silica.
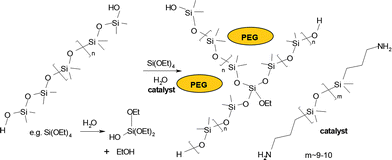 | ||
| Fig. 3 General chemical equation for RTV silicone crosslinking. | ||
Hydrophilic domains were to be introduced into the elastomer using two strategies: silica formed from TEOShydrolysis, as noted above, and the use poly(ethylene glycol), which was optionally added into the pre-elastomer formulation. It was recognized that efficient distribution of the PEG into the silicone, particularly as channels rather than droplets, might require the presence of surface-active agents because of the near immiscibility of the two materials. Therefore, a siliconesurfactant based on PEG and silicones (DC 3225C) was optionally added to the pre-elastomer mixture. Thus, mixing the precursors (Table 1, Table 2), placing them in an open vessel lined with a Teflon® film and allowing them to cure at ambient temperature for up to 4 days led to translucent or opaque silicone elastomers.
| Entry | Elastomera | Translucent or opaque | Phase separation | Homogeneous (Y/N) | Surface hardnessb | Bottom hardnessb |
|---|---|---|---|---|---|---|
| a For more detailed formulations, see Table 1. b Arbitrary hardness scale where 1 = viscous liquid, 5 = shore A hardness of 40, 10 = hard/brittle rubber. | ||||||
| Effect of TEOS | Humidity = 20–30% | |||||
| Samples | Without PEG | |||||
| 1 | 5% TEOS | T | Liquid on surface | Y | 3 | 3 |
| 2 | 10% TEOS | T | None | N | 5 | 3 |
| 3 | 15% TEOS | T | None | N | 7 | 4 |
| 4 | 20% TEOS | T | None | N | 8 | 2 |
| 5 | 25% TEOS | T | None | N | 10 | 1 |
| Samples | Containing PEG | |||||
| 6 | 5% TEOS | O | Liquid on surface | Y | 4 | 4 |
| 7 | 10% TEOS | O | None | N | 5 | 4 |
| 8 | 15% TEOS | O | None | N | 7 | 4 |
| 9 | 20% TEOS | O | None | N | 8 | 4 |
| 10 | 25% TEOS | O | None | N | 10 | 5 |
| Effect of catalyst | Humidity = 20–30% | |||||
| 11 | 10% Cat. | O | None | N | 7 | 4 |
| 12 | 7% Cat. | O | None | N | 6 | 4 |
| 13 | 4% Cat. | O | None | Y | 5 | 5 |
| Effect of humidity | Humidity = 90% | |||||
| Samples | Without PEG | |||||
| 14 | 5% TEOS | T | Liquid on surface | Y | 4 | 4 |
| 15 | 10% TEOS | T | None | Y | 5 | 5 |
| 16 | 15% TEOS | T | None | Y | 5 | 5 |
| 17 | 20% TEOS | T | None | N | 6 | 5 |
| 18 | 25% TEOS | T | None | N | 8 | 4 |
| Samples | Containing PEG | |||||
| 19 | 5% TEOS | O | Liquid on surface | Y | 4 | 4 |
| 20 | 10% TEOS | O | None | Y | 5 | 5 |
| 21 | 15% TEOS | O | None | Y | 5 | 5 |
| 22 | 20% TEOS | O | None | N | 6 | 5 |
| 23 | 25% TEOS | O | None | N | 6 | 5 |
| Effect of surfactant | Humidity = 20–30% | |||||
| 24 | 1% S, 10% Cat. | O | None | N | 7 | 5 |
| 25 | 5% S, 10% Cat. | O | None | N | 7 | 5 |
| 26 | 5% S, 7% Cat. | O | None | N | 6 | 5 |
| 27 | 5% S, 4% Cat. | O | None | Y | 5 | 5 |
Controlling cure asymmetry
RTV silicones are usually formulated with all required reagents, except water, which is provided by humidity in the environment. As a consequence, cure in such silicones initially takes place at the outer, air-contacting surface, and then proceeds further into the uncured body as moisture migrates through the silicone. Formulation is normally optimized so that a homogeneous elastomer is produced.Our objective was to create surface-modified silicones with hydrophilic domains, which could be accomplished by controlling the relative rates of crosslinking, silica formation, and the rates of diffusion of reagents through the uncured mixture. The silicones formulated here cured slowly and, only exceptionally, led to homogenous materials. In general, the elastomer cured asymmetrically, generating an elastomer with a higher modulus proximal to the air-contacting surface, and softer elastomers beneath. The natures of the silicones produced were examined as a function of aminopropylsilicone catalyst, water, and TEOS concentrations.
