A facile route for the preparation of azide-terminated polymers. “Clicking” polyelectrolyte brushes on planar surfaces and nanochannels
Basit
Yameen
a,
Mubarak
Ali
b,
Marta
Álvarez
a,
Reinhard
Neumann
c,
Wolfgang
Ensinger
b,
Wolfgang
Knoll
d and
Omar
Azzaroni
*e
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany
bDarmstadt University of Technology, Department of Materials Science, Petersenstraße 23, D-64287 Darmstadt, Germany
cGSI Helmholtzzentrum für Schwerionenforschung GmbH, Planckstr. 1, D-64291, Darmstadt, Germamy
dAustrian Institute of Technology, Donau-City-Straße 1, 1220 Vienna, Austria
eInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas (INIFTA), Universidad Nacional de La Plata – CONICET, CC 16 Suc. 4, 1900, La Plata, Argentina. E-mail: azzaroni@inifta.unlp.edu.ar; Web: http://softmatter.quimica.unlp.edu.ar
First published on 25th November 2009
Abstract
In this work we describe the facile preparation of azide-terminated polymers by conventional radical polymerization (cRP) using azo initiators bearing azide groups. We show that cRP provides a convenient avenue for the preparation of azide end-functional polymers in a one-step process. The versatility of this chemical methodology was demonstrated by the synthesis of unprecedented azide end group-functionalized sodium polystyrene sulfonate (PSSNa) and poly(2-methacryloyloxyethyl-trimethylammonium chloride) (PMETAC) which were then “clicked” onto alkyne-terminated silicon surfaces and polyethylene terephthalate nanochannels to form polyelectrolyte brush layers. The facile synthesis of the end-functionalized macromolecular building blocks will enable the creation of a wide variety of “clickable” architectures using very simple synthetic tools. We are confident that these results will constitute a key element in the “click” chemistry toolbox and, as such, will have strong implications for the molecular design of interfaces using macromolecular architectures.
1. Introduction
The manipulation of the chemical composition of surfaces plays a pivotal role in different technological areas such as wetting, adhesion, lubrication and catalysis, just to name a few examples.1 One of the most common procedures for modifying substrates and achieving accurate control over their surface properties consists of anchoring tailored macromolecular architectures with pre-defined functionalities. An appropriate method is the attachment of end-functionalized polymer chains to surface-modified substrates, thus implying the covalent attachment of the macromolecules having functional groups that can react with surface functionalities anchored on the substrate.2 This strategy is often called the “grafting-to” method. It is well-known that surface modification via a grafting-to method involves a relatively low grafting density of polymer chains. This is due to steric hindrance introduced by the grafted polymer chains that precludes the further tethering of other chains.3 Notwithstanding this limitation, the “grafting-to” method is widely used for surface modification since it is simple, practical and can be conducted under mild conditions. It is also worthwhile noting that some applications, as is the case of bioseparations or functionalized biointerfaces, the polymer chains do not require a very high density because they are often derivatized with large-sized moieties or conjugated to bulky building blocks, like proteins.4,5Silica, glass and gold surfaces can be easily modified with different chemical functionalities by using self-assembled monolayers.6 Then, these chemical groups can undergo condensation reactions with polymer chains end-functionalized with adequate reactive counterparts. For example, Tran and Auroy reported the grafting of polystyrene sulfonate chains on surfaces by using a sequence of chemical reactions. First, they polymerized polystyrene chains terminated by a reactive trichlorosilane group and covalently tethered them to the silicon surface, Then the PS chains were converted to polystyrene sulfonate by a soft sulfonation reaction.7
In a similar way, Sirard et al. grafted poly(dimethysiloxane) chains on silicon surfaces by spin coating the polymer chains modified with silanol groups onto the hydroxyl-terminated substrate.8 Finally, the Minko and Luzinov groups reported the grafting of carboxy-terminated polymer chains onto 3-glycidoxytrimethoxysilane-modified silicon surfaces by spin coating and thermal annealing in order to obtain robust responsive macromolecular layers.9
One of the important aspects of grafted polymer films relies on the stability of the tethered chains since many applications require stable films in liquid media. This demands the use of robust covalent attachment as a route to anchor the polymer chains. In this context, “click” chemistry emerged as a versatile strategy to generate robust covalent bonds with remarkable stability to hydrolysis, oxidation or reduction.
“Click” chemistry refers to a chemical procedure for creating stable covalent bonds through azide/alkyne reactions. In particular, the very well documented copper(I) catalyzed azide-alkyne reaction cycloaddition, also termed as the “Sharpless click reaction”, has demonstrated to be a versatile method of C–N formation in organic chemistry.10 To date, this chemical methodology has been implemented in the synthesis, modification and/or functionalization of a wide range of materials, such as metal11 and oxidenanoparticles,12 liposomes,13layer-by-layer assemblies,14dendrimers,15 micelles,16dendronized polymers17 and so on. There are many advantages that make “click” chemistry particularly attractive in fields like macromolecular chemistry or materials science.18,19 These particular features are: (a) the reaction is solvent-insensitive and proceeds with high yields with no by-products, (b) the chemical strategy exhibits functional group orthogonality, which indicates good tolerance to different chemical groups, and (c) the reaction also works in heterogeneous conditions with high yields.18 The latter one is a key feature indicating that “click” chemistry is also a powerful toolbox to deal with colloidal and surface science problems. During last years, considerable progress has been made in the field of “click” chemistry related to surface science aspects.20–24 Seminal works demonstrated that “click” reactions can occur on different substrates with quantitative yields. Chidsey and co-workers were able to “click” electroactive functionalities onto metal surfaces enabling the facile modification of the electrode surface.25 Later on, similar surface “click” chemistry approaches were extended to different substrates like silicon, gold, glass or graphitic surfaces.26 This straightforward strategy enabled the covalent surface attachment of different molecules like redox centers,27fluorescent dyes28 or even PEGylated oligomers29 in a simple manner.
