Aliphatic polyester elastomers derived from erythritol and α,ω-diacids†
Devin G.
Barrett
,
Wei
Luo
and
Muhammad N.
Yousaf
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: mnyousaf@email.unc.edu; Fax: +1 (919) 962-2388; Tel: +1 (919) 966-5074
First published on 5th January 2010
Abstract
Soft polyester elastomers have emerged as a promising family of biodegradable materials for drug delivery and tissue engineering. Specifically referring to soft tissue engineering, potential biomaterials should be elastic and flexible, so as to mimic the mechanical properties of natural tissue. Herein, we report the design of several elastomers based on the polycondensation of erythritol, a sugar substitute that is approved for human consumption by the Food and Drug Administration, and one of eight dicarboxylic acids: glutaric, adipic, pimelic, suberic, azelaic, sebacic, dodecanedioic, and tetradecanedioic acids. By varying the length of the diacid and the curing conditions, several elastomers were designed with a range of physical and mechanical properties. Poly(erythritol glutarate), poly(erythritol adipate), poly(erythritol pimelate), poly(erythritol suberate), poly(erythritol azelate), poly(erythritol sebacate) poly(erythritol dodecanedioate), and poly(erythritol tetradecanedioate) achieved Young's modulus, ultimate tensile stress, and rupture strain values of 0.08–80.37 MPa, 0.14–16.65 MPa, and 22–466%, respectively. Additionally, as tissue engineering may require the use of complex 3-dimensional designs, embossed films and porous films were designed in order to demonstrate the ease of processing. Hydrolytic degradation rates ranging from 100% in 3 weeks to 6.4% in 6 weeks were obtained in phosphate-buffered saline solutions at 37 °C. Finally, in vitro cytotoxicity was studied with Swiss albino 3T3 fibroblasts and human mesenchymal stem cells. Based on these results, we believe that the poly(erythritol dicarboxylate) series are excellent candidates for potential soft biomaterials.
1. Introduction
The demand for biodegradable polymers in medical applications, such as tissue engineering and drug delivery, has motivated the materials community to develop new polymers.1–3 Recently, elastomeric materials have become popular for soft tissue scaffolds due to their mechanical similarities to blood vessels, heart valves, cartilage, bladder, and other soft tissues.4,5 While several examples of polyester scaffolds have shown extensive promise, many of these materials suffer from complex or expensive synthetic designs.5 A simple and facile synthesis would allow for easy scale-up for industrial or medical applications.Within the class of polyester scaffolds, many examples of both thermoplastics and thermosets have been described.4 Thermosets offer a number of advantages for biomedical applications, when compared to thermoplastic materials. For example, thermoplastic elastomers often have crystalline regions, causing heterogeneous degradation and a non-linear loss of mechanical strength.6,7 Additionally, the three-dimensional (3D) geometry of a thermoplastic material is commonly lost during the hydrolysis period.8 Alternatively, thermosets can be designed from completely amorphous precursors, allowing for linear degradation and a predictable loss of mechanical strength.7 Polyester thermosets degrade by a combination of bulk and surface erosion, which often results in the retention of the 3D structure throughout the hydrolytic process.8
Therefore, we believe that the ideal polymer for biomedical applications should possess several characteristics. Biocompatibility and biodegradation are critical to minimize negative biological reactions to the material. The polymer should also be a thermoset with a glass transition temperature that is lower than physiological temperature so that no crystalline regions are present, minimizing the chances of heterogeneous degradation. Also, a wide range of degradation rates and mechanical properties should be accessible from a single facile and inexpensive design strategy. This would allow the ability to tailor a material's properties to best mimic those of the natural tissue.
Herein, we describe the synthesis and characterization of the poly(erythritol dicarboxylate) family of elastomers (PErD). Poly(erythritol glutarate) (PErGl), poly(erythritol adipate) (PErAd), poly(erythritol pimelate) (PErPi), poly(erythritol suberate) (PErSu), poly(erythritol azelate) (PErAz), poly(erythritol sebacate) (PErSe), poly(erythritol dodecanedioate) (PErDo), and poly(erythritol tetradecanedioate) (PErTe) were designed by thermal polycondensation of erythritol and 5-, 6-, 7-, 8-, 9-, 10-, 12-, and 14-carbon α,ω-diacids, respectively. By varying the length of the diacid, the rigidity and hydrophobicity of the resulting materials can be tuned, allowing for control over mechanical properties and degradation rates. The non-cured pre-polymers are easily melted or dissolved for facile processing, demonstrated by embossed films and porous scaffolds. Finally, cytotoxicity was studied in vitro with human mesenchymal stem cells (hMSCs) and Swiss albino 3T3 (SA) fibroblasts.