Effect of varying crosslinker concentration
A series of elastomers were prepared with and without PEG in which the crosslinker wt% was varied (Table 2). At low crosslinker concentrations (5 wt%), a viscous liquid was extruded onto the surface of the elastomer after cure. The nature of this separated liquid will be discussed below. Otherwise, the greater the concentration of TEOS, the greater was the difference in modulus between the outer surface (from 10 μm to 2 mm) and the remainder of the elastomer. At the extremes of the materials prepared, the outer surface was a brittle, friable layer, and the underlying silicone an almost uncured gel -like material. When PEG was also included in a given formulation, the elastomers cured more homogeneously, with lower differences in moduli between exposed and underlying elastomers (Table 2).Effect of varying catalyst concentration
Elastomers were prepared using high catalyst concentrations between 4 and 10%, with the crosslinker fixed at 15%. The asymmetry of cure increased with increasing catalyst concentration: only elastomers prepared with 4% catalyst were homogeneous (Table 2).Effects of Humidity
Atmospheric water is a co-catalyst for silicone crosslinking and a reagent for TEOShydrolysis. The cure rate, not surprisingly, was directly affected by moisture in the environment. More significant was the interplay between humidity, TEOS concentration and asymmetry with which the elastomers cured. At high humidity (e.g., 90% relative humidity, RH) and TEOS concentrations below 15%, the resulting elastomers were homogenous: less water or more TEOS led to asymmetric elastomers that were more highly cured at the air interface. Lower surface/volume ratios of the curing vessel will reduce the efficiency with which water can penetrate the curing pre-elastomer. For example, a recipe that led to homogenous elastomers prepared in a high surface area/volume Petri dish (e.g., 15% TEOS, 90% RH), was asymmetric when cured in a 1 × 1 × 4.5 cm cuvette; the top 0.5 cm was much harder than the remaining 4 cm depth.Relative rates: cure vs. reagent diffusion in the silicone
The ingress of atmospheric water controls the asymmetry of cure. Under conditions of high water concentration (e.g., 90% RH), diffusion of water through the curingsilicone occurred more rapidly than crosslinking and diffusion of TEOS to the air interface, such that the entire body underwent nearly simultaneous cure. PEG, when added into the pre-elastomer mixture, acts to wick or transmit water into the silicone, leading to a lower degree of cure asymmetry than when PEG is omitted from formulae. Any factor that reduces the rate of water ingress, when compared to diffusion or crosslinking processes through the silicone, will lead to an asymmetric cure. At higher catalyst or TEOS loadings, for instance, the rates of cure/silica formation at the interface overwhelm the ability of water to penetrate the silicone, leading to asymmetry and, at the extremes, brittle resins on the surface, and lightly cured gels beneath. Asymmetric RTV systems are avoided in commerce by controlling the viscosity of the uncured silicone, for example by the use of fillers.Structuring silicone interfaces
The development of more biocompatible surfaces requires control of a variety of different structural features including surface geometry, hydrophilicity, and internal structuring. Control of the hydrophilic silica/PEG domains within the elastomer is important for the delivery of bioactives.9 In the remainder of the manuscript, we examine in detail the structural features of the more highly cured, upper elastomer interface.Surface chemistry
Asymmetric cure is a consequence of higher reaction rates at the air interface. The distribution of amine groups, from the RTV catalyst, aminopropylsilicone, within the silicone matrix was investigated colorimetrically, and by XPS. After submersing elastomer disks into a ninhydrin solution, asymmetric elastomers exhibited the characteristic purple color, arising from reaction of ninhydrin with amines,15 preferentially at the air interface (Fig. 4): a gradient in color existed from air interface inwards and, therefore, catalyst concentration decreased from the air surface to the silicone interior. Note that sides of the elastomer disks cut from a larger sample, and bottom surfaces that contacted the Teflon® sheet exhibited no color.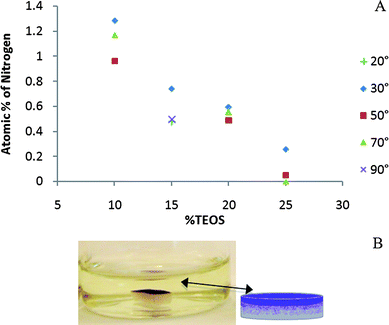 | ||
| Fig. 4 A: Nitrogen (catalyst) concentration (XPS) at surface with varying TEOS concentrations at different take-off angles (the 15% sample was examined at 2 different angles than the other samples). B: Ninhydrin staining showing presence of catalyst preferentially at the air interface. | ||
Angular-dependent XPS measurements also showed that the amine preferentially resided at the air interface. As noted above, asymmetry of the elastomer increased with TEOS concentration. Asymmetry correlated inversely with the amine concentration at the air interface (Fig. 4, see also ESI† ): more highly asymmetric materials exhibited lower surface levels of amine. In the case of the elastomer prepared with only 5 wt% TEOS (Table 2, entry 6), a viscous liquid phase separated from the elastomer. 1H NMR spectra showed that the phase-separated liquid contained ∼40 mg of catalyst (of 250 mg, 16%), ∼12.5 mg of PEG (of 125 mg, 10%) and less than ∼21.3 mg of PDMS (of 2.0 g, Table 1, Table 2, about 1%).