The high versatility of “click” chemistry for tailoring surface properties and design materials with molecular accuracy relies on our ability to generate the adequate building blocks to “click” together, i.e.: azide- and alkyne-functionalized molecules, in a predictable manner. In this regards, the further evolution and application of “click” chemistry in scientific disciplines different to organic or polymer chemistry will greatly depend on finding new routes to create azide- and alkyne-terminated building blocks in a low-cost, straightforward and simple manner. Macromolecular science provided “click” chemistry with a more ambitious outlook through the incorporation of more sophisticated and powerful methods to achieve tailored molecular design of the “clickable” building blocks. The click chemistry approach has been successfully combined with many controlled radical polymerization reactions including atom transfer radical polymerization (ATRP),30nitroxide-mediated polymerization (NMP),31reversible addition fragmentation transfer (RAFT)32 or ring-opening metathesis polymerization (ROMP),33 among others. These methods of controlled radical polymerization have been demonstrated to be a powerful tool to generate well-defined macromolecular architectures; however, in some cases they also present incompatibilities among the participating species in the reaction mixture. In other words, the interplay between catalysts, monomers and solvent can play a decisive role to successfully achieve the end-functionalized macromolecule suitable for the “click” reaction.
On the other hand, conventional radical polymerization (cRP) initiated by azo initiators is still the most robust synthesis route for the preparation of polymers which is applicable to almost any vinyl monomer. Within this framework, we consider that merging the fidelity of alkyne–azide “click” reactions with the robustness of cRP will give a new dimension to the molecular design of materials by opening a gate to nearly unlimited “clickable” polymeric architectures obtained by a traditional synthetic procedure.
We hereby report the use of an azo initiator bearing azide groups for the facile synthesis of azide-terminated polymers. Central to our work is to demonstrate that cRP provides a one-step process for preparation of azide end-functional polymers that can be used for surface functionalization of different substrates. To illustrate the versatility of this approach unprecedented azide end group-functionalised sodium polystyrene sulfonate (PSSNa) and poly(2-methacryloyloxyethyl-trimethylammonium chloride) (PMETAC) were prepared. The azide–alkyne click reaction of the telechelic polymers functionalized with azide end groups was demonstrated by grafting them onto alkyne-terminated silicon surfaces and polyethylene terephthalate nanochannels.
2. Experimental section
2.1 Materials and methods
Sodium 4-vinylbenzenesulfonate (technical, ≥90%, Fluka), [2-(methacryloyloxy)ethyl]trimethylammonium chloride (METAC) as 75% aqueous solution, 4,4′-azobis(4-cyanopentanoic acid) (purum, ≥98.0%, Fluka), phosphorus(V) chloride (purum, ≥98.0%, Fluka), methyltrichlorosilane (99%), hexamethylphosphoramide (HMPA, ≥98.0%, Fluka), 1,3-dichloro-1,1,3,3-tetramethyldisiloxane (≥97.0%, Fluka), ethynylmagnesium chloride (0.5 M in THF), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 98%, Fluka), pentafluorophenol (PFP, +99%,) were obtained from Sigma-Aldrich, Schnelldorf, Germany. Propargylamine (99%), dry dichloromethane was obtained from Acros Organics, Geel, Belgium. Triethylamine was refluxed overnight with calcium hydride, distilled and stored under argon. Dimethylsulfoxide was distilled prior to use. All other chemicals were used as received. Infrared (IR) spectra were recorded as neat films using a Nicolet FT-IR 730 spectrometer. 1H NMR was performed on a Bruker Spectrospin 250 MHz NMRspectrometer (Fallanden, Switzerland). Polyethylene terephthalate (PET) (Hostaphan RN 12, Hoechst) membranes of 12 µm thickness were irradiated at the linear accelerator UNILAC (GSI, Darmstadt) with single swift heavy ions (Pb, U and Au) of energy 11.4 MeV/nucleon.2.2 Synthesis of acyl chloride-terminated azo initiator (2)
5 g (0.01784 mole) of 4,4′-azobis(4-cyanopentanoic acid) were suspended and stirred with 150 mL dried dichloromethane in a 3-necked round bottom flask equipped with a stopper and nitrogen inlet and outlet. The white suspension was cooled to 0 °C in an ice bath. 9.29 g of PCl5 (0.0446 mole, 2.5 equiv.) were added progressively over 30 min. At the end of the addition, the solution was completely clear, following solubilization of 4,4′-azobis(4-cyanopentanoic acid) in dichloromethane. The reaction mixture was allowed to warm up to room temperature and the dichloromethane was evaporated under reduced pressure on a rotary evaporator at room temperature until the volume of the remaining solution was approximately 30 mL. Then, 70 mL n-hexane (dried overnight over molecular sieves) was added to the reaction mixture and the flask was cooled down to 0 °C to allow the complete crystallization of 2.34 The solid was then filtered, washed with 10 mL cold n-hexane and dried overnight under vacuum (yield: 90%).2.3 Synthesis of 3-amino-1-azide propane (4)