2. Experimental
2.1 Materials
All chemicals were purchased from Fisher Scientific (Philadelphia, PA), and used without further purification.2.2 Poly(erythritol dicarboxylate) synthesis
The synthesis of PErD was performed based on methods previously reported.9,10 Equimolar amounts of a dicarboxylic acid (glutaric, adipic, pimelic acid, suberic, azelaic, sebacic, dodecanedioic, or tetradecanedioic acid) and erythritol were combined in a round bottom flask, which was then sealed. Under an inert environment of argon, monomers were stirred at 145 °C. After a homogenous melt formed, the mixture was stirred for an additional 2 h. The pressure was then reduced to 2 Torr, and the materials continued stirring for 7 h. The round bottom flasks were immediately placed into a water bath (room temperature) to quench polycondensation. The pre-polymer was then dissolved in a minimal amount of tetrahydrofuran, purified by precipitation in −78 °C methanol, and dried by rotary evaporation. To form elastomeric films, different procedures were followed, depending on the desired properties of the material. Pre-polymers were cured at either 120 °C for 3 d or at 140 °C for 3 d. Additionally, oligo(erythritol glutarate) was cured at 120 °C for 2 days, resulting in an extremely flexible material.2.3 Polyester characterization
1H NMR spectra of pre-polymers were acquired on a Bruker 400 MHz AVANCE spectrometer in deuterated dimethyl sulfoxide (DMSO). Molecular weights were measured, compared to polystyrene standards, on a Waters gel permeation chromatography (GPC) system with detection based on refractive index values. The measurements were taken at 40 °C with tetrahydrofuran as the mobile phase on three columns in series (Waters Styragel HR2, HR4, and HR5). Tensile tests were conducted with dry samples on an Instron 5566 at a crosshead speed of 10 mm min−1 at 25 °C.Samples were cured in a dog-bone-shaped mold with approximate dimensions of 10 mm × 3 mm × 1.5 mm. The Young's modulus (E; Pa) was calculated according to the linear segment of the stress–strain curve. Three trials were performed and the average was reported. Using
| n = E(3RT)−1 | (1) |
Sol–gel analysis was conducted by soaking films in THF for 24 h at room temperature. After solvent removal, the films were dried, and the percent soluble fraction (Qs) was calculated by
| Qs = (mi − mf)mi−1(100) | (2) |
Water swelling (WS) experiments were performed by swelling films in double-distilled H2O (ddH2O) for 24 h at room temperature. After removing surface water, the WS was calculated by
| WS = (mf − mi)mi−1(100) | (3) |
Contact angle measurements were obtained using flat polyester films on glass slides. Measurements were performed on a KSV CAM200 contact angle meter with water as the wetting liquid.
2.4 Complex film fabrication
Porous films were prepared by previously reported methods.11 Embossed films were designed by adding a small amount of pre-polymer onto a glass slide and heated to 120 °C in a vacuum oven. After 10 min of reduced pressure (∼50 Torr) to remove any gas or solvent, a PFPE mold was placed directly on the pre-polymer.12–14 The material was allowed to cure as described above, followed by the removal of the mold. To image the embossed films, a Hitachi model 2-4700 scanning electron microscope (SEM) was used after coating the polyesters in ∼2 nm of a Pd/Au alloy (Cressington 108 auto sputter coater, Cressington Scientific Instruments Ltd.).2.5 In vitro degradation
To obtain standard degradation rates, approximately 75 mg samples of elastomers were placed in scintillation vials. The elastomers were submerged in phosphate-buffered saline (PBS) and samples were then stored in an incubator (37 °C).At weekly intervals, samples were removed from the incubator, rinsed thoroughly, dried, and weighed again. To prevent saturation, PBS was replaced every 7 days. Values were obtained using
| mass loss = (mi − mf)mi−1(100) | (4) |
2.6 In vitro cytotoxicity