The preferential presence of the amine at the interface, a consequence of efficient migration to the interface which has previously been seen with aminopropyltrialkoxysilanes in epoxypolymers,19 manifests itself in various ways. The aminopropylsilicone, while less effective than the traditional and more efficient tin- or titanium-based RTV catalysts, still facilitates both curing of the silicone and formation of silica. Appropriate formulation thus permits simultaneous control of surface morphology, roughness and hydrophilicity: at pH values near neutrality, amine groups will be protonated leading to surface charge and, as discussed below in more detail, surface active species that may assist in geometric control of the silica formed.
Surface roughness
At low humidity levels there was a clear trend between increasing TEOS concentration and increasing surface roughness‡ up to 20% TEOS, at which point the roughness then decreases (Fig. 5A): the presence of PEG led to slightly smoother surfaces (see ESI† ).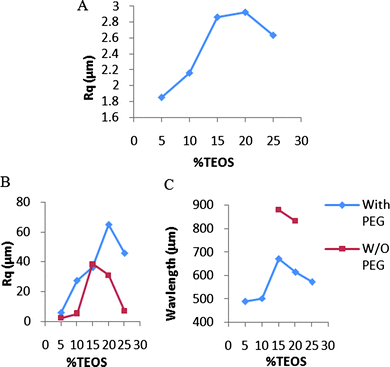 | ||
| Fig. 5 A: Roughness of surfaces with increasing crosslinker by profilometry (Table 2, entries 6–10). B: Roughness of surfaces with increasing crosslinker at 90% RH by profilometry (Table 2, entries 14–23). C: Wavelength of elastomers cured at 90% RH (Table 2, entries 16, 17, 19–23). | ||
There was an inverse trend between catalyst concentration and roughness. With the crosslinker TEOS concentration fixed at 15 wt%, elastomers were prepared using catalyst concentrations between 4 and 10%. The highly inhomogeneous elastomers prepared at 10% and 7% catalyst, respectively, exhibited roughnesses of 2.86 μm and 2.16 μm (Fig. 6A, ESI† ). By contrast, the homogenous elastomer prepared with 4% catalyst had an Rq value of 364 nm, making it significantly smoother than those prepared with higher catalyst levels (Fig. 6B).
 | ||
| Fig. 6 Profilometry images of elastomers prepared with 15% TEOS and A: 10% catalyst (Table 2, entry 11), B: 4% catalyst (Table 2, entry 13). Scale bars represent 50 μm. C: SEM image of elastomer prepared with 25% TEOS, 10% catalyst, and 90% RH (entry 23). Scale bar represents 1 mm. | ||
Atmospheric moisture also had a significant impact on the surface roughness of the elastomers. When the same formulations as those just described were cured at high humidity (90% RH) random structures on the surface did not form and instead highly corrugated, macroscopic wave structures formed on elastomers (Fig. 5B,C). These waves could be readily seen by eye, with wavelengths of up to 880 μm (Fig. 6C, ESI† ). In the absence of PEG, only pre-elastomers containing 15% or 20% crosslinker developed these structures. In the presence of PEG, however, all elastomer formulations developed waves but with shorter wavelengths and greater amplitudes than when PEG was absent (Fig. 5B,C).