In a 1 L Schlenk flask connected to a dropping funnel, 1-bromo-3-aminopropane hydrobromide3 (0.15 mol, 32 g) was dissolved in water (100 mL) followed by the addition of NaN3 (0.5 mol, 3.2 g) in 150 mL of water. After the addition was complete, the dropping funnel was replaced with a reflux condenser, and the reaction mixture was heated to reflux for 16 h followed by the removal of 2/3 of the water on a rotary evaporator. The resulting mixture was cooled in an ice bath and 500 mL of diethyl ether was added to it. Followed by addition of 40 g of KOH pellets in small portion while keeping the temperature below 10 °C. The organic layer was separated and the aqueous phase was extracted with diethyl ether (2 × 300 mL). The combined organic layers were dried over K2CO3 and concentrated to give 12.45 g of clear yellow oil (yield: 85%).IR (film): 3305 cm−1 (N–H stretching vibration), 2933, 2866 cm−1 (C–H stretching vibration), 2091 cm−1 (–N3 antisymmetric stretching vibration). 1H NMR (250 MHz, CDCl3) δ [ppm]: 3.31 (t, J = 6.8 Hz, 2H, –CH2N3), 2.74 (t, J = 6.8 Hz, 2H, –CH2NH2), 1.66 (5, J = 6.68 2H, –CH2–), 1.13 (s, 2H, –NH2). 13C NMR (250 MHz, CDCl3) δ [ppm]: 28.2 (–CH2–) 39.2 (–CH2–NH2), 49.1 (–CH2–N3).35
2.4 Synthesis of azide functionalized azo initiator (5)
Acyl chloride 2 (5.49 g, 0.017 mole) was dissolved in dry CH2Cl2 (45 mL) and transferred to a dry 250 mL Schlenk flask under N2(g). The solution was cooled in an ice/salt bath. To this solution a five times excess of amine1 (8.66 g, 0.086 mole) dissolved in dry CH2Cl2 (30 mL) was added dropwise. After the complete addition the reaction mixture was allowed to warm up to room temperature and stirred overnight. The reaction mixture was then extracted with 1% HCl (3 × 70 mL) followed by washing with brine until the aqueous extract was neutral. The solvent was removed at room temperature on rotary evaporator and solid residue obtained was suspended in small volume of ethanol. Hexane was added to this suspension to get clear precipitates, which were allowed to settle, and supernatant liquid was decanted. This procedure was repeated several times till a white solid was obtained (in 92% yield). The thus obtained azide-functionalized azo initiator was of reasonable purity as reflected by 1H and 13C–NMR spectra, listed in the following:IR (neat): 3256 (NH) 3056–2869 (CH), 2097 (–N3) 1628, 1561 (amide linkage). 1H NMR (d6-DMSO): δ [ppm] = 8.05 (1H, NH, br-s), 3.33–3.39 (2H HN–CH2, t, J = 6.8), 3.10–3.12 (2H, HNCO–CH2, q, J = 6.42 Hz) 1.91–2.40 (4H, 2H from N3–CH2–CH2–CH2–NHCO and 2H from CH2 attached to the quarternary carbon, m) 1.66–1.70 (5H, 3H from –CH3 and 2H form N3–CH2, br). 13C NMR (d6-DMSO): δ [ppm] 170.3, 118.5, 72.3, 48.7, 36.2, 33.5, 30.3, 28.6, 23.6.
2.5 Synthesis of azide-terminated polyelectrolytes
As an example we will describe the synthetic procedure for the preparation of azide-terminated sodium polystyrene sulfonate, as described in Table 1 (entry 3). In a Schlenk tube, 0.12 g (0.58 mmole) of sodium 4-vinylbenzenesulfonate monomer and 0.26 g (0.58 mmole) initiator 5 were dissolved in DMSO, and the solution was stirred and degassed by N2(g) bubbling for an hour. The Schlenk tube was closed under a positive pressure of N2(g) and polymerization was carried out at 60 °C for 20 min. Polymerization was quenched by rapid cooling in an ice bath and exposure to air. The polymerization solution was then poured into a non-solvent for the polymer, i.e.THF. The polymers were purified by repeated precipitations from their water solution into THF. The purified polymer was characterized by FTIR spectroscopy and GPC. In case of PMETAC–N3acetone was used as a non-solvent to precipitate out the polymer from polymerization solution and for subsequent re-precipitations from water for purification purposes.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 559
5592.6 Click chemistry on the silicon surface
![[triple bond, length half m-dash]](https://www.rsc.org/images/entities/char_e007.gif) C–H stretching vibration), 2957, 2919, 2861 cm−1 (C–H stretching vibration), 2036 cm−1 (–CC– stretching vibration). 1H-NMR (250 MHz, CDCl3) δ [ppm] 0.24 (s, 12H, –CH3) and 2.36 (s, 2H, –CCH). 13C NMR (250 MHz, CDCl3) δ [ppm]: 1.89 (–CH3), 88.96 (–CCH), 92.3 (–CCH). A mixture of 3.01 g (16.5 mmole) 9, 1.7 g (11.4 mmole) of methyltrichlorosilane, 0.131 µL of HMPA and 11.7 µL of water was stirred at 60 °C for 4 h. The reaction mixture was then subjected to distillation to give ethynyldimethylchlorosilane (10) as a colourless liquid (boiling point: 38–42 °C). IR (film): 3277 cm−1 (C–H stretching vibration), 2962, 2903 cm−1 (C–H stretching vibration), 2035 cm−1 (–CC– stretching vibration). 1H-NMR (250 MHz, CDCl3) δ [ppm] 0.54 (s, 6H, –CH3) and 2.56 (s, 1H, –CCH). 13C NMR (250 MHz, CDCl3) δ [ppm]: 3.4 (–CH3), 85.83 (–CCH), 95.19 (–CCH).36,37
C–H stretching vibration), 2957, 2919, 2861 cm−1 (C–H stretching vibration), 2036 cm−1 (–CC– stretching vibration). 1H-NMR (250 MHz, CDCl3) δ [ppm] 0.24 (s, 12H, –CH3) and 2.36 (s, 2H, –CCH). 13C NMR (250 MHz, CDCl3) δ [ppm]: 1.89 (–CH3), 88.96 (–CCH), 92.3 (–CCH). A mixture of 3.01 g (16.5 mmole) 9, 1.7 g (11.4 mmole) of methyltrichlorosilane, 0.131 µL of HMPA and 11.7 µL of water was stirred at 60 °C for 4 h. The reaction mixture was then subjected to distillation to give ethynyldimethylchlorosilane (10) as a colourless liquid (boiling point: 38–42 °C). IR (film): 3277 cm−1 (C–H stretching vibration), 2962, 2903 cm−1 (C–H stretching vibration), 2035 cm−1 (–CC– stretching vibration). 1H-NMR (250 MHz, CDCl3) δ [ppm] 0.54 (s, 6H, –CH3) and 2.56 (s, 1H, –CCH). 13C NMR (250 MHz, CDCl3) δ [ppm]: 3.4 (–CH3), 85.83 (–CCH), 95.19 (–CCH).36,37
2.7 Asymmetric conical nanochannels
After etching, the diameter of the large opening (D) of the channel was determined by field emission scanning electron microscopy (FESEM) using a PET sample containing 107 pores/cm2 which was etched simultaneously with the single channel under the same conditions. The diameter of the small opening (d) was estimated by assuming the conical geometry of the channel from its conductivity using the following relation:38