Growth medium and hMSCs were obtained from Lonza. Stem cell culture protocol and staining were performed as described elsewhere.15 Two different cell lines were used to assess in vitro cytotoxicity: hMSCs and SA fibroblasts. Medium elution tests, according to ISO 10993-5 standards, were performed with both cell lines. Briefly, elastomer samples were sterilized in ethanol for 30 min and washed in PBS for 2 h while under a germicidal lamp. Polyester segments were then added to culture medium and maintained at 37 °C in an incubator for 24 h. The medium–extract solution was then used to culture cells for at least 48 h. After the culture period, cell confluence was examined and toxicity was assessed. Fibroblasts were imaged 48 h after addition of extracted medium. hMSCs were exposed to medium–extract solution for 48 h, followed by induction of differentiation. Images were taken 10 d after addition of the differentiation induction medium using a Nikon Eclipse TE2000-E inverted microscope.Results and discussion
3.1 Motivation
We recently described the properties of the poly(triol α-ketoglutarate) (PTK) family of polyester elastomers.16,17 Although these materials have many interesting features, their degradation rates were particularly of note. In terms of designing potential tissue scaffolds, the PTK series hydrolyzed in PBS too rapidly to be useful. We aimed to design similar materials with degradation rates that were more relevant to future applications in soft tissue engineering. Based on our prior experience, the critical factor to achieving this goal was to eliminate the presence of a ketone immediately adjacent to an acid in the monomer.The foundation of our current material design is based on erythritol, a sugar alcohol that may be endogenous to the human metabolism.18,19 As a sugar substitute that is approved by the Food and Drug Administration, erythritol was an attractive monomer due to human tolerance of large amounts of the sugar alcohol with no negative side effects.19–22 Additionally, erythritol can be absorbed by the body and excreted unchanged, allowing normal metabolic pathways to continue unaffected.19–22 We therefore chose several aliphatic diacids of increasing length to polymerize with erythritol: glutaric, adipic, pimelic, suberic, azelaic, sebacic, dodecanedioic, and tetradecanedioic acid. By altering the length of the diacids, the polyester elastomers were expected to achieve a range of hydrophobicities, degradation rates, and mechanical characteristics. Furthermore, several of these monomers are either naturally occurring small molecules or have previously been used in biomaterials.23–30
3.2 Design and synthesis
The synthesis of the PErD series was designed for biomedical applications and, therefore, elastomers were synthesized without any catalysts or co-reagents (Fig. 1). After forming a homogenous melt in an inert environment of argon at 145 °C, monomers were stirred while heating for 2 h. The pressure of the reaction was then reduced to ∼2 Torr for an additional 7 h, allowing random ester-based cross-linking to form pre-polymers. Materials were either amorphous or semi-crystalline, depending on the length of the diacid; diacids with less than 10 carbons combined with erythritol to form amorphous pre-polymers, while sebacic, dodecanedioic, and tetradecanedioic acids led to the synthesis of semi-crystalline materials that were solid and opaque at room temperature. The monomers were heated only to the extent of cross-linking that would create oligomeric pre-polymers with Mn ≥ 500 g mol−1 (Table 1). Thermoset formation was intended to occur during a second heating phase, enabling molding and shaping of elastomeric films.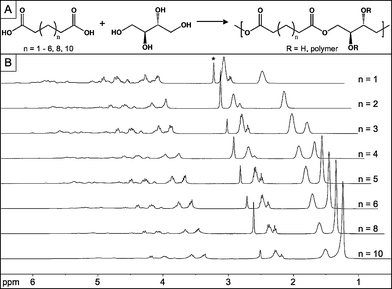 | ||
| Fig. 1 Synthesis and NMR spectra of PErD using glutaric, adipic, pimelic, suberic, azelaic, sebacic, dodecanedioic, and tetradecanedioic acids. (A) Thermal polycondensation of erythritol and dicarboxylic acids of differing lengths led to the catalyst-free design of pre-polymers. Further heating of the pre-polymers allowed for the preparation of polyester thermosets. (B) 1H NMR spectra of the pre-polymers OErGl (n = 1), OErAd (n = 2), OErPi (n = 3), OErSu (n = 4), OErAz (n = 5), OErSe (n = 6), OErDo (n = 8), and OErTe (n = 10). The asterisk designates the solvent peak (d6-DMSO). | ||
3.3 Pre-polymer characterization