Internal structural organization
Internal hydrophilic structures could arise from the presence of PEG, when present, and silica formed by the hydrolysis and condensation of TEOS.20 In asymmetric elastomers, the harder layer near the air interface exhibited a different distribution of hydrophilic structures than the softer underlayer. The greater organization in the more highly crosslinked upper layer could be readily seen, after staining with fluorescein, using fluorescence microscopy (Fig. 7).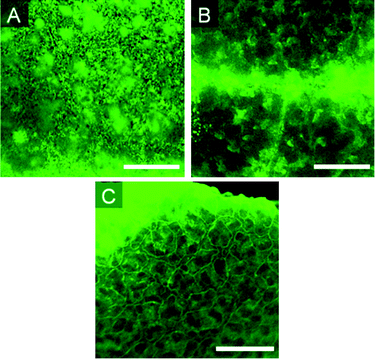 | ||
| Fig. 7 Fluorescence images of elastomers with varying amount of crosslinker. A: 5%. B: 15%. C: 25% (Table 2, entries 6, 8 and 10). Scale bar = 500 μm. | ||
Effect of crosslinker concentration
The structure of hydrophilic domains in PEG-containing samples changed significantly with TEOS concentration (Table 2, entries 6–10). At 5 wt% TEOS, the dispersed PEG/silica globules were approximately 10 μm in diameter and relatively monodisperse. With the higher concentration of 15% TEOS, the hydrophilic structures appear as thick ribbons on the order of 100–500 μm at their widest point, more than 10 times larger than the globules seen at 5%. At 25% TEOS, a highly reticulated 3D structure had evolved with structures of 50–100 μm in width. Other factors that increase asymmetry in the elastomer, such as increased catalyst concentration, similarly lead to more internal organization (see ESI† ).Addition of surfactant
It will be proposed below that internal structuring is facilitated by surface-active materials, including the aminopropylsilicone catalyst. This hypothesis was tested by adding a PEG/silicone rake surfactant, DC 3225C, to the pre-elastomer. Using a 15% TEOS formula, the surfactant was added at levels of 1% and 5%, respectively; three different catalyst loadings were used (Table 2, entries 24–27, Fig. 8). It can be seen that increased concentrations of surfactant and of catalyst both contribute to the formation of greater interconnectivity of hydrophilic channels. The sample containing 5% surfactant and 10% catalyst has hydrophilic domains of sizes below the resolution of light microscopy. | ||
| Fig. 8 Fluorescence images of elastomers prepared with 15% TEOS and A: 1% surfactant (S), 10% catalyst (C), B: 5% (S) 10% (C), C: 5% (S) 7% (C), D: 5% (S) 4% (C) (Table 2, entries 24–27). Scale bar = 500 μm. | ||
Discussion
Asymmetric cure and surface modification
RTV silicone elastomers are normally designed to be homogeneous throughout the polymer body. Commercial samples are pre-filled, normally with silica, but also with less expensive, non-reinforcing fillers such as calcium carbonate.21 In such cases, the only reaction designed to occur during cure of a one-part RTV is hydrolysis/condensation between silicones terminated with silanols and TEOS; silica formation is not anticipated. The crosslinking reaction occurs first at the air interface, which is normally the only source of water (water is a co-catalyst in the case of tin- or titanium-based catalysts22). The air-presenting surface thus cures and becomes tack-free much more rapidly than the layer beneath. The high viscosity of a filled silicone pre-elastomer normally leads to homogenous materials, consistent with diffusion of water through the matrix being faster than diffusion of other materials within the formulation, and because cure (silanols + crosslinker) occurs more rapidly than TEOShydrolysis/condensation with the activated catalysts.In the silicone elastomers described above, the use of an atypical (slow) catalyst, aminopropylsilicone, in low viscosity, unfilled RTV systems, alters the cure profile significantly. Only in cases where water ingress is appreciably faster than either crosslinking or silica formation is a homogeneous silicone formed. Such cases occurred at high RH, low TEOS concentrations and/or low catalyst concentrations. Even in such cases, however, inhomogeneity can result if the water must travel relatively long distances through the silicone. For example, the low surface area/volume sample (1 × 1 × 4.5 cm deep) gave a highly cured elastomer only to a depth of about 0.5 cm even with cure at 90% RH. Thus, in general, crosslinking and silica formation occurred faster, and to a greater extent, at the air interface of these silicones.