| d = 4LI/µDkV |
2.8 Gel permeation chromatography (GPC)
Molecular weight distributions were measured using GPC and the experimental conditions depended on the studied polyelectrolyte. For azide-terminated sodium polystyrene sulfonate a series of columns, TSK Gel G6000 PWXL- TSK Gel G5000 PWXL and TSK Gel G3000, supplied by TosoHaas (Stuttgart) was used. The GPC was equipped with a Waters 590 pump, and with UV (S-3702, SOMA) and RI (ERC 7519, Erma Inc.) detectors. The eluent was a solution consisting of 80% (0.1 M NaNO3) : 20% (CH3CN) at a flow rate of 1 mL/min, and the column temperature was 23 °C. Sodium polystyrene sulfonate was used as a calibration standard. The experimental conditions for the azide-terminated poly(2-methacryloyloxyethyl-trimethylammonium chloride) were similar to those described above, except for the eluent (97% (0.8 M NaNO3) : 3% (CH3CN)), the column temperature (60 °C), and the calibration standard (polyethylene oxide, PEO).3. Results and discussion
3.1 Synthesis of the azide-terminated azo initiator
As previously discussed, cRP provides a robust alternative for growing a wide variety of polymers bearing different chemical groups. This simple route relies almost exclusively on the generation of reactive free radicals originating from the thermally induced dissociation of an adequate initiator. In our case, the free radical initiator was an azo compound bearing terminal azide groups synthesized via a three-step route (Scheme 1). First, 2,2′azobis(4-cyanovaleric acid) (1) was treated with a slight excess of PCl5 in CH2Cl2 at 0 °C to obtain an acyl halide-terminated azoinitiator (2). Then, the azide-terminated linker (4) was obtained by reacting 1-bromo-3-aminopropane hydrobromide with sodium azide.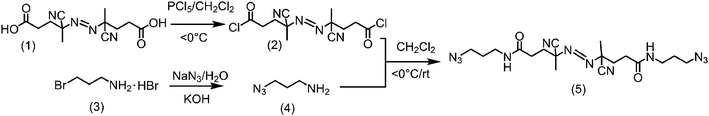
Finally, the synthesis of the initiator was accomplished by the reaction of acyl chloride2 with an excess of 4. A CH2Cl2 solution of 4 was added drop-wise to a cooled (<0 °C) solution of 2 in CH2Cl2. After the complete addition, the reaction was allowed to proceed at RT under stirring during 12 hours. The reaction mixture was then extracted with 1% HCl, followed by rotary evaporation to remove the solvent. The solid residue obtained was re-suspended in ethanol and precipitated by adding hexane to the solution. This procedure enabled the preparation of the azide-terminated azo initiator (5) with reasonable yield (92%) and purity as indicated by the 1H-NMR and 13C-NMR spectra (Fig. 1).
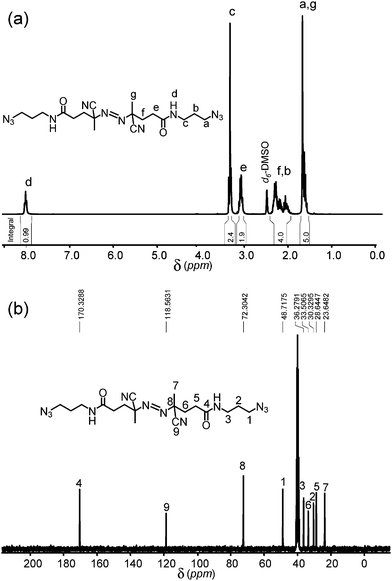 | ||
| Fig. 1 (a) 1H-NMR (b) 13C-NMR spectra in d6-DMSO of the azide-terminated azo initiator. | ||
FT-IR characterization of the purified solid also corroborated the presence of the chemical entities corresponding to the azide-terminated initiator (Fig. 2).
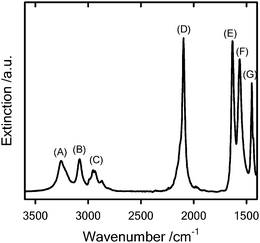 | ||
Fig. 2
FT-IR spectrum of the azide-terminated azo initiator 5. The different labels correspond to: (A and B) 3257 and 3079 cm−1 (N–H) stretching vibration, (C) 2987–2865 cm−1(C–H) stretching vibration, (D) 2097 cm−1 (–N3) antisymmetric stretching vibration, (E) 1631 cm−1 (C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O) stretching vibration – amide I, (F) 1564 cm−1 (N–H) bending vibration, amide II, (G) 1447 cm−1 (C–H) bending vibration. O) stretching vibration – amide I, (F) 1564 cm−1 (N–H) bending vibration, amide II, (G) 1447 cm−1 (C–H) bending vibration. | ||
3.2 Synthesis of azide-terminated polyelectrolytes
The characterization of the azo initiator was followed by the synthesis of the azide-terminated polymers. In our case, to illustrate the potentials of the approach we synthesized sodium polystyrene sulfonate (PSSNa) and poly(2-methacryloyloxyethyl-trimethylammonium chloride) (PMETAC) functionalized with azide terminal groups (Scheme 2).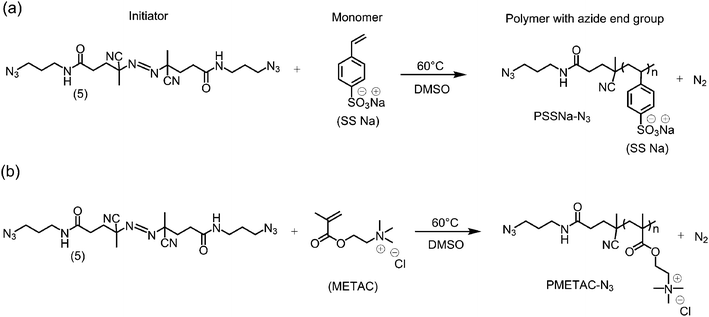 | ||
| Scheme 2 Scheme describing the polymerization of (a) azide-terminated sodium polystyrene sulfonate (PSSNa–N3) and (b) azide-terminated poly(2-methacryloyloxyethyl-trimethylammonium chloride) (PMETAC–N3). | ||
The synthesis was carried out in a Schlenk tube in the absence of oxygen. The monomer and the initiator were dissolved in , and the solution was stirred and degassed by N2 (g) bubbling for one hour. Then, the Schlenk tube was closed under a positive pressure of N2, and the polymerization was carried out at 60 °C. After a preset polymerization time the solution was quenched by rapid cooling in an ice bath and exposure to ambient conditions. Finally, the purification of the azide-terminated polyelectrolytes was accomplished by precipitation in THF or acetone for PSSNa–N3 or PMETAC–N3, respectively. Once purified, the polymers were characterized by gel permeation chromatography to elucidate their molecular weight. Table 1 summarizes the results obtained from both polyelectrolytes. Polymerization times between 20 min and 2 hours enabled the facile synthesis of azide-terminated polyelectrolytes with number average molecular weights ranging from a few kDa to ∼20 kDa. Moreover, it can be seen that polydispersities are within the expected values for cRP initiated from typical free radical initiators. This indicates that the chemical modifications introduced in the 2,2′azobis(4-cyanovaleric acid) and leading to the synthesis of the azide-terminated initiator, do not have detrimental effects on its free radical initiation characteristics.