Polycondensation of erythritol and diacids (5–10, 12, 14 carbons) led to pre-polymers in the form of oligo(erythritol glutarate) (OErGl), oligo(erythritol adipate) (OErAd), oligo(erythritol pimelate) (OErPi), oligo(erythritol suberate) (OErSu), oligo(erythritol azelate) (OErAz), oligo(erythritol sebacate) (OErSe), oligo(erythritol dodecanedioate) (OErDo), and oligo(erythritol tetradecanedioate) (OErTe). These pre-polymers were characterized by 1H NMR and GPC. The 1H NMR spectra, as seen in Fig. 1, show that all of the pre-polymers have very similar chemical signatures. The pre-polymer compositions were approximately 1 : 1 (erythritol : diacid), with the main difference in the 1H NMR spectra being the location of the methylene groups. The presence of signals around 4 and 5 ppm indicates that alcohols and acids combined to form esters; peaks d′ or e′ represents the free alcohols that did not participate in cross-linking (Fig, S1–S8, ESI†). The signals associated with hydrophobic methylene groups are observed between 1.2 and 1.7 ppm. The molecular weights of the polyester networks are based on a combination of monomer reactivity and mass of the monomers. The lowest molecular weight was 710 g mol−1 (OErGl) while the largest was 1470 g mol−1 (OErDo).3.4 Complex film design
One way to enhance the versatility of a material is to demonstrate processing compatibility with embossing of topologies and microstructures. Recent literature has focused on the importance of nano- and micro-features related to interfacial properties.31–33 Therefore, to determine how easily embossed films could be fabricated, thermal curing was conducted in the presence of a perfluoropolyether (PFPE) mold. The molding material, PFPE, was used due to its low surface energy, allowing for simple release from the polyester film after the completion of the cross-linking period.11–13 A small amount of the pre-polymer was placed on a glass slide and placed in an oven for 10 min, reducing the viscosity. The PFPE mold was then placed in direct contact with the pre-polymer and returned to the oven. Curing conditions were carried out as mentioned in Table 2. After the cross-linking period, the mold was removed and the result was a micro-embossed film (Fig. 2). The features (2 μm × 2 μm × 6 μm) were inversely transferred from the PFPE mold with high fidelity over a large area. We will continue to explore the potential for micro-textured adhesive films or embossed tissue scaffolds to control cell behavior.| Material | Diacid | T/°C | Duration/days |
|---|---|---|---|
| 1 | Glutaric | 120 | 2 |
| 2 | Glutaric | 120 | 3 |
| 3 | Adipic | 120 | 3 |
| 4 | Pimelic | 120 | 3 |
| 5 | Suberic | 120 | 3 |
| 6 | Azelaic | 120 | 3 |
| 7 | Sebacic | 120 | 3 |
| 8 | Dodecanedioic | 120 | 3 |
| 9 | Tetradecanedioic | 120 | 3 |
| 10 | Glutaric | 140 | 3 |
| 11 | Adipic | 140 | 3 |
| 12 | Pimelic | 140 | 3 |
| 13 | Suberic | 140 | 3 |
| 14 | Azelaic | 140 | 3 |
| 15 | Sebacic | 140 | 3 |
| 16 | Dodecanedioic | 140 | 3 |
| 17 | Tetradecanedioic | 140 | 3 |
 | ||
| Fig. 2 Scanning electron microscopy (SEM) images of PErD films. Curing pre-polymer in the presence of a perfluoropolyether mold allows for the facile design of micro-patterned polyester films. The pattern was (A) transferred with high fidelity, (B) creating features with an approximate dimensions of 2 μm × 2 μm × 6 μm. Additionally, porous scaffolds (C) were fabricated using standard salt-leaching techniques. Both of these fabrication methods were possible with all pre-polymers. | ||
Porous materials are also commonly used in tissue engineering because they allow cells and nutrients to easily pervade the interior of scaffolds.34–36 In order to demonstrate that micro-porous PErD thermosets could be designed, pre-polymers were dissolved in tetrahydrofuran and combined with sieved sodium chloride (NaCl) crystals (< 400 μm). The mixture was added to a poly(tetrafluoroethylene) mold and, after solvent evaporation, the polyester–salt system was cured as described above. The cured elastomers were then swelled in ddH2O for 96 h in order to leach out the NaCl porogen. After drying at room conditions, the films were fully dried under vacuum. We will utilize these porous scaffolds in future in vitro and in vivo biological studies.
3.5 Mechanical testing
The ability to design elastomers with control over the mechanical properties is attractive for materials used in biomedical applications. Therefore, several curing conditions were used to create materials with a range of structural properties (Table 2). With the exception of elastomer 1, all pre-polymers were cured for 3 days to form thermosets (elastomer 1 cured for 2 days). As expected, cross-linking these materials at 140 °C led to more rigid materials than curing at 120 °C. The mechanical properties exhibited by various PErD elastomers varied over a wide range (Fig. 3; Table 3). Elastomers 1–17 had Young's modulus (YM), ultimate tensile stress (UTS), and rupture strain (RS) values of 0.08–80.37 MPa, 0.14–16.65 MPa, and 22–466%, respectively.| Material | T g (Tm)a/°C | Young's Modulusb/MPa | UTSb/MPa | Rupture Strainb (%) | n /mmol L−1 | Q s (%) | Contact angle/°) | WSe (%) |