The structural features of greatest interest of these asymmetric cured silicones are: interfacial hydrophilicity, roughness, and internal structuring. The synthetic parameters that allowed their control (Table 2) are examined in turn. The amine catalyst (Fig. 3) has a nitrogen content about 4.4 atom%. As seen from Fig. 4, the surfaces of the cured silicone elastomer can be highly enriched in catalyst (nearly 25% of the material present) at the upper interface.19 The degree to which the upper interface is populated by amines is a function of the catalyst concentration, and of the TEOS and water concentrations. More rapid cure and higher concentrations of TEOS were associated with decreased catalyst presence at the interface. The catalyst thus diffuses to the interface unless extensive silica formation occurs more rapidly. Amine groups near neutral pH are protonated in aqueous media, which will impact both on the cure chemistry (see below) and the chemistry of the interface. Cationic surfaces have been proposed to be beneficial with biomaterials because they support cell growth (following adsorption of plasma proteins).
Higher concentrations of TEOS, and of catalyst, but lower water concentrations led to high degrees of crosslinking and of silica formation at the air interface (Fig. 9). At ambient humidity, the reactions at the interface, and diffusion of the reagents within the silicone to the surface, occur more rapidly than water vapor can diffuse into the silicone. At the extreme, with high levels of catalyst and TEOS under relatively dry conditions, rough, brittle layers form on top of nearly uncured silicone elastomers. Decreasing concentrations of either TEOS or catalyst, or increased water concentrations enhanced the thickness of the more highly cured and filled upper layer. Note that thicker overlayers were formed when PEG was present in the formulations. The hydrophilic nature of PEG provides a mechanism to deliver water further into the silicone than in its absence. That is, PEG lowers the barrier for water transmission such that the ratio of internal vs. surface crosslinking/silica formation is increased. Thus, as a consequence of the clear differences in reaction rates between cure/silica formation and the diffusion of moisture, it is possible to control both the modulus of the silicone at the air interface and the interfacial hydrophilicity.
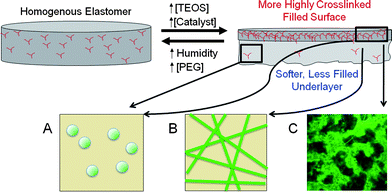 | ||
| Fig. 9 Asymmetry caused by increasing crosslinker. With increasing TEOS concentration, A: dispersed silica particles, B: assembled into ribbons, and then, C: into three-dimensional networks. | ||
At low humidity, very rough surfaces were observed at high concentrations of TEOS. Highly reticulated brittle surfaces, comprised mostly of silica, are unable to anneal during cure, leading to rough, kinetically formed structures. By contrast, when excess water was present, and particularly when PEG was present to deliver water into the core of the elastomer, well-defined waves could be clearly seen even by eye at the external interface. The wavelength (up to nearly 1 mm) and amplitude (up to 250μm, see ESI† ) of these waves were affiliated with much lower surface crosslink densities, but higher than the underlayers. Such waves, particularly on silicone elastomer surfaces, have previously been examined by a variety of authors.23–27 During cure, two elastomer layers form. The higher modulus elastomer at the air interface shrinks and the resulting waves are a consequence of yielding of the lower modulus underlayer. As seen from Fig. 5, the amplitude and wavelength of the surface can be manipulated over a large range particularly by changing the TEOS concentration, which directly correlates both with chemical crosslinking, and physical crosslinking via the silica particles.
Internal structural organization
The soft underlayer of the silicone elastomer contained a homogeneous dispersion of silica as isolated or slightly aggregated particles and, when present, of PEG as droplets. By contrast, as shown using fluorescence microscopy, highly organized hydrophilic domains presented in the upper layer except for very lightly crosslinked materials (Fig. 7A).Two features in particular controlled the nature of the internal hydrophilic structuring: TEOS concentration and the presence of surface-active species. As the concentration of TEOS increased, isolated particles first became linear ‘ribbons’ and then three-dimensional network structures (Fig. 7B,C, Fig. 9, ESI† ). The facility for 3D structuring increased with catalyst concentration (data not shown) and when silicone/PEGsurfactants were added (3225C, Fig. 8).