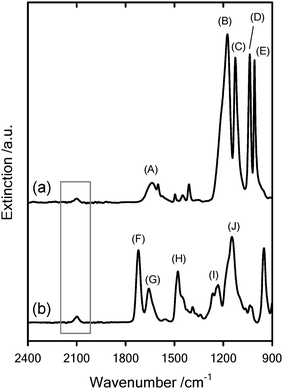
The azide-terminated polyelectrolytes were also characterized by FTIR (fig. 3). The spectra depict the typical IR frequencies encountered in PMETAC and PSSNapolymers plus the band associated to the antisymmetric stretching of the azide group. Hence, this typical fingerprint, corresponding to the –N3, corroborates the end functionalization of the polyelectrolyte chains.
 | ||
| Fig. 3
FTIR spectra corresponding to: (a) azide-terminated sodium polystyrene sulfonate (PSSNa–N3) and (b) azide-terminated poly(2-methacryloyloxyethyl-trimethylammonium chloride) (PMETAC–N3). The different labels of the infrared spectra are: (A) 1640 cm−1 (C–C) stretching vibration of aromatic skeleton, (B) 1175 cm−1 SO3− group symmetric vibration, (C) 1127 cm−1 in-plane skeleton vibration of phenyl ring, (D) 1036 cm−1 SO3− group antisymmetric vibration, (E) 1008 cm−1 in-plane bending vibration of phenyl ring, (F) 1719 cm−1 (CO) stretching vibration, (G) 1655 cm−1 asymmetric bending vibration of the quaternary amine cation (QA+), (H) 1477 cm−1 (C–H) in-plane bending vibration of QA+, (I) 1232 cm−1 (C–N) stretching vibration, (J) (OC–O–) stretching vibration. The gray frame indicates the region corresponding to the antisymmetric stretching vibration of the –N3 groups (∼2100 cm−1). | ||
3.4 “Clicking” polyelectrolyte chains on planar Si surfaces
Solid substrates modified with polyelectrolyte chains have encountered a wide variety of applications in different technological fields like lubrication or colloidal stabilization.1,2 In this regard, anchoring polymer chains to surfaces using a “grafting-to” approach has proven to be an effective strategy for creating highly functional interfaces. As previously described, the “click” reaction involves two reactants, the azide- and the alkyne-modified building blocks. The polyelectrolytes were functionalized with terminal azide groups, so we modified the substrates with alkyne functionalities (Fig. 4). This was readily accomplished by derivatization of the Si substrates with alkyne-terminated self-assembled monolayers (SAMs). The procedure for assembling the alkyne functionalities was based on the condensation of ethynyldimethylchlorosilane with silanols from the surface of plasma-treated silicon wafers. The use of monochlorosilane enables attaining SAMs with better homogeneity and improves the stability of the self-assembling solution, which in our case was in toluene. As is well-known, trichlorosilanes are prone to hydrolysis and crosslinking in the presence of traces of water, leading to the formation of aggregates in solution that are deposited on the substrates.39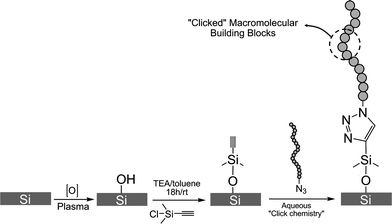 | ||
| Fig. 4 Scheme describing the functionalization of the silicon surface with alkyne groups followed by the “clicking” of the polyelectrolyte chains. | ||
After an 18 h assembly period, the substrates were rinsed with toluene, and the wettability was examined. A water contact angle of ∼90° corroborated the effectiveness of the surface functionalization with the alkyne-functionalized SAM (Fig. 5). The macromolecular building blocks were then “clicked” on the alkyne functionalized Si surface. The alkyne-terminated surfaces were immersed in a water solution containing the azide-terminated polymer, CuSO4·5H2O, and sodium ascorbate and left overnight at RT (Fig. 4). The modified silicon wafers were rinsed thoroughly with water and dried under a stream of N2.
 | ||
| Fig. 5 Atomic force imaging and contact angle measurements corresponding to: (a) alkyne-functionalized Si surface, (b) PSSNa–N3-modified Si surface and (c) PMETAC–N3-modified Si surface. | ||
Once the polyelectrolytes were “clicked” on the surfaces, the wetting characteristics were studied (Fig. 5). Both PSSNa- and PMETAC-modified silicon surfaces showed a marked increase in hydrophilicity when compared to the alkyne-terminated platforms. The contact angles corresponding to the PSSNa- and PMETAC-modified substrates were 42° and 49°, respectively. This change in wettability indicated that the grafting of the polyelectrolyte chains had a noticeable impact on the macroscopic properties of the substrate. To further corroborate the successful surface functionalization of the substrates we performed X-ray photoelectron spectroscopy (XPS) analysis of the “clicked” samples. XPS data indicated the presence of the elements constituting the macromolecular building blocks (Fig. 6). In addition, detailed XPS analysis of the C1s signal revealed the presence of different chemical environments for carbon, which is in close agreement with that expected for PSSNa and PMETACmacromolecules.