|---|---|---|---|---|---|---|---|---|
| a Determined by DSC. b Determined by Instron (crosshead speed of 10 mm min−1). c eqn (1). d Extracted in THF for 24 h at 25 °C; eqn (2). e Swelled in water for 24 h; eqn (3). | ||||||||
| 1 | −4.1 | 0.21 ± 0.04 | 0.39 ± 0.03 | 418 ± 38 | 29 ± 5 | 37.3 ± 2.0 | 64.5 ± 3.3 | 26.4 ± 2.0 |
| 2 | 0.9 | 0.48 ± 0.01 | 1.03 ± 0.17 | 314 ± 45 | 64 ± 2 | 24.0 ± 1.3 | 83.1 ± 3.2 | 21.8 ± 2.1 |
| 3 | −10.2 | 0.39 ± 0.07 | 0.44 ± 0.05 | 208 ± 14 | 53 ± 10 | 32.0 ± 2.0 | 74.9 ± 1.3 | 15.3 ± 2.0 |
| 4 | −14.5 | 0.19 ± 0.05 | 0.29 ± 0.07 | 319 ± 56 | 26 ± 78 | 36.6 ± 3.3 | 75.9 ± 1.6 | 14.7 ± 1.2 |
| 5 | −16.8 | 0.22 ± 0.07 | 0.24 ± 0.02 | 315 ± 24 | 30 ± 9 | 44.5 ± 1.8 | 66.9 ± 0.7 | 9.8 ± 0.6 |
| 6 | −17.9 | 0.35 ± 0.09 | 0.38 ± 0.07 | 182 ± 34 | 47 ± 12 | 27.5 ± 1.6 | 79.7 ± 3.2 | 8.9 ± 1.9 |
| 7 | −20.4 | 0.08 ± 0.01 | 0.14 ± 0.03 | 466 ± 41 | 10 ± 2 | 51.1 ± 3.4 | 58.6 ± 1.7 | 8.6 ± 0.6 |
| 8 | −32.2 (50.1) | 46.26 ± 3.33 | 10.80 ± 0.44 | 407 ± 15 | 6224 ± 448 | 23.4 ± 0.9 | 80.9 ± 0.8 | 3.6 ± 0.6 |
| 9 | −35.8 (63.2) | 80.37 ± 2.85 | 16.65 ± 0.21 | 149 ± 41 | 10813 ± 383 | 29.1 ± 2.2 | 79.8 ± 1.0 | 2.3 ± 1.0 |
| 10 | 3.8 | 18.36 ± 3.03 | 1.61 ± 0.11 | 63 ± 10 | 2470 ± 408 | 17.1 ± 2.9 | 91.4 ± 1.4 | 20.4 ± 0.7 |
| 11 | −4.7 | 0.47 ± 0.17 | 0.55 ± 0.12 | 198 ± 4 | 63 ± 22 | 7.4 ± 5.3 | 96.2 ± 1.6 | 13.5 ± 1.8 |
| 12 | −6.6 | 1.90 ± 0.26 | 0.86 ± 0.25 | 60 ± 16 | 256 ± 35 | 14.9 ± 2.6 | 84.5 ± 0.7 | 10.7 ± 2.5 |
| 13 | −13.5 | 1.08 ± 0.12 | 0.79 ± 0.14 | 100 ± 8 | 145 ± 16 | 17.3 ± 3.7 | 96.9 ± 2.1 | 9.5 ± 0.6 |
| 14 | −15.9 | 12.50 ± 1.46 | 1.31 ± 0.15 | 22 ± 4 | 1682 ± 196 | 11.6 ± 2.5 | 93.5 ± 2.6 | 5.2 ± 0.8 |
| 15 | −16.6 | 1.92 ± 0.12 | 1.03 ± 0.19 | 85 ± 14 | 258 ± 17 | 10.8 ± 6.2 | 100.0 ± 2.4 | 5.6 ± 0.4 |
| 16 | −17.0 | 1.95 ± 0.31 | 1.48 ± 0.06 | 146 ± 47 | 263 ± 42 | 2.7 ± 0.7 | 98.6 ± 1.0 | 3.0 ± 1.8 |
| 17 | −27.4 (53.8) | 38.89 ± 4.46 | 13.00 ± 0.61 | 446 ± 75 | 5232 ± 601 | 22.4 ± 0.8 | 105.1 ± 0.5 | 2.4 ± 0.6 |
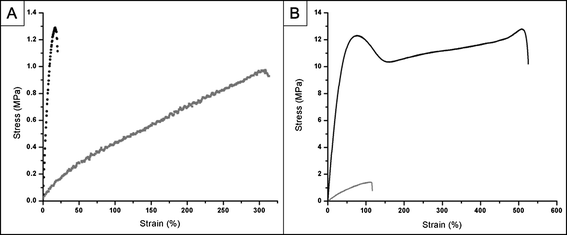 | ||
| Fig. 3 Representative stress versus strain curves for PErD. (A) For PErD films derived from odd-numbered carbon diacids, adjusting the curing temperature from 120 °C to 140 °C led to a large increase in the Young's modulus. Interestingly, the thermosets synthesized from even-numbered carbon diacids did not experience as marked an increase in the moduli. The grey curve represents elastomer 2 while the black curve displays the stress–strain relationship for elastomer 14. (B) Semi-crystalline thermosets displayed irregular stress–strain curves when compared to elastomeric materials. Elastomer 16 (grey) was used as the example for elastomers while material 17 (black) was shown to represent semi-crystalline thermosets. The irregular shape of the red curve (semi-crystalline materials) is most likely due to the presence of an elastomeric network containing semi-crystalline oligomers. | ||
As expected, after the stress was released, the elongated dog-bones composed of amorphous materials immediately reverted to their original dimensions. However, the three semi-crystalline polyesters (materials 8, 9, and 17) experienced semi-permanent deformations; the original shape was not recovered unless these materials were heated above their transition temperatures. Presumably, this is due to the presence of an amorphous polyester encasing semi-crystalline oligomers. This hypothesis was strengthened after conducting the sol content analysis of these three semi-crystalline polyesters because the dried, sol-free materials were amorphous. The interesting composition of the materials 8, 9, and 17 also led to unique and reproducible stress vs. strain curves (Fig. 3). These polymers had higher YM and UTS values when compared to the amorphous polymers. The stress vs. strain curves were uniquely shaped, achieving a local maximum, then a local minimum, and finally steadily increasing before rupturing.