Two independent but closely related processes are occurring during hydrolysis and condensation: crosslinking of the siliconepolymer, and formation of silica due to the large excess of TEOS present. Of course, these processes are not mutually exclusive. While a single TEOS atom could lead to a 3- or 4-armed branch point (Fig. 3), condensation of the silicone can occur with silicic acid oligomers or silica ribbons/particles. Reinforcement of the silicone can also occur through physical interactions with silica. We focus now on the formation of silica.
The hydrolysis of TEOS is very slow at neutral pH. By contrast, rates increase rapidly away from pH 7. This pH sensitivity has been exploited to control silica structuring in various ways, perhaps most famously in the formation of model silicacolloids by Stöber, in which TEOS is hydrolyzed in ethanol with ammonia catalysis.28 We propose that analogous pH control is provided by the amino group on the aminopropylsilicone catalyst to facilitate hydrolysis/condensation of TEOS, yielding silica, primarily in particulate form.
The organization of the hydrophilic silica/PEG domains can be understood by considering the templated synthesis of meso- and macroporous silica from TEOS using surfactants. Perhaps best known of these are the syntheses of zeolites such as MCM-41 in which surfactants such as CTAB create cylinders around which silica synthesis occurs.29 Normally, such syntheses are undertaken in hydrophilic solvents, which may be aqueous or alcohol based, although strategies for assembly in organic solvents have been elegantly reviewed by Morris and Weigel.30
As with any of the templating processes used to prepare zeolites and other mineral species, a surface-active material guides the assembly of these structures by interactions occurring, in part, between complementary charged species. We propose, in the silicone elastomers formed using aminopropylsilicone catalysts, that the ammonium ions on the catalyst can interact with silanolates on silica surfaces (see model in Fig. 10). The polar silica interface is stabilized in the hydrophobic silicone elastomer matrix by such an interaction. Similar types of interactions, although based on non-ionic/polar interactions can occur when the siliconesurfactant 3225C is present (Fig. 8, Fig. 10). Such interactions have been used to direct the assembly and porosity of silica particles31 and silica monoliths.32As the concentration of surfactant is increased, the hydrophilic silica domain size is decreased (Fig. 8).
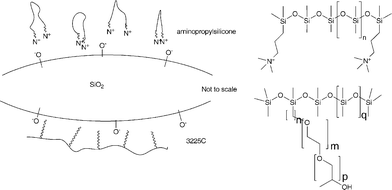
The biocompatibility of silicone species relies on interactions that take place at the interface of the device. As shown above, the use of a surface active catalyst in an RTV system, in combination with control of crosslinker and water concentrations, permits the preparation of elastomer surfaces with controlled geometry and with internal hydrophilic structuring. We shall report in due course on the biocompatibility of such systems per se, and the ability to release hydrophilic bioactive species from the structured silicones.
Conclusion
The density and organization of hydrophilic silica/PEG domains within silicone RTV elastomers can be controlled by manipulating the relative rates of hydrolysis/condensation with respect to diffusion of water through the matrix. At high TEOS (crosslinker and silica precursor) concentrations, preferential reactions occur proximal to the air interface. Surface-active materials, which include the aminopropylsilicone cure catalyst and, optionally, PEG-modified siliconesurfactants act to assemble the hydrophilic domains into ribbons and three-dimensional structures.Acknowledgements
We acknowledge with gratitude the financial support of the Natural Sciences and Engineering Research Council of Canada and 20/20: NSERC Ophthalmic Materials Network, and thank Renita D'souza for assistance with the profilometry measurements.Notes and references
- D. Jones, Elastomerics, 1988, 120, 12–16 Search PubMed.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials Science, Academic Press, New York, 1999 Search PubMed.
- M. J. Owen, in Silicon-Based Polymer Science: A Comprehensive Resource, ed. J. M. Zeigler and F. W. G. Fearon, American Chemical Society, Washington, D.C., 1990, pp. 705–739 Search PubMed.
- M. J. Owen, in Siloxane Polymers, ed. S. J. Clarson and J. A. Semlyen, Prentice Hall, Englewood Cliffs, 1993, p. 309 Search PubMed.
- W. Sanchez, J. Evans and G. George, Aust. J. Chem., 2005, 58, 447–450 CrossRef CAS.
- L. Jones, M. Senchyna, M. A. Glasier, J. Schickler, I. Forbes, D. Louie and C. May, Eye Contact Lens Sci. Clin. Pract., 2003, 29, S75–79 Search PubMed , discussion S83–74, S192–194.
- L. Werner, Curr. Opin. Ophthalmol., 2008, 19, 41–49 CrossRef.