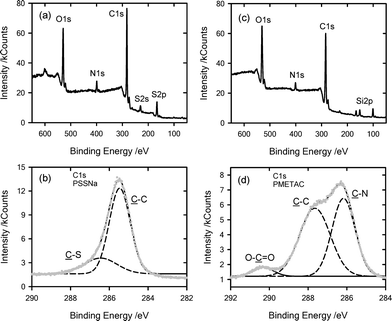 | ||
| Fig. 6 XPS analysis corresponding to: (a) PSSNa-modified silicon, broad spectrum, (b) PSSNa-modified silicon, 1Cs region, (c) PMETAC-modified silicon, broad spectrum, (d) PMETAC-modified silicon, C1s region. | ||
In a similar fashion, AFM imaging revealed significant topographic changes after grafting the polyelectrolyte changes as a consequence of the appearance of nodular-like aggregates evenly distributed on the surface (Fig. 5). On average, grafting polyelectrolyte chains of ∼15 kDa led to ∼2 nm thick films (dry thickness). Considering the grafting density as given by eqn 1:
| σ = (hρNA)/Mn | (1) |
3.5 Functionalization of single conical polymer nanochannels via a “click” chemistry approach
There is a growing interest in developing nanodevices based on synthetic nanopores because they are considered to be promising candidates for a wide variety of applications, including separation techniques and chemical sensing. Tailoring the chemical characteristics of nanopore surfaces is of great interest as it means that the surface composition is no longer fixed by the choice of the substrate material. Within this framework, Siwy et al.41 demonstrated that a polymeric membrane containing a single conical channel can act as an ion rectifier, and this rectifying behaviour depended on the nature of the fixed charges of the nanochannel wall.This experimental evidence clearly indicates that developing strategies to manipulate the surface charges of nanochannels is of paramount importance in order to control and regulate the transport of ions and molecules through nanopores.42 To monitor the “clicking” of the polyelectrolytes on the nanochannels, we carried out all the surface modification steps with the single channel membrane mounted in the electrochemical cell. This enabled us to track the changes occurring at the nanochannel surface due to consecutive chemical modifications (Fig. 7). We first modified the PET surface with alkyne groups. This was accomplished by first activating the carboxyl groups on the etched surface with EDC and PFP for one hour at room temperature (Fig. 7a,b). Afterwards, the alkyne groups were introduced on the surface by coupling between the activated esters and propargylamine. This step required an overnight reaction time to achieve a successful coupling of the alkyne moieties on the pore PET surface. This chemical modification was evidenced by a change from a rectifying to a non-rectifying behaviour (Fig. 8). This was due to the fact that the –COO− groups imparting negative surface charges were reacted with propargylamine, which neutralized the pore surface. Subsequently, the alkyne-modified membrane was subjected to “click” chemistry by immersing the membrane in a solution containing azide-terminated polyelectrolytes and the corresponding catalyst system i.e.CuSO4·5H2O and sodium ascorbate (Fig. 7c). After washing the “clicked” membranes several times with water the corresponding I–V curves were measured (Fig. 8). As expected, clicking the polyelectrolyte chains on the nanochannel surface promoted drastic changes of their rectifying characteristics. It is clearly observed that incorporating the charged building blocks into the PET nanochannels enables a straightforward manipulation of their permselective properties. Nanochannels modified with PSSNa–N3 described a rectifying behaviour (i.e. rectification characteristic of the channel was recovered) similar to that displayed by PET nanochannels with surface –COO− groups, but achieving lower rectified currents at similar voltages (Fig. 8a).
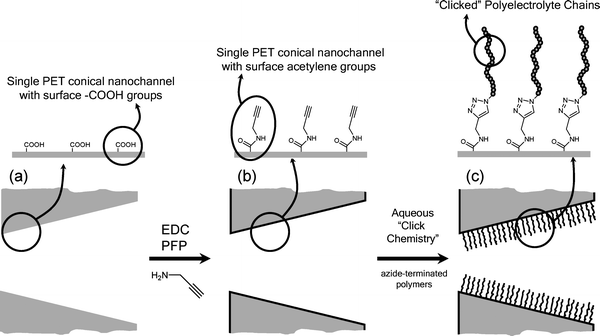 | ||
| Fig. 7 A scheme showing the surface chemical modification of the COOH-functionalized PET conical nanochannels (a) with alkyne groups (b) followed by the “clicking” of the polyelectrolyte chains (c). | ||
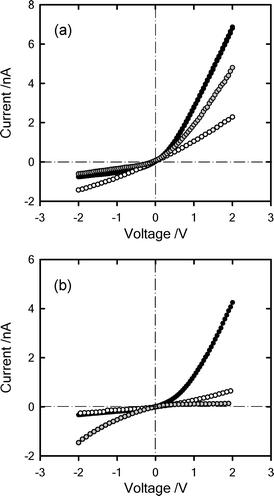 | ||
| Fig. 8 Current–voltage curves corresponding to nanochannels in 0.1 M KCl modified with: (a) PSSNa–N3 having tip diameter d = 8 nm and (b) PMETAC–N3 having tip diameter d = 5 nm. The different colours indicate: (black) carboxylated nanochannel, (white) alkyne-modified nanochannel and (gray) polyelectrolyte-modified nanochannel. | ||
This observation can be attributed to the fact that grafting the polyelectrolyte chains do not solely change the surface charge but also alter the effective cross-section of the nanochannels. It is well known that these two contributions have an impact on the magnitude and characteristics of the rectified currents.42 In a similar fashion, PMETAC–N3-modified nanochannels displayed a well-defined rectifying behaviour in which anions were permselectively transported across the positively charged channels. This leads to the inversion of rectification as expected for a positively charged surface (Fig. 8b). Applying a positive transmembrane potential (2 V) revealed very low ionic transport through the pore, while at −2 V the ionic transport suffered significant changes and currents of nearly −1.46 nA were detected. This experimental evidence strongly supports the idea that “clicking” polyelectrolytes on the nanochannel can be an avenue for the facile creation of permselective macromolecular gates.