Extending the potential of PErD elastomers, the mechanical properties of several biological materials fall within the ranges observed by these materials.4,5 Furthermore, the design of materials with many more combinations of mechanical characteristics should be possible by adjusting the curing conditions. An advantage of using thermally cured materials is the ease with which the rigidity and flexibility can be tuned.
3.6 Degradation studies
As stated previously, our earlier work included a series of polyketoester elastomers that degraded rapidly in PBS at 37 °C (complete hydrolysis in 2–28 days).16,17 The design of the PErD family of polyester elastomers was motivated by the need for biodegradable materials that hydrolyze on a timescale that is more relevant to tissue engineering applications. The only materials that were studied during in vitro degradation experiments were those that were amorphous, due to the need for materials that can recover from the dynamic stresses associated with in vivo tissue engineering. However, elastomer 7 was not studied because it turned opaque when submerged in aqueous solution. The degradation rates of PErD seem to be controlled by two factors: the extent of cross-linking within the polymer network and the hydrophobicity of the elastomer. As such, the fastest degrading material should be soft and hydrophilic (elastomer 1), while hard, hydrophobic materials should survive longer in hydrolytic conditions (elastomers 16). This logical trend was observed in the relative rates of degradation for our materials. Due to the presence of fewer methylene groups, glutaric acid is more hydrophilic than adipic acid, which is more hydrophilic than pimelic acid, etc. Therefore, materials composed of these monomers should degrade according to their hydrophilic natures, with the fastest degrading elastomer being composed of the most hydrophilic diacid. Also, curing at 140 °C for 3 d should lead to more extensive ester formation than cross-linking at 120 °C for 3 d. As esters are more hydrophobic than acids, the materials resulting from the 140 °C, 3 day curing should be more hydrophobic than those cross-linked at 120 °C for 3 days. By combining these two trends, intuitive deduction would suggest the following trend in degradation rates (fastest to slowest degradation): 1 > 2 > 3 > 4 > 5 > 6 > 10 > 11 > 12 > 13 > 14 > 15 > 16. As seen in Fig. 4, the materials degrade according to the expected trend. By altering the diacid and the curing conditions, the degradation rate could be controlled, allowing for the ability to tune the hydrolysis rate to match the needs of a potential application. Overall, hydrolytic degradation rates ranged from 100% in 3 weeks to 6.4% in 4 weeks; the PErD series of polyesters offers a wide range of degradation rates, which could increase the versatility of these materials in regards to tissue engineering applications.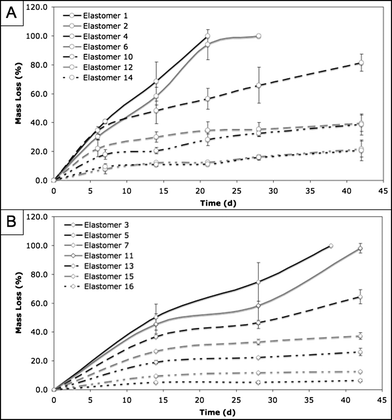 | ||
| Fig. 4 In vitro PErD degradation rates. Films were incubated in PBS at 37 °C for four weeks. At pre-determined time points, the percent mass loss was recorded. The PErD series achieved in vitro degradation ranging from 100% in 3 weeks to 6.4% in 6 weeks. | ||
3.7 In vitro cytotoxicity
These materials were primarily designed for tissue engineering applications. Therefore, because biocompatibility is a requisite of tissue scaffolds, the cytotoxicity of these elastomers was studied using an elution-based method. Only those polyesters that demonstrated low water swelling values were chosen for cell studies (elastomers 4, 5, 6, 11, 12, 13, 14, 15, 16) because of potential complications that could arise from structural deformations caused by hydration. The first method consisted of culturing SA fibroblasts in polymer-extracted medium. Polyester thermosets were extracted in cell medium for 24 h at 37 °C. The medium–extract solution was then used to culture cells on tissue culture plastic that were not yet confluent. As seen in Fig. 5, within 48 h of extract addition, cells were able to reach confluence, regardless of the polymer extract used. In the case of fibroblasts, the presence of the extracts did not prevent the cells from achieving and maintaining confluence; all cells were viable for 1 week, at which time the study was stopped. Cells that were exposed to polymer extracts were virtually indistinguishable from cells that were not exposed to any material extracts.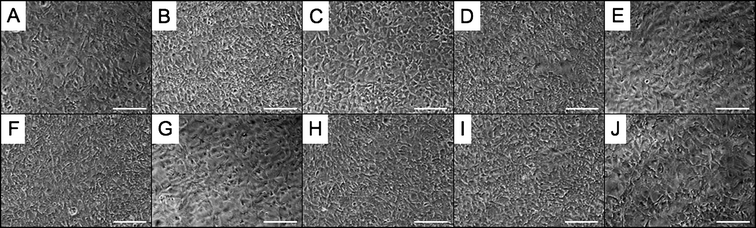 | ||