- M. Kajihara, T. Sugie, T. Hojo, H. Maeda, A. Sano, K. Fujioka, S. Sugawara and Y. Urabe, J. Controlled Release, 2001, 73, 279–291 CrossRef CAS.
- M. A. Brook, A. C. Holloway, K. K. Ng, M. Hrynyk, C. Moore and R. Lall, Int. J. Pharm., 2008, 358, 121–127 CrossRef CAS.
-
B. Ratner, C. Kwok, K. Walline, E. Johnston and R. J. Miller, Genzyme Corporation, USA, Silicone blends and composites for drug delivery, Int. Patent Application, WO 2004000382, 2003 Search PubMed.
- G. Di Colo, Biomaterials, 1992, 13, 850–856 CrossRef CAS.
- J. H. Lee, H. B. Lee and J. D. Andrade, Prog. Polym. Sci., 1995, 20, 1043–1079 CrossRef CAS.
- J. S. Turner and Y. L. Cheng, Macromolecules, 2000, 33, 3714–3718 CrossRef CAS.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 CrossRef CAS.
- T. W. G. Solomons and C. B. Fryle, Organic Chemistry, John Wiley and Sons, Hoboken, NJ, 2008 Search PubMed.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA 300 Database, John Wiley & Sons, Chichester, 1992 Search PubMed.
- S. J. Clarson and J. E. Mark, in Siloxane polymers, ed. S. J. Clarson and J. A. Semlyen, PTR Prentice Hall, Englewood Cliffs, NJ, 1993, p. 637 Search PubMed.
- J. M. Pujol and C. Prébet, J. Adhes. Sci. Technol., 2003, 17, 261–275 CrossRef CAS.
- R. T. Foister, J. Colloid Interface Sci., 1984, 99, 568–585 CrossRef CAS.
- M. A. Brook, in Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, pp. 309–339 Search PubMed.
- W. J. Noll, Chemistry and Technology of Silicones, Academic Press, New York, 1968 Search PubMed.
- M. A. Brook, in Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000, pp. 256–308 Search PubMed.
- N. Bowden, S. Brittain, A. G. Evans, J. W. Hutchinson and G. M. Whitesides, Nature, 1998, 393, 146–149 CrossRef CAS.
- W. T. S. Huck, N. Bowden, P. Onck, T. Pardoen, J. W. Hutchinson and G. M. Whitesides, Langmuir, 2000, 16, 3497–3501 CrossRef CAS.
- D. Breid and A. J. Crosby, Soft Matter, 2009, 5, 425–431 RSC.
- E. P. Chan and A. J. Crosby, Soft Matter, 2006, 2, 324–328 RSC.
- N. Patrito, C. McCague, P. R. Norton and N. O. Petersen, Langmuir, 2007, 23, 715–719 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- C. Brinker and G. Scherer, Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing, Academic Press, San Diego, 1990 Search PubMed.
- R. E. Morris and S. J. Weigel, Chem. Soc. Rev., 1997, 26, 309–317 RSC.
- R. Cademartiri, M. A. Brook, R. Pelton and J. D. Brennan, J. Mater. Chem., 2009, 19, 1583–1592 RSC.
- M. A. Brook, Y. Chen, K. Guo, Z. Zhang and J. D. Brennan, J. Mater. Chem., 2004, 14, 1469–1479 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR of liquid separated from the 5% TEOS elastomer (Table 2, entry 6), N atm% XPS data for over and underlayers as a function of TEOS concentration (Table 2, entries 7–10), surface profilometry images (Table 2, entries 6–10), Table of amplitudes and SEM images of surface waves (Table 2, entries 19–21), fluorescence microscope images (Table 2, entries 11–13). See DOI: 10.1039/b9py00220k |
‡ Surface profilometry provides roughness as Rq, 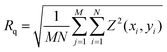 , the root mean square roughness (Wyko Surface Profilers Technical Reference Manual, Veeco Metrology Group, 1999, 16), where x and y = spatial axes, M and N = number of data points in the x and y direction, respectively, and Z = surface height relative to the reference mean plane. , the root mean square roughness (Wyko Surface Profilers Technical Reference Manual, Veeco Metrology Group, 1999, 16), where x and y = spatial axes, M and N = number of data points in the x and y direction, respectively, and Z = surface height relative to the reference mean plane. |
| This journal is © The Royal Society of Chemistry 2010 |