4. Conclusions
In summary, in this work we described a new route for the facile one-step preparation of azide-terminated polymers. The strategy is based on the use of conventional radical polymerization in combination with a novel azide-terminated azo initiator which permitted the straightforward synthesis of different polymers end-functionalized with azide groups. The facile synthesis of the functionalized macromolecular building blocks enabled us to merge the versatility of “click” chemistry with the robustness and ease of cRP to pave the way to a wide variety of “clickable” architectures. This was demonstrated by the “clicking” of azide-terminated polyelectrolytes on alkyne-terminated surface via a “grafting-to” approach. In particular, we exploited these “clickable” macromolecular building blocks to tailor the chemical characteristics of planar silicon surfaces; and to tune the surface charge of conical PET nanochannels in order to control their permselectivity. We envision that these results will not only appeal to organic and polymer chemists but also to materials scientists willing to explore the use “click” chemistry in different colloidal and surface science applications. As such, we consider that this approach will have strong implications for the molecular design of interfaces using macromolecular architectures.Acknowledgements
B.Y. acknowledges support from the Higher Education Commission (HEC) of Pakistan and Deutscher Akademischer Austauschdienst (DAAD) (Code #A/04/30795). M.A. thanks the Higher Education Commission (HEC) of Pakistan, on receiving partial financial support. O.A. is a CONICET fellow and acknowledges financial support from the Max Planck Society (Germany), the Alexander von Humboldt Stiftung (Germany) and the Centro Interdisciplinario de Nanociencia y Nanotecnología (CINN) (ANPCyT – Argentina). We are grateful to Sandra Seywald and Ute Heinz (MPIP – Mainz, Germany) for helpful assistance in GPC measurements.References
- A. M. Urban, M. W. Urban, in “Stimuli-Responsive Polymeric Films and Coatings”, M. W. Urban, (ed.) (American Chemical Society, Washington, 2005) Ch. 1, pp. 1–25 Search PubMed.
- (a) S. Reichelt, U. Gohs, F. Simon, S. Fleischmann, K.-J. Eichhorn and B. Voit, Langmuir, 2008, 24, 9392–9400 CrossRef CAS; (b) J. Huang, R. R. Koepsel, H. Murata, W. Wu, S. B. Lee, T. Kowaleski, A. J. Russell and M. Matyjaszewski, Langmuir, 2008, 24, 6785–6795 CrossRef CAS; (c) K. S. Iyer, B. Zdyrko, H. Malz, J. Pionteck and I. Luzinov, Macromolecules, 2003, 36, 6519–6526 CrossRef CAS; (d) S. Minko, S. Patil, V. Datsyuk, F. Simon, K.-J. Eichhorn, M. Motornov, L. Tokarev and M. Stamm, Langmuir, 2002, 18, 289–296 CrossRef CAS; (e) H. Huang and L. S. Penn, Macromolecules, 2005, 38, 4837–4843 CrossRef CAS.
- A. M. Granville, and W. J. Brittain, in “Polymer Brushes: Synthesis, Characterization and Applications”, R. A. Advincula, W. J. Brittain, K. C. Caster, and J. Ruhe, (eds.) (VCH-Wiley, Weinheim, 2004) pp. 35–50 Search PubMed.
- F. Denizli, Y. Arica and A. Denizli, React. Funct. Polym., 2000, 44, 207–217 CrossRef CAS.
- M. Slater, M. Snauko, F. Svec and J. M. J. Frechet, Anal. Chem., 2006, 78, 4969–4975 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kreibel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- Y. Tran and P. Auroy, J. Am. Chem. Soc., 2001, 123, 3644–3654 CrossRef CAS.
- S. M. Sirard, R. R. Gupta, T. P. Russell, J. J. Watkins, P. F. Green and K. P. Johnston, Macromolecules, 2003, 36, 3365–3373 CrossRef CAS.
- (a) O. Burtovyy, V. Klep, H. C. Chen, R. K. Hu, C. C. Lin and I. Luzinov, J. Macromol. Sci., Part B: Phys., 2007, 46, 137–154 Search PubMed; (b) I. Luzinov, D. Julthongpiput, A. Liebmann-Vinson, T. Cregger, M. D. Foster and V. V. Tsukruk, Langmuir, 2000, 16, 504–516 CrossRef CAS; (c) L. Ionov, A. Sidorenko, M. Stamm, S. Minko, B. Zdyrko, V. Klep and I. Luzinov, Macromolecules, 2004, 37, 7421–7423 CrossRef CAS; (d) L. Ionov, A. Sidorenko, K. H. Eichhorn, M. Stamm, S. Minko and K. Hinrichs, Langmuir, 2005, 21, 8711–8716 CrossRef CAS; (e) S. Minko, S. Patil, V. Datsyuk, F. Simon, K. J. Eichhorn, M. Motornov, D. Usov, I. Tokarev and M. Stamm, Langmuir, 2002, 18, 289–296 CrossRef CAS.
- (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Shapless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS; (b) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS; (c) H. C. Kolb, M. G. Finn and K. B. Kolb, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- (a) J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, N. Dirvianskyte, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373–1375 CrossRef CAS; (b) D. A. Fleming, C. J. Thode and M. E. Williams, Chem. Mater., 2006, 18, 2327–2334 CrossRef CAS.
- M. A. White, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS.
- F. S. Hassane, B. Frisch and F. Schuber, Bioconjugate Chem., 2006, 17, 849–854 CrossRef CAS.
- (a) G. Such, J. F. Quinn, A. Quinn, E. Tjipto and F. Caruso, J. Am. Chem. Soc., 2006, 128, 9318–9319 CrossRef CAS; (b) G. K. Such, E. Tjipto, A. Postma, A. P. R. Johnston and F. Caruso, Nano Lett., 2007, 7, 1706–1710 CrossRef CAS.
- (a) K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS; (b) M. J. Joralemon, R. K. O'Reilly, J. B. Matson, A. K. Nugent, C. J. Hawker and K. L. Wooley, Macromolecules, 2005, 38, 5436–5443 CrossRef CAS; (c) P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 5775–5777 RSC.
- (a) R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef CAS; (b) M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892–16899 CrossRef CAS.
- (a) B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS; (b) M. Malkoch, K. Schleicher, E. Drockenmuller, C. J. Hawker, T. P. Russell, P. Wu and V. V. Fokin, Macromolecules, 2005, 38, 3663–3678 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- (a) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–34 CrossRef CAS; (b) H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef CAS.