| Fig. 5 Elastomer extract-based in vitro cytotoxicity of PErD with fibroblasts. SA fibroblasts were cultured on tissue culture plastic in the presence of extracts from elastomer 4 (A), 5 (B), 6 (C), 11 (D), 12 (E), 13 (F), 14 (G), 15 (H), and 16 (I) for 48 h. As a positive control, cells were cultured in medium containing no polymer extracts (J). Fibroblasts were able to reach and maintain confluence, allowing these polyesters to be deemed non-cytotoxic. All scale bars represent 200 μm. | ||
To study the effect of polymer extracts on complex cellular functions, not simply the viability, hMSCs were also cultured in the presence of the extracts. Stem cells were brought to confluence in the presence of polymer-extracted medium, followed by the addition of differentiation induction medium. hMSCs were imaged 10 days after induction of differentiation, enabling ample time for the transition from stem cells to adipocytes.15 The presence of adipose cells can be confirmed by the formation of large lipid vacuoles. After staining with Oil Red O, the vacuoles (red) can easily be seen throughout the cells (Fig. 6). Based on comparisons with hMSCs that were not exposed to any polymer extracts, PErD materials were deemed non-cytotoxic. The stem cells that were cultured with polymer extracts from diacids with an even number of carbon exhibited noticeably more differentiation. As seen in Fig. 6 (images B, D, F, H, and I), the presence of more red demonstrates that more vacuoles were produced during differentiation. Future studies will focus on further observing the relationship between differentiation and the number of carbons in the diacids.
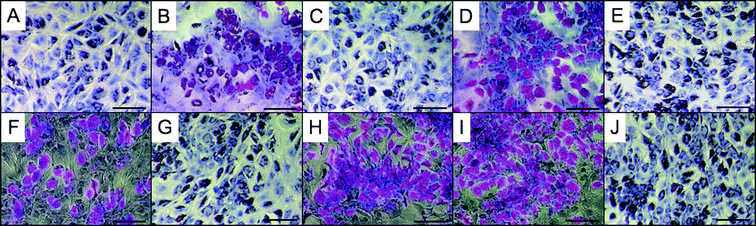 | ||
| Fig. 6 Elastomer extract-based in vitro cytotoxicity of PErD with stem cells. hMSCs were cultured on tissue culture plastic in the presence of extracts from elastomer 4 (A), 5 (B), 6 (C), 11 (D), 12 (E), 13 (F), 14 (G), 15 (H), and 16 (I) for 48 h. As a positive control, cells were cultured in medium containing no polymer extracts (J). After reaching confluence, induction medium was added to begin the differentiation process. As seen by the presence of vacuoles (red), hMSCs were able to differentiate into adipocytes after exposure to PErD extracts. All scale bars represent 200 μm. | ||
Conclusions
We have designed a novel series of polyester elastomers based on aliphatic diacids and erythritol, a sugar alcohol that is FDA-approved for human consumption. Thermal polymerization offers a simple avenue for tuning the mechanical properties, hydrophobicity, and the degradation rates of the poly(erythritol dicarboxylate) family of materials. Future work will focus on in vivo characterization and applications (tissue engineering and drug delivery) of this family of thermosets. The pre-polymers are compatible with imprint lithography and emulsions (data not shown), enabling multiple methods of obtaining particles or embossed films. We will also continue to explore the cause of the odd–even effect and how it relates to the biocompatibility of these materials. However, based on our preliminary results, we believe that the poly(erythritol dicarboxylate) series of polyester elastomers may find wide use in a range of biomedical applications.Acknowledgements
The authors thank the Ashby and DeSimone research groups for insightful discussions, as well as Professor Wei You. This work was supported by the Carolina Center for Cancer Nanotechnology Excellence (NCI), the Burroughs Wellcome Foundation (Interface Career Award), and the National Science Foundation (Career Award).Notes and references
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS
.
- E. Lavik and R. Langer, Appl. Microbiol. Biotechnol., 2004, 65, 1–8 CAS
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS
- A. R. Webb, J. Yang and G. A. Ameer, Expert Opin. Biol. Ther., 2004, 4, 801–812 Search PubMed
- B. Amsden, Soft Matter, 2007, 3, 1335–1348 RSC
- C. G. Pitt, M. M. Gratzl, G. L. Kimmel, J. Surles and A. Schindler, Biomaterials, 1981, 2, 215–220 CrossRef CAS
- R. F. Storey and T. P. Hickey, Polymer, 1994, 35, 830–838 CrossRef CAS
- Y. Wang, Y. M. Kim and R. Langer, J. Biomed. Mater. Res. A, 2003, 66, 192–197
- J. P. Bruggeman, B.-J. de Bruin, C. J. Bettinger and R. Langer, Biomaterials, 2008, 29, 4726–4735 CrossRef CAS
- J. P. Bruggeman, C. J. Bettinger, C. L. E. Nijst, D. S. Kohane and R. Langer, Adv. Mater., 2008, 20, 1922–1927 CrossRef CAS
- A. G. Mikos, G. Sarakinos, S. M. Leite, J. P. Vacanti and R. Langer, Biomaterials, 1993, 14, 323–330 CrossRef CAS
- J. P. Rolland, R. M. Van Dam, D. A. Schorzman, S. R. Quake and J. M. DeSimone, J. Am. Chem. Soc., 2004, 126, 2322–2323 CrossRef CAS
- J. P. Rolland, E. C. Hagberg, G. M. Denison, K. R. Carter and J. M. DeSimone, Angew. Chem., Int. Ed., 2004, 43, 5796–5799 CrossRef CAS
- S. E. A. Gratton, S. S. Williams, M. E. Napier, P. D. Polhaus, Z. Zhou, K. B. Wiles, B. W. Maynor, C. Shen, T. Olafsen, E. T. Samulski and J. M. DeSimone, Acc. Chem. Res., 2008, 41, 1685–1695 CrossRef CAS
- W. Luo, S. R. Jones and M. N. Yousaf, Langmuir, 2008, 24, 12129–12133 CrossRef CAS
- D. G. Barrett and M. N. Yousaf, Macromolecules, 2008, 41, 6347–6352 CrossRef CAS
- D. G. Barrett, B. M. Lamb and M. N. Yousaf, Langmuir, 2008, 24, 9861–9867 CrossRef CAS
- O. Touster, S. O. Hecht and W. M. Todd, J. Biol. Chem., 1960, 235, 951–953 CAS
- W. O. Bernt, J. F. Borzelleca, G. Flamm and I. C. Munro, Reg. Toxicol. Pharmacol., 1996, 24, S191–S197 CrossRef CAS
- F. R. J. Bornet, A. Blayo, F. Dauchy and G. Slama, Reg. Toxicol. Pharmacol., 1996, 24, S280–S285 CrossRef CAS
- W. Tetzloff, F. Dauchy, S. Medimagh, D. Carr and A. Bär, Reg. Toxicol. Pharmacol., 1996, 24, S286–S295 CrossRef CAS
- F. R. J. Bornet, A. Blayo, F. Dauchy and G. Slama, Reg. Toxicol. Pharmacol., 1996, 24, S296–S302 CrossRef CAS
- A. Pourjavadi, N. Rezai and M. J. Zohuriaan, J. Appl. Polym. Sci., 1998, 68, 173–183 CrossRef CAS
- S. Ben-Shabat, E. Abuganima, A. Raziel and A. J. Domb, J. Polym. Sci. A: Polym. Chem., 2003, 41, 3781–3787 CrossRef CAS
- M. Okada, K. Tsunoda, K. Tachikawa and K. Aoi, J. Appl. Polym. Sci., 2000, 77, 338–346 CrossRef CAS
- P. Siriphannon, P. Monvisade, S. Jinawath and K. Hemachandra, J. Biomed. Mater. A, 2007, 81, 381–391 Search PubMed
- M. A. Carnahan and M. W. Grinstaff, Macromolecules, 2006, 39, 609–616 CrossRef CAS
- L. Sheihet, K. Piotrowska, R. A. Dubin, J. Kohn and D. Devore, Biomacromolecules, 2007, 8, 998–1003 CrossRef CAS
- G. Liu, B. Hinch and A. D. Beavis, J. Biol. Chem., 1996, 271, 25338–25344 CrossRef CAS
- A. V. Grego and G. Mingrone, Clin. Nutr., 1995, 14, 143–148 CrossRef CAS
- A. K. Geim, S. V. Dubonos, I. V. Grigorieva, K. S. Novoselov, A. A. Zhukov and S. Y. Shapoval, Nat. Mater., 2003, 2, 461–463 CrossRef CAS
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS
- A. Mahdavi, L. Ferreira, C. Sundback, J. W. Nichol, E. P. Chan, D. J. D. Carter, C. J. Bettinger, S. Patanavanich, L. Chignozha, E. Ben-Joseph, A. Galakatos, H. Pryor, I. Pomerantseva, P. T. Masiakos, W. Faquin, A. Zumbuehl, S. Hong, J. Borenstein, J. Vacanti, R. Langer and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2307–2312 CrossRef CAS
- J. Yang, G. Shi, J. Bei, S. Wang, Y. Cao, Q. Shang, G. Yang and W. Wang, J. Biomed. Mater. Res. A, 2002, 62, 438–446 Search PubMed
- J. Yang, A. R. Webb and G. A. Ameer, Adv. Mater., 2004, 16, 511–516 CrossRef CAS
- J. Yang, A. R. Webb, S. J. Pickerill, G. Hageman and G. A. Ameer, Biomaterials, 2006, 27, 1889–1898 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Fully labeled 1H NMR spectra for pre-polymers. See DOI: 10.1039/b9py00226j |
| This journal is © The Royal Society of Chemistry 2010 |