- S. Ciampi, T. Böcking, K. Kilian, J. B. Harper and J. J. Gooding, Langmuir, 2008, 24, 5888–5892 CrossRef CAS.
- S. Ciampi, T. Böcking, K. Kilian, M. James, J. B. Harper and J. J. Gooding, Langmuir, 2007, 23, 9320–9329 CrossRef CAS.
- C. Haensch, C. Ott, S. Hoeppener and U. S. Schubert, Langmuir, 2008, 24, 10222–10227 CrossRef CAS.
- R. V. Ostaci, D. Damiron, S. Capponi, G. Vignaud, L. Leger, Y. Grohens and E. Drockenmuller, Langmuir, 2008, 24, 2732–2739 CrossRef CAS.
- S. Prakash, T. M. Long, J. C. Selby, J. S. Moore and M. A. Shannon, Anal. Chem., 2007, 79, 1661–1667 CrossRef CAS.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051–1053 CrossRef CAS.
- (a) A. Devadoss and C. E. D. Chidsey, J. Am. Chem. Soc., 2007, 129, 5370–5371 CrossRef CAS; (b) T. Lummerstorfer and H. Hoffmann, J. Phys. Chem. B, 2004, 108, 3963–3966 CrossRef CAS; (c) J. K. Lee, Y. S. Chi and I. S. Choi, Langmuir, 2004, 20, 3844–3847 CrossRef CAS.
- J. P. Collman, N. K. Devaraj, T. P. A. Eberspacher and C. E. D. Chidsey, Langmuir, 2006, 22, 2457–2464 CrossRef CAS.
- S.-Y. Ku, K. T. Wong and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 2392–2393 CrossRef CAS.
- L. Britcher, T. J. Barnes, H. J. Griesser and C. A. Prestidge, Langmuir, 2008, 24, 7625–7627 CrossRef CAS.
- (a) N. V. Tsarevsky, K. V. Bernaerts, B. Dufour, F. E. Du Prez and K. Matyjaszeski, Macromolecules, 2004, 37, 9308 CrossRef CAS; (b) G. Mantovani, V. Ladmiral, L. Lao and D. M. Haddleton, Chem. Commun., 2005, 2089 RSC; (c) B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszeski, Macromolecules, 2005, 38, 7540 CrossRef CAS.
- (a) M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, B. T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS; (b) R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776 CrossRef CAS.
- (a) R. J. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 CrossRef CAS; (b) R. K. O'Reilly, M. J. Joralemon, W. Lui, C. J. Hawker and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5203 CrossRef CAS; (c) D. Quemener, T. P. Davis and M. H. Stenzel, Chem. Commun., 2006, 5051 RSC.
- (a) W. H. Binder and C. Kluger, Macromolecules, 2004, 37, 9321 CrossRef CAS; (b) W. H. Binder, C. Kluger, M. Josipovic, C. Straif and G. Friedbacher, Macromolecules, 2006, 39, 8092 CrossRef CAS.
- A. Koenig, U. Ziener, A. Schaz and K. Landfester, Macromol. Chem. Phys., 2007, 208, 155–163 CrossRef CAS.
- N. S. Hatzakis, H. Engelkamp, K. Velonia, J. Hofkens, P. C. M. Christianen, A. Svendsen, S. A. Patkar, J. Vind, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2006, 2012–2014 RSC.
- W.-Y. Wong, C. K. Wong and C.-L. Lu, J. Organomet. Chem., 2003, 671, 27–34 CrossRef CAS.
- M. Sukeda, S. Ichikawa, A. Matsuda and S. Shuto, J. Org. Chem., 2003, 68, 3465–3475 CrossRef CAS.
- (a) M. Ali, V. Bayer, B. Schiedt, R. Neumann and W. Ensinger, Nanotechnology, 2008, 19, 485711 CrossRef; (b) P. Y. Apel, Y. E. Korchev, Z. Siwy, R. Spohr and M. Yoshida, Nucl. Instrum. Methods Phys. Res., Sect. B, 2001, 184, 337–346 CrossRef CAS.
- B. C. Bunker, R. W. Carpick, R. A. Assink, M. L. Thomas, M. G. Hankins, J. A. Voight, D. Sipola, M. P. de Boer and G. L. Gulley, Langmuir, 2000, 16, 7742 CrossRef CAS.
- L. S. Penn, H. Huang, R. P. Quirk, and T. H. Cheong, in “Polymer Brushes: Synthesis, Characterization and Applications”, R. A. Advincula, W. J. Brittain, K. C. Caster, J. Ruhe, (eds.) (VCH-Wiley, Weinheim, 2004) Ch. 16, pp. 317–330 Search PubMed.
- Z. Siwy, E. Heins, C. C. Harrell, P. Kohli and C. R. Martin, J. Am. Chem. Soc., 2004, 126, 10850–10851 CrossRef CAS.
- (a) L. T. Sexton, L. P. Horne and C. P. Martin, Mol. BioSyst., 2007, 3, 667–685 RSC; (b) R. Karnik, R. Fan, M. Yue, D. Li, P. Yang and A. Majundar, Nano Lett., 2005, 5, 943–948 CrossRef CAS; (c) F. H. J. van der Heyden, D. J. Bonthuis, D. Stein, C. Meyer and C. Dekker, Nano Lett., 2007, 7, 1022–1025 CrossRef CAS; (d) H. Daiguji, P. Yang and A. Majumdar, Nano Lett., 2004, 4, 137–142 CrossRef CAS; (e) I. Vlassiuk and Z. S. Siwy, Nano Lett., 2007, 7, 552–556 CrossRef CAS; (f) L. A. Baker and S. P. Bird, Nat. Nanotechnol., 2008, 3, 73–74 CrossRef CAS; (g) M. Ali, B. Schiedt, K. Healy, R. Neumann and W. Ensinger, Nanotechnology, 2008, 19, 085713 CrossRef; (h) B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Nano Lett., 2009, 9, 2788–2793 CrossRef CAS; (i) B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Small, 2009, 5, 1287–1291 CrossRef CAS; (j) B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, J. Am. Chem. Soc., 2009, 131, 2070–2071 CrossRef CAS; (k) M. Ali, B. Yameen, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, J. Am. Chem. Soc., 2008, 130, 16351–16358 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |