Micelles with surface conjugated RGDpeptide and crosslinked polyurea core viaRAFT polymerization†
Hien T. T.
Duong
,
T. L.
Uyen Nguyen
and
Martina H.
Stenzel
*
Centre of Advanced Macromolecular Design (CAMD), School of Chemical Sciences and Engineering, The University of New South Wales, Sydney NSW 2052, Australia. Web: http://www.camd.unsw.edu.auE-mail: M.Stenzel@unsw.edu.au; Fax: +61-2-93856250; Tel: +61-2-93854344
First published on 25th November 2009
Abstract
An improved drug delivery system was developed, which enhances the cellular uptake by stabilizing the micelle via core crosslinking resulting in the formation of stable core–shell particles and by decorating the micelles with arginine-glycine-aspartic acid (RGD) containing peptides. In order to generate stable core–shell nanoparticles, block copolymers composed of poly(oligo(ethylene glycol) methyl ether methacrylate)-block-polystyrene-co-poly(3-isopropenyl-α,α-dimethylbenzyl isocyanate) (POEGMA-block-P(STY-co-TMI)) were synthesized viareversible addition fragmentation chain transfer (RAFT) polymerization. Depending on the length of the hydrophobic block, self-assembled aggregates such as micelles, rods and large compound micelles were created in the aqueous solutions. The use of a protected aldehyde containing RAFT agent (CTA) allowed the formation of endfunctional block copolymers , producing micelles bearing aldehyde groups incorporated onto the surface allowing further conjugation with a RGD containing peptide after deprotection. The micelles were stabilized by core crosslinking through the reaction of reactive isocyanate groups with hexamethylenediamine forming stable urea containing core–shell particles. Conjugation with a RGD containing linear peptide, GRGDS, resulted in the formation of micelles bearing peptide groups on the surface. Cytotoxicity tests confirmed the biocompatibility of the synthesized crosslinked micelles revealing efficient cell uptake without causing any signs of cell damage. The conjugation of GRGDS to polymeric micelles and the core crosslinking was shown to significantly enhance the cellular uptake.
Introduction
Polymeric micelles, self-assembled block copolymers with core–shell structures and diameters of 10–100 nm, are well-known as promising vehicles for the targeted delivery of drugs, proteins, genes and imaging agents. During the past few decades, many research groups have focused their attention on the development and the feasibility of polymeric micelles for therapeutic and diagnostic medicine applications.1–11When self-assembled in aqueous solution, these core–shell structures have a hydrophobic inner core, which can serve as a nanocontainer for hydrophobic drugs, while the hydrophilic outer shell stabilizes the micelles in the aqueous dispersion. Since the molecular weight of polymeric micelles may reach more than 106 g mol−1, they surpass the typical renal threshold (ca. 40 kDa).12 In addition, their nanoscopic size is typically less than 200 nm and they are therefore less recognizable by the phagocytic cell of the reticuloendothelial system (RES). The uptake is, however, also dependent on the surface chemistry of the particles. Thus, polymeric micelles usually lead to a prolonged blood circulation.13,14 The “stealth” property of micelles, which leads to a delayed detection by the RES, in combination with the passive accumulation in the solid tumour through the enhanced permeability and retention (EPR) mechanism15 make these core–shell particles a suitable choice for drug delivery purposes.
However, recently it has been suggested that not only polymeric micelles, but also other self-assembled structures such as “crew-cut” aggregates can be advantageous for drug delivery purposes. Recent literature has highlighted that a rod-like architecture can enhance the circulation time of the drug carrier within the body.16 Vesicles with their aqueous interior could be advantageous in the delivery of water-soluble drugs or protein based drugs such as vaccines. Eisenberg and coworkers have pioneered the field of micellar morphology control with so-called “crew-cut” micelles and have proved that the observed morphologies of micelles were strongly dependent on the copolymer composition, preparation method and also copolymer polydispersity.7,17,18 The morphological transition behaviour from spheres to rods to vesicles has been investigated in detail on series of PS-PAA19 and PS-PEO20copolymers . Various morphologies of the amphiphilic multiblock copolymer [poly(ethylene oxide)-block-polystyrene]n micelles were observed, which were strongly dependent on the length of the PS block chains while the chain length of PEO was fixed.21
The attraction of polymeric micelles and other aggregates as drug delivery systems is further enhanced by the flexibility of the underlying block copolymer chemistry allowing the modification and functionalization of the core and shell of the nanostructure for the targeted delivery purpose. Various polymeric micelle systems based on different amphiphilic block-copolymers composed of a variety of hydrophilic and hydrophobic chains have been investigated. Poly(ethylene oxide) (PEO)-based block copolymers (e.g.PEO-b-PPO-b-PEO, Pluronics, PEO-b-poly(L-amino acid)s, PEO-polyesters) are the most widely investigated systems.22PEO as building block promotes significant hydration of the drug delivery device leading to excellent biocompatibility.23
Research in the field of polymeric micelles for drug delivery has been booming, the development of highly efficient, targeted drug delivery systems is, however, still challenging due to poor stability and inefficient cellular uptake. It is well-known that because of their dynamic nature, polymeric micelles will disintegrate into unimers at low concentration or under certain changes in their environment due to changes in temperature, pH values or ionic strength.24 This will cause the undesired premature release of the drug from the micellar nanocontainer before the carrier reaches the intended targets. Crosslinking between the polymer chains of the core or corona has been demonstrated as an easy and promising approach to reinforce the micellar structure. Wooley and coworkers have developed the corona crosslinking strategy and obtained the so-called shell-crosslinked (SCL) “knedel-like” micelles.25–27 There are also numerous reports on the stabilization of the polymeric micelles by core crosslinking (CCL).28–34 Unfortunately, core crosslinking often affects the loading capacity of the micelle and the uptake of drugs is limited.
As mentioned earlier, the poor cellular uptake still remains a major challenge when utilizing polymeric micelles for targeted drug delivery. Several studies focused on the covalent attachment of bioactive molecules such as peptides, folic acid-based ligands or sugars to improve the interaction between the cell and the synthetic substrates.35–39Arginine-glycine-aspartic acid (RGD) is recognized by integrin and therefore constitutes a major recognition system for cell attachment. To conjugate RGD to the surface of a micelles, reactive groups are a prerequisite. Aldehydes can easily undergo bioconjugation with the RGD containing peptideGRGDS under mild conditions with high coupling efficiency via the formation of Schiff bases.
Herein, we have designed a versatile block copolymer that can promote fast and efficient crosslinking whilst carrying reactive end groups for further conjugation to a peptide, which can advantageously enhance the cell-uptake of the crosslinked micelle. Therefore, we investigated the synthesis of core crosslinked poly(oligo(ethylene glycol) methyl ether methacrylate)-block-poly(styrene)-co-poly(3-isopropenyl-α,α-dimethylbenzyl isocyanate) (POEGMA-block-P(STY-co-TMI)) micelles viareversible addition fragmentation chain transfer (RAFT) polymerization using a functional RAFT aldehyde containing agent (Scheme 1). The chosen POEGMA block is closely related to PEO and it is suggested that similar low-fouling properties can be achieved despite the less flexible polymethacrylate backbone.40 The RAFT processhas been demonstrated to be a versatile and robust technique, suitable for generation of well-defined complex macromolecules, applicable to a large range of monomers in a wide variety of solvents (including water), under broad range of experimental conditions.41–43 The proposed system here uses a core domain composed of a random copolymer of styrene and TMI. TMI is a particularly attractive monomer because of its inherent dual functionality allowing it to take part in free radical processes and in polyadditions such as polyurea formation (Scheme 2). Barner et al. observed that TMI is reluctant to homopolymerize to any significant extent due to the steric hindrance of its α-methyl group, thus, TMI can only be co-polymerized with other monomers such as styrene.44 The reaction kinetics of the radical copolymerization of TMI and styrene has already been studied in detail as well as the thermal properties of the resulting polymer.44–46
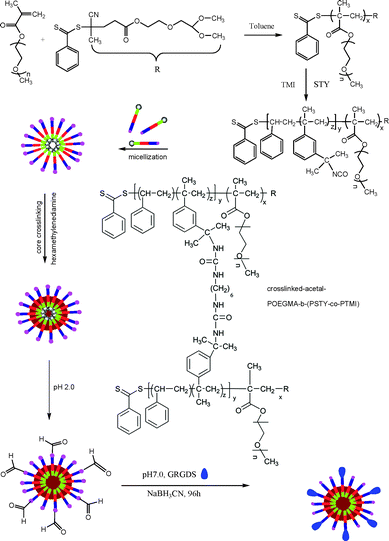 | ||
| Scheme 1 Synthetic route for the preparation of crosslinked GRGDS-POEGMA-block-P(STY-co-TMI) | ||
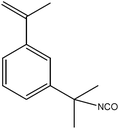 | ||
| Scheme 2 Molecular structure of TMI | ||
Materials and methods
Materials
All reagents were purchased from Sigma-Aldrich with the highest purity and were used as supplied unless otherwise noted. Oligo(ethylene glycol) methyl ether methacrylate (OEGMA) (Mn = 300 g mol−1) and styrene were destabilized by passing them through a column of basic alumina. 2,2-Azobisisobutyronitrile (AIBN) (Fluka, 98%) was purified by recrystallization from methanol.Synthesis
1H NMR (300 MHz, CDCl3) δ = 2.10 (bs, 1H, OH), 3.40 (s, 6H, OCH3), 3.55 (d, J = 5.3 Hz, 2H, CHCH2O), 3.61–3.63 (m, 2H, OCH2CH2), 3.72–3.74 (m, 2H, CH2OH), 4.51 (t, J = 5.3 Hz; 1H, CH(OCH3)2; 13C NMR (300 MHz, CDCl3) δ = 54 (2C, OCH3), 61 (1C, CH2OH), 71 (1C, CHCH2O), 73.00 (1C, OCH2CH2), 103 (1C, CH(OCH3)2).
1
H NMR (300 MHz, CDCl3) δ = 1.84 (s, 3H, -CH3), 2.39–2.79 (m, 4H, CH2CH2CO, 3.4 (s, 6H, OCH3), 3.54 (d, J = 5.0 Hz, 2H, CHCH2O), 3.74–3.71 (m, 2H, OCH2CH2), 4.29–4.24 (m, 2H, CH2OH), 4.51 (t, J = 5.0 Hz; 1H, CH(OCH3)2, 7.92–7.89 (m, 2H, CH aro.), 7.60–7.55 (m, 1H, CH aro.), 7.42–7.37 (m, 2H, CH aro.)
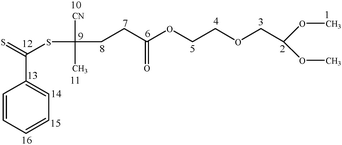
13C NMR (300 MHz, CDCl3) δ (ppm): 23.73(C-11), 29.65 (C-7), 33.27 (C-8), 45.74 (C-9), 53.98 (C-1), 63.66 (C-5), 69.10 (C-3), 70.79 (C-4), 102.66 (C-2), 118.36 (C-10), 127.0 (C-15), 128.28 (C-14), 133.08 (C-16), 144.60 (C-13), 171.47 (C-6), 221.89 (C-12)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1 (PDI = 1.13).
900 g mol−1 (PDI = 1.13).
The polymer was purified three times by precipitation in petroleum spirits (boiling range of 40–60 °C). After centrifugation (7000 rpm for 15 min), the polymer was dried under reduced pressure at room temperature. The samples were stored in a freezer prior to modification
900 g mol−1, Mn(SEC) = 26800 g mol−1) was used as a so-called macroRAFT agent for chain extension with styrene and TMI. The number of repeating units of POEGMA was calculated from the monomer conversion obtained from 1H NMR. The POEGMA macroRAFT agent (2.10 × 10−1 g, 9.3 × 10−6 mol) was dissolved in 8 mL (7.27 g, 6.98 × 10−2 mol) of STY and 2 mL (2.04 g, 1.02 × 10−2 mol) of TMI. The reaction mixture was divided into 6 vials for the block copolymerization kinetic study and then purged with nitrogen for one hour in an ice bath to avoid the evaporation of styrene. The polymerizations were carried out in an oil bath at 100 °C. The vials were taken out at different time intervals over a period of 24 h. The polymerizations were terminated by placing the samples in an ice bath for 5 min. The copolymer was purified using membrane dialysis for 2 days against acetone, to remove unreacted styrene and TMI. The prepared polymer solutions (in acetone) were kept at 2 °C prior to further experiments.
Analysis
(1) 100% A for the first 1 min, (2) a linear gradient from 100% A to 60% A for 20 min, a linear gradient from 60% A to 0% A for 4 min, (4) 0% A for 2 min, (5) 0% A to 100% A in 4 min, and (6) 100% A for 5 min. The micellar solutions (20 µL) were injected into the HPLC system. Free peptide was detected with a UV detector at a wavelength of 214 nm.
Results and discussions
RAFT polymerization
The conjugation of peptides or proteins to polymer chains can be carried out using a variety of synthetic pathways.48 A potential synthetic avenue is the reaction between the aldehyde and amino functionalities of the peptide/protein. The Schiff base can be formed under mild conditions avoiding potential destruction of the biological molecule. For the synthesis of α-aldehyde terminally functional block copolymers , an acetal containing RAFT agent was synthesized in a two step reaction as shown in Scheme 1. The incorporation of the aldehyde group on the R-group of the RAFT agent was targeted to avoid loss of conjugated peptide in the final product as a result of the hydrolysis of the sensitive thiocarbonyl group. The RAFT agent was synthesized in a two step procedure starting with the synthesis of 2-(2,2-dimethoxy-ethoxy)-ethanol (1) by a conventional substitution reaction from the commercially available 3-bromo-1-propanol. In the next step, the final RAFT agent (2) was prepared by the coupling reaction between 2-(2,2-dimethoxy-ethoxy)-ethanol and 4-cyanopentanoic acid dithiobenzoate (CPADB) yielding conversions as high as 83% (Scheme 3). CPADB was prepared according to the procedure described in the literature.47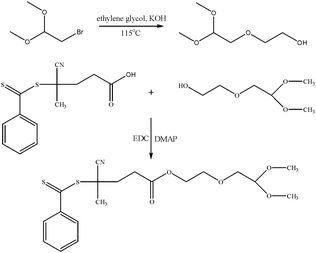 | ||
| Scheme 3 Synthesis of RAFT agent 4-cyano-4-methyl-4-thiobenzoylsulfonyl-butyric acid 2-(2,2-dimethoxy-ethoxy)-ethyl ester (acetal-RAFT) (2) | ||
The RAFT agent (2) was subsequently employed in the polymerization of POEGMA in toluene at 70 °C. The conversion of the monomer was determined using NMR spectroscopy comparing the intensity of vinyl proton peaks (6.1 and 5.6 ppm) to that of aliphatic proton peaks (1.1–1.3 ppm). The results are summarized in Table 1. As displayed in Fig. 1(A), the monomer conversion increased with reaction time and the radical concentration was constant during the course of polymerization as evidenced by a pseudo first order plot. A slight deviation from the linear plot was seen at a polymerization time of 17 h, which is probably due to the significant AIBNdecomposition at 70 °C. The theoretical and experimental molecular weights are proportional with the monomer conversion as shown in Table 1. The experimental molecular weight increased linearly with the monomer conversions and the polydispersity index (PDI) remained low (<1.20) during the course of polymerization indicating a well-controlled RAFT process. The SEC profiles in Fig. 1(B) reveal the shift of polymer peaks towards higher molecular weight with increasing conversion whilst the formation of shoulders, which may indicate the formation of a substantial amount of terminated by-product, is clearly absent, confirming the high degree of control exerted over the polymerization in this system.
| Time/h | Conversion (%)a | Mn (theo)/g mol−1b | Mn (SEC)/g mol−1c | PDI |
|---|---|---|---|---|
| a The monomer conversion was calculated from the 1H NMR of the reaction mixture by comparing the intensity of vinyl proton peaks (at ca. 6.1 and 5.6 ppm) to that of aliphatic proton peaks (at ca.1.2 ppm). b Theoretical molecular weight was calculated from linear relationship between molecular weight and conversion according to according to Mn = ([M]o/[RAFT]o) * x * Mwmonomer + MwRAFT, where [M]o, [RAFT]o), x, Mwmonomer, and MwRAFT are monomer and RAFT agent concentration, monomer conversion, molecular weight of monomer and RAFT agent, respectively. c The experimental Mn and PDI were measured by SEC using dimethyl acetamide (DMAc, 0.05% w/v BHT and 0.03% w/v LiBr) as eluent and polystyrene standards with the molecular weight ranging from 168 to 106 g mol−1). | ||||
| 1 | 17 | 5600 | 6800 | 1.13 |
| 3 | 39 | 12300 |
12500 |
1.12 |
| 5 | 53 | 16400 |
17300 |
1.15 |
| 8 | 69 | 21200 |
21900 |
1.11 |
| 17 | 88 | 26800 |
26900 |
1.13 |
![RAFT polymerization of acetal-POEGMA using acetal-RAFT (2) in toluene at 70 °C ([M] : [RAFT] : [AIBN] = 100 : 1 : 0.2). (A) Monomer conversion at different time intervals and pseudo-first-order kinetic plot for the homopolymerization; (B) SEC profiles of acetal-POEGMA at 17, 39, 53, 69, 88% monomer conversions.](/image/article/2010/PY/b9py00210c/b9py00210c-f1.gif) | ||
| Fig. 1 RAFT polymerization of acetal-POEGMA using acetal-RAFT (2) in toluene at 70 °C ([M] : [RAFT] : [AIBN] = 100 : 1 : 0.2). (A) Monomer conversion at different time intervals and pseudo-first-order kinetic plot for the homopolymerization; (B) SEC profiles of acetal-POEGMA at 17, 39, 53, 69, 88% monomer conversions. | ||
The POEGMA macroRAFT agent used for the block copolymer synthesis and subsequent micellization, core crosslinking and peptide conjugation experiments was obtained after a monomer conversion of 88% (Mn (theo) = 26900 g mol−1, Mn (SEC) = 26800 g mol−1, PDI = 1.11). The medium-range conversion was chosen to avoid the formation of a significant amount of dead polymer, which is frequently observed at very high monomer conversion of more than 90%.24
To confirm the presence of the acetal and therefore the aldehyde functionality, the resulting polymer was subjected to acid treatment, which hydrolysed the acid-sensitive acetal group, forming the aldehyde. Fig. 2 shows the 1H NMR spectrum of prepared aldehyde-POEGMA in CDCl3. The intensity of peaks corresponding to the methine protons of the acetal group at δ = 4.6 ppm completely disappeared and the related peak for the aldehyde proton appeared at δ = 9.8 ppm.
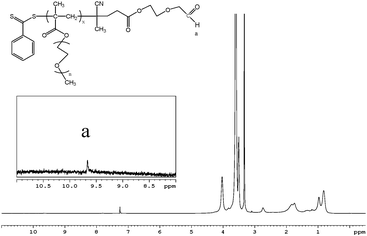 | ||
| Fig. 2 1H NMR spectrum of aldehyde-POEGMA in CDCl3. | ||
As mentioned earlier, TMI does not homopolymerize to any significant extent by a radical mechanism due to the low ceiling temperature of derivatives of α-methyl styrene, thus the chain extension of POEGMA was carried out with the mixture of TMI and styrene. Copolymerization with styrene is advantageous. By adjusting the ratio of the two monomers the crosslinking density in the core can be carefully tailored. In this work, the synthesis of a hydrophobic block which was significantly larger than the hydrophilic POEGMA block was targeted in order to not only attain the micelle structures, but also crew-cut aggregates. Crew-cut aggregates, such as rods or vesicles, are typically formed when the core-forming block exceeds the shell-forming block in size. The ratio between monomer and thiocarbonylthio concentration was therefore set to 8000, which is clearly pushing the limits of the RAFT process, and it is expected that a certain amount of termination products will emerge with increasing monomer conversion.
1H NMR spectroscopy was employed to monitor the consumption of TMI and styrene individually through their non-overlapping vinylic proton signals (see ESI† ).44 The analysis of the 1H NMR spectra using the vinylic proton signals of TMI (5.7 ppm) and styrene (6 ppm) results in the individual monomer conversion of both monomers as a function of the reaction time.
The POEGMA sample with 93 repeating units Mn (theo) = 26900 g mol−1, Mn (SEC) = 26800 g mol−1, PDI = 1.13) was used as a macroRAFT agent for the chain extension with TMI and STY. Fig. 3A shows the two traces of the individual monomer conversion versus reaction time for a copolymerization with [TMI] : [STY] = 1 : 4 in the initial reaction mixture. The rate of consumption of TMI is significantly higher in the early stages of the polymerization. This behaviour is in stark contrast to earlier results where the consumption of styrene was continuously higher than that of TMI.44 However, the fact that a hydrophilic macroRAFT agent was used needs to be taken into consideration. This macroRAFT agent influences the microenvironment of the RAFT endgroup encouraging a local concentration of hydrophilic TMI monomers. This bootstrap effect is known to significantly influence reactivity ratios leading to the preferred consumption of one monomer depending on the microenvironment.49
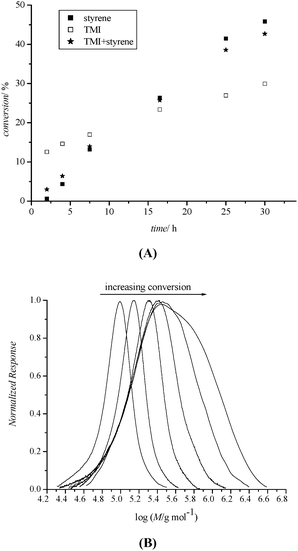 | ||
| Fig. 3 Chain extension of acetal-POEGMA with STY and TMI at 100 °C. (A) Monomer conversion vs. time for STY and TMI in the RAFT polymerization at 100 °C in bulk. (B) SEC profiles of acetal-POEGMA-block-P(STY-co-TMI) at different conversion. | ||
The molecular weight was found to increase with increasing conversion (Table 2, see ESI† for detailed analysis). However, after a reaction time of 7.5 h, the molecular weight distribution of purified polymer started to broaden (Fig. 3B) accompanied by a deviation of the experimental molecular weight from the theoretical molecular weight. As mentioned previously, the RAFT concentration in this system was uncommonly low since the synthesis of hydrophobic blocks with high molecular weights was targeted. The low RAFT agent concentration could therefore not efficiently suppress termination reactions resulting in the formation of a significant amount of dead polymer. Disregarding the high polydispersity index, the synthesis of block copolymer with a long hydrophobic block could be achieved.
900 g mol−1, Mn (SEC) = 26800 g mol−1, PDI = 1.13) at 100 °C in bulk, [styrene] = 6.98 g mol−1, [TMI] = 1.02 g mol−1, [POEGMA macroRAFT] = 1.02 × 10−3 g mol−1
| Time/h | STY + TMI conversion (%)a | Mn (theo) g mol−1b | Mn(SEC) g mol−1 | PDI |
|---|---|---|---|---|
| a The monomer conversion was calculated from the 1H NMR of the reaction mixture from the decrease of the intensity of STYvinyl proton peaks (6 ppm). b The theoretical molecular weight was calculated using monomer conversion from NMR and molar ratio between monomer and macroRAFT agent concentration; see ESI for details. | ||||
| 2 | 2.9 | 67282 |
71700 |
1.23 |
| 4 | 6.4 | 80704 |
108500 |
1.18 |
| 7.5 | 14 | 171620 |
127000 |
1.35 |
| 16.5 | 26 | 314489 |
133000 |
1.66 |
| 25 | 39 | 468265 |
162700 |
1.9 |
| 30 | 43 | 518067 |
204000 |
2.05 |
It should be noted here that the amount of TMI or the ratio of TMI and styrene will determine the crosslinking density of the core. The crosslinking density will furthermore have an influence on the loading capacity of the micelle. High crosslinking densities usually limit the amount of drug taken up by a micelle.
Self-assembly of POEGMA-block-P(STY-co-TMI) in aqueous solution
Soo and Eisenberg have done extensive research on the factors that may control the morphology of self-assembled aggregates such as absolute and relative block length, the water content of the solvent mixture, the nature and the presence of additives (ions, homopolymers, and surfactants) and polydispersity (PDI).50 It was also found that micellization is, to a significant extent, controlled by the length of the hydrophobic block.51 The block copolymers acetal-POEGMA-block-P(STY-co-TMI), where the length of the hydrophilic block was fixed and the length of the hydrophobic block was varied (Table 3), were dissolved in acetone. An aqueous phosphate buffer solution was slowly added followed by the evaporation of acetone. The isocyanate group was found to be stable under these conditions for a short period of time and premature crosslinking viaurethane formation was negligible. The solution was then investigated viaDLS and TEM.| Time/h | Number of repeating unit NSTY | Number of repeating unit NTMI | Number of repeating unit MTMI + STY |
|---|---|---|---|
| 2 | 56 | 167 | 222 |
| 4 | 399 | 194 | 593 |
| 7.5 | 1212 | 225 | 1437 |
| 16.5 | 2420 | 310 | 2731 |
| 25 | 3809 | 359 | 4167 |
| 30 | 4211 | 398 | 4609 |
As depicted in Fig. 4, the hydrodynamic diameter increased initially with the hydrophobic (TMI and STY) chain length. The length of the hydrophobic block needs to be considered as it has a significant influence on the measured hydrodynamic diameter. A short hydrophobic block compared to the hydrophilic block usually leads to the formation of micelles. Extending the length of the hydrophobic block may result in an increase in the size of the micelles, but leads to increasingly entropically unfavourable stretching of polymer chains, which needs to be accommodated. As a result, a transition of morphology to rods and vesicles may occur. These significantly different morphologies can have hydrodynamic diameters, which are substantially different. A morphology study using TEM is therefore necessary to complement any DLS data. However, once the number of hydrophobic repeating unit was very high (in this case, higher than 4000 units), the hydrodynamic diameter decreased again (Fig. 4). This unexpected result can be explained by the polydispersity of the block copolymer used. Terreau et al.52 investigated the effect of the PAA block polydispersity on the aggregate morphology on a series of PS-block-PAAcopolymers and found that the vesicle sizes generally decreased as the PAA polydispersity index (PDI) increased. Luo and Eisenberg suggested that the decrease in size was due to the segregation of the long chains preferentially to the outside whist the short chains segregated to the inside of the vesicle.18 This study was done using a block copolymer with a broad molecular weight distribution of the hydrophilic block, however, similar mechanism may take place when the hydrophobic block has a broad molecular weight distribution.
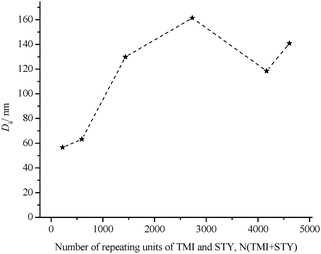 | ||
| Fig. 4 Hydrodynamic diameter Dhvs NSTY + TMI of micelles viaDLS (c = 4 g L−1 in phosphate buffer solution). | ||
Self-assembled micelles composed of amphiphilic diblock copolymers such as PS-block-PAA and PS-block-PEO in solution have been investigated extensively and morphologies such as spheres, rods, lamellae, tubules, vesicles, large compound micelles (LCMs) and large compound vesicles (LCVs) have been observed in the various systems.50,53 In this work, we hoped to achieve a similar catalogue of morphologies from POEGMA-block-P(STY-co-TMI) in aqueous media just by simply modifying the length of the hydrophobic block. As can be seen from Fig. 5, the increase in particle sizes on the basis of DLS analysis is accompanied by the transition of one different morphological structure to another one. As the hydrophobic chain length increases the type of self-assembled aggregate formed passes from spherical micelles (Fig. 5A and B) to rods (Fig. 5C) and to large compound micelles (LCM) (Fig. 5D), finally reaching aggregates that seem to form bilayer structures around LCM (Fig. 5E and F). However, the origin of Fig. 5E and F is not confirmed and need to be supported by further investigations. For a catalogue of structures the reader is referred to body of work by Eisenberg and coworkers.17–20,50,52
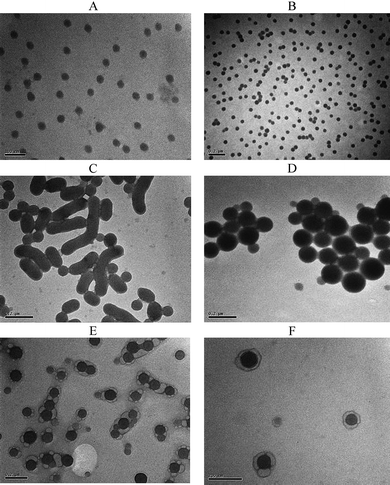 | ||
| Fig. 5 TEM analysis of the micelles (concentration of micelles 1 mg ml−1 in water) of (A) POEGMA93-block-P(STY56-co-TMI167), (B) POEGMA93-block-P(STY399-co-TMI194), (C) POEGMA93-block-P(STY1212-co-TMI225), (D) POEGMA93-block-P(STY2420-co-TMI310), (E) POEGMA93- block-P(STY3809-co-TMI359), (F) POEGMA93- block-P(STY4211-co-TMI398). | ||
Core crosslinked acetal-POEGMA-block-P(STY-co-TMI)
Self-assembled structures such as the ones depicted in Fig. 5 are dynamic structures with a tendency to dissociate at very low concentrations. The trend to dissociate is usually in correlation with the length of the hydrophobic block, with a short core-forming block forming less stable structures, whilst long block length can result in thermodynamically and kinetically very stable aggregates. The micellar structure formed from POEGMA93-block-P(STY56-co-TMI167) (Mn,theo = 67300 g mol−1, Mn,SEC = 71700 g mol−1, PDI = 1.23), which is the sample taken at 2 h during the block copolymerization benefits from further core crosslinking in order to achieve stable core–shell structures.
The pendant isocyanate group represents a reactive anchor for a difunctional group for crosslinking using amino or alcoholic functionalities, as well as water, leading to urethane formation. The reaction is usually fast and efficient in the presence of amine. However, since the crosslinking process is carried out in an aqueous environment, a preliminary experiment was necessary to determine the amount of crosslinking occurring in a competing process with water. FTIR analysis was employed to evaluate the amount of isocyanate reacting with water. Within the time frame investigated, the polymers did not show any significant signs of crosslinking in the aqueous environment. The hydrophobic core—in which the isocyanate is embedded—is therefore efficiently repelling any water penetration. It also needs to be considered that the reaction between isocyanates and water is usually slow in the absence of catalyst, especially at room temperature.54 The crosslinking reaction was carried out using hexamethylenediamine, a compound that is water-soluble but is also able to penetrate into the core of the micelle. Hexamethylenediamine was therefore added to the micellar solution of POEGMA93-block-P(STY56-co-TMI167). The reaction between isocyanate groups and the crosslinker hexamethylenediamine was monitored by ATR-FTIR spectroscopy and SEC. FTIR recorded the absorption of purified block copolymer POEGMA93-block-P(STY56-co-TMI167) before and after the crosslinking reaction (Fig. 6). The peak at 2250 cm−1 in the FT-IR spectra, which can be assigned to the NCO group, completely disappeared after a reaction time of 12 h, while the urea peak at around 3340 cm−1 confirmed the complete conversion of isocyanates into urea (Fig. 6).
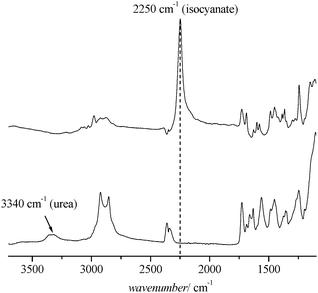 | ||
| Fig. 6 FT-IR spectra of POEGMA93-block-P(STY56-co-TMI167) before (top) and after (bottom) crosslinking reaction. | ||
The crosslinked polymer was also characterized by SEC by dissolving the copolymer after the crosslinking reaction in DMAc, which is a good solvent for both the hydrophobic and hydrophilic sections of the copolymer . The SEC chromatograms of the prepared crosslinked polymers and the underlying block copolymers are displayed in Fig. 7. A substantial shift in molecular weight between the block copolymer and the crosslinked micelle, 71700 g mol−1 compared to 900000 g mol−1, provides further evidence for the successful crosslinking of the micelles. The crosslinked sample (PDI = 1.23) shows only one peak with a narrow molecular weight distribution indicating not only the complete crosslinking with the absence of free block copolymer , but also the formation of stable core–shell particles with a narrow size distribution. It should be noted that the SEC system was calibrated with linear polystyrene standards and the number average molecular weight obtained underestimates the actual molecular weight of the micelles.
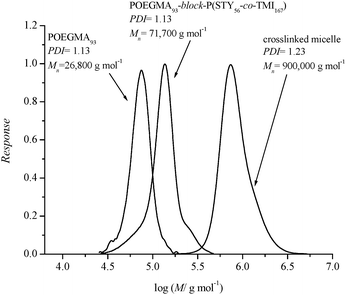 | ||
| Fig. 7 Comparison of SEC chromatograms of POGEMA homopolymer, uncrosslinked block copolymer and crosslinked micelle. | ||
The typical surface active behaviour of an amphiphilic block copolymer disappears with crosslinking of the micelle. Determination of the critical micelle concentration (CMC) is a means by which the stability of a micelle can be evaluated. Amphiphilic molecules display distinct behaviour when interacting with water by arranging themselves at the water–air interface such that the polar section interacts with the water and the non-polar part is held above the surface. The presence of these molecules on the surface disrupts the cohesive energy at the surface and thus lowers the surface tension until the surface is filled and micelle formation occurs. With increasing concentration of the surfactant, the surface tension plateaus at a value which is indicative for the density of the surface layer. POEGMA93-block-P(STY56-co-TMI167) was thoroughly purified by dialysis against acetone for 2 days and subsequently transferred into an aqueous environment. The CMC of uncrosslinked micelle solution was determined to be at 6.2 × 10−4 g L−1 (0.62 mg L−1). For comparison, a block copolymer based on polystyrene with around 107 repeating units and poly(ethylene oxide) with 400 repeating units (Mn = 28000 g mol−1) was measured to have an apparent critical micelle concentration of 1.0 mg L−1.55 The typical behavior observed using amphiphilic molecules disappears when using the crosslinked micelle. The surface tension gradually decreases with increasing concentration indicative of a light enrichment of the crosslinked micelle at the water–air interface. The distinctive drop of the surface tension with the onset of the micelle formation is, however, absent (Fig. 8).
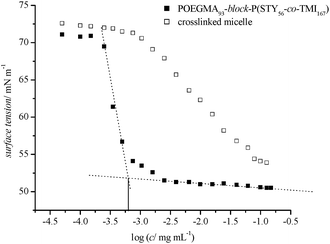 | ||
| Fig. 8 Surface tension vs. concentration of uncrosslinked and crosslinked copolymer POEGMA93-block-P(STY56-co-TMI167). | ||
The formation of crosslinked micelles was evident from the DLS analysis. The particle sizes of uncrosslinked micelles in water is slightly smaller than that of crosslinked micelles (ca. 35 nm for the crosslinked micelle compared to ca. 45 nm for the uncrosslinked micelle) (Fig. 9A and C). Crosslinking seems to cause the contraction of the micelle, which has frequently been observed in earlier studies. DMAc, which is a good solvent for both blocks of the polymer, led to the disassociation of the micelle into unimers (free block copolymers ) indicating the structural instability (Fig. 9B). In contrast, unimers are fully absent when the crosslinked copolymer was dissolved in DMAc revealing the stabilization of micelles by core crosslinking (Fig. 9D).
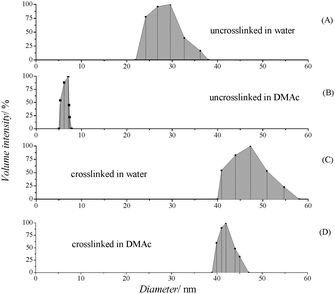 | ||
| Fig. 9 Hydrodynamic diameter analysis viaDLS (concentration of micelles solution of 1 mg mL−1) of copolymer POEGMA93-block-P(STY56-co-TMI167) (A) uncrosslinked in water, (B) uncrosslinked in DMAc, (C) crosslinked in water, (D) crosslinked DMAc. | ||
The formation of micelles was also evidenced by TEM analysis in Fig. 10A and B. The nanoparticles have spherical morphology with low polydispersity. The particle sizes measured by TEM and DLS are in good agreement with the sizes obtained from DLS being slightly larger than the sizes from TEM due to the hydration of the corona.
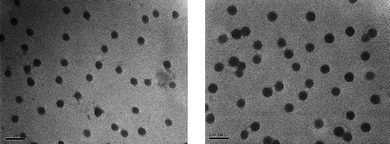 | ||
| Fig. 10 TEM analysis of the micelles (concentration of micelles 1 mg mL−1 in water) of copolymer POEGMA93-block-P(STY56-co-TMI167). Left: uncrosslinked, right: crosslinked, scale bar = 100 nm. | ||
Formation of crosslinked micelles with aldehyde functionalities on the surface and conjugation to GRGDS
In a subsequent step, the acetal group—which was introduced to protect the aldehyde group—was deprotected to yield micelles with a high concentration of aldehyde groups on the surface. This synthesis step needs to be carried as the last step prior to the attachment of peptides. The reasons include the potential side reactions of aldehydes with the diamine crosslinker, the possible interference of aldehydes during the polymerization and the formation of other products such as aldols in alkaline media, which could potentially result in crosslinking between micelles.Arg-Gly-Asp (RGD) peptide recognition sites can be found on numerous adhesive proteins present in the extracellular matrix (ECM) and in blood. They belong to the family of integrins and they represent a fundamental cell-binding domain for natural RGD containing ligands, but also for viruses and bacteria. These foreign objects obtain entry to the cell by binding to the cell surface followed by endocytosis. Attachment of RGD to drug delivery vectors was therefore shown to enhance the cellular adhesion by mimicking the behaviour of pathogens such as bacteria and viruses.56 The Gly-Arg-Gly-Asp-Ser (GRGDS) pentapeptide is related to RGD, with both compounds able to stimulate the same integrin receptor sites.57
The crosslinked micelles bearing aldehyde functionalities were obtained after stirring the micelles in acidic conditions. The micelles were then conjugated to GRGDS through the reaction between the aldehyde group on the micelle with the terminal amino group of the peptide at pH = 7 followed by the reduction of the Schiff base (Scheme 4). The successful conjugation has been confirmed using HPLC monitored at λ = 214 nm. The free GRGDS was eluted at about 8 mins while the crosslinked micelle POEGMA93-block-P(STY56-co-TMI167) had a retention time of around 14 min (Fig. 11). As can be seen from Fig. 10, while the signal of free peptideGRGDS incubated with aldehyde-POEGMA93-block-P(STY56-co-TMI167) almost disappeared after 90 h, the signal of a new peak corresponding to the conjugates increased. It is noted that the free polymer shows a weak signal at λ = 214 nm. This result reveals the high conjugation efficiency.
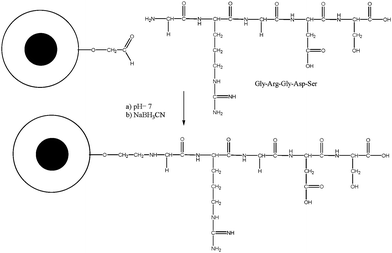 | ||
| Scheme 4 Conjugation of GRGDS onto the surface of the crosslinked micelle via reaction between aldehyde and amino groups followed by the subsequent reduction of the Schiff base. | ||
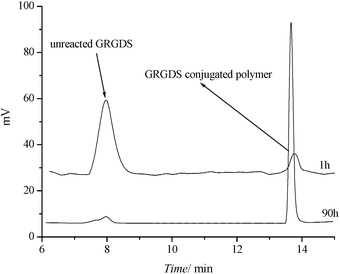 | ||
| Fig. 11 SEC-HPLC analysis of the conjugation reaction mixture of core crosslinked micelles POEGMA93-block-P(STY56-co-TMI167). | ||
In vitro cytotoxicity and cellular uptake
To evaluate the cytotoxicity of the prepared polymeric micelles, a L929 cell line was exposed to uncrosslinked micelles, crosslinked micelles and RGD containing peptide conjugated micelles at a concentration of 1.25 mg mL−1 for 24 h and 48 h. The cytotoxicity was examined using the cell assay of Australian Standards protocol for Biological Evaluation of Medical Devices: Tests for in vitro cytoxicity S/ISO10993.5-2002 (see ESI† ). Compared to the control (null) the presence of all the samples tested did not inhibit cell growth suggesting that the micelles synthesized were non-toxic (Fig. 12).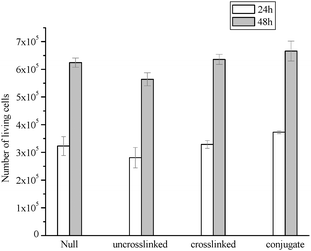 | ||
| Fig. 12 Number of living cells after being exposed to a solution containing uncrosslinked, crosslinked, and GRGDS conjugated micelles POEGMA93-block-P(STY56-co-TMI167) compared to the control experiment without added polymer (null). | ||
To examine whether the prepared micelles were internalized by cells, fluorescein-labeled uncrosslinked, crosslinked, and conjugated crosslinked were synthesized as described in the experimental part. The average diameter of prepared fluorescein-labeled micelles was 40 nm, as determined by DLS. Cells (L929) were exposed to a solution of the different fluorescein-labelled micelles in media using the same procedure and concentration as in the cytotoxicity tests. After treatment for 24 and 48 h, respectively, the media was extracted Therefore, cells and solution were separated and the cell-free solution was analyzed using fluorescence spectroscopy. The decline in fluorescence intensity is expected to be an indirect measure for the amount of fluorescing micelles taken up by cells. The percentage of micelles taken up by the cells was calculated by the difference between the fluorescence intensity from the solution prior to treatment and after being exposed to the cell line for a preset time. A more common technique to determine the uptake of the micelle by cells is the direct observation of the fluorescence intensity of the cells. This technique should complement the results from the solution fluorescence intensity. In this study, the uncrosslinked micelles were found to have the lowest cellular uptake. While the mechanism of the uptake in this particular case is unknown, there is a possibility that the micelle structure is partly depleted during the uptake process. With crosslinking and the formation of highly stable core–shell particle the internalization is enhanced. Since crosslinked micelles are stable and cannot dissociate, they can only be taken up as a whole by endocytosis. As expected, the GRGDS conjugated micelle shows the highest uptake (Fig. 13). With the uptake of the micelles into the cell, the cells start fluorescing allowing the distribution of the micelles in the cells to be effectively monitored (Fig. 14).
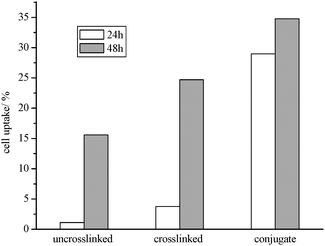 | ||
| Fig. 13 Cell uptake of uncrosslinked, crosslinked, and GRGDS conjugated micelles POEGMA93-block-P(STY56-co-TMI167) determined by the comparison of fluorescence intensity of micelle solution before and after treatment with cells. | ||
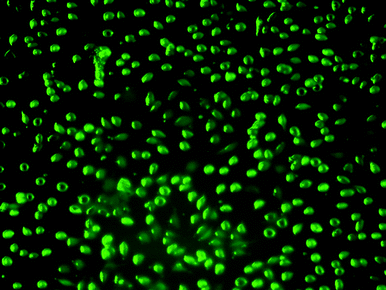 | ||
| Fig. 14 Fluorescence images of L929 cells incubated with micelles POEGMA93-block-P(STY56-co-TMI167) at 37 °C for 48 h. | ||
Conclusion
Novel core crosslinked micelles with aldehyde reactive groups on the surface were prepared in order to generate a drug carrier with structural stability, facilitating cell attachment and showing inherent biocompatibility due to the non-toxicity of the POEGMA shell. The reactive micelles were conjugated to GRGDS in order to achieve an enhanced cellular uptake relative to unfunctionalized or uncrosslinked systems. Cell experiments confirm that both procedures—core-crosslinking and the decoration of the micelle with GRGDSpentapeptides—improve the internalization of micelles into cells.Acknowledgements
MS acknowledges funding from the ARC (Australian Research Council). HD thanks the Australian government for Endeavour Postgraduate Award. The authors would like to thank the Centre for Advanced Macromolecular Design (CAMD) for support.Notes and references
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169 CrossRef CAS.
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295 CrossRef CAS.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169 CrossRef CAS.
- N. S. Cameron, M. K. Corbierre, and A. Eisenberg. 1998 E.W.R. Steacie Award Lecture Asymmetric amphiphilic block copolymers in solution: a morphological wonderland. in 81st Annual Conference of the Canadian-Society-for-Chemistry. 1998. Whistler, Canada Search PubMed.
- L. Zhang, T. L. U. Nguyen, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2007, 8, 2890 CrossRef CAS.
- Z. F. Jia, L. J. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS.
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630 CrossRef CAS.
- J. Q. Liu, H. Y. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899 CrossRef CAS.
- S. Bontha, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2006, 114, 163 CrossRef CAS.
- G. Kwon, S. Suwa, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1994, 29, 17 CrossRef CAS.
- H. Liu, S. Farrell and K. Uhrich, J. Controlled Release, 2000, 68, 167 CrossRef.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249 CrossRef CAS.
- L. B. Luo and A. Eisenberg, Langmuir, 2001, 17, 6804 CrossRef CAS.
- L. B. Luo and A. Eisenberg, J. Am. Chem. Soc., 2001, 123, 1012 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168 CrossRef CAS.
- K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6359 CrossRef CAS.
- Z. F. Jia, X. W. Xu, Q. Fu and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6071 CrossRef CAS.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS.
- K. B. Thurmond II and K. L. Wooley, Shell cross-linked knedels: amphiphilic core–shell nanospheres with unique potential for controlled release application, in Tailored polymeric materials for controlled delivery systems, ed. I. McCulloch and S. W. Shalaby, 1998 Search PubMed.
- M. H. Stenzel, Chem. Commun., 2008, 3486 RSC.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068 RSC.
- Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642 CrossRef CAS.
- K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1397 CrossRef CAS.
- F. Henselwood and G. Liu, Macromolecules, 1997, 30, 488 CrossRef CAS.
- M. Iijima, Y. Nagasaki, T. Okada, M. Kato and K. Kataoka, Macromolecules, 1999, 32, 1140 CrossRef CAS.
- Ji-Heung Kim, K. Emoto, M. Iijima, Y. Nagasaki, T. Aoyagi, T. Okano, Y. Sakurai and K. Kataoka, Polym. Adv. Technol., 1999, 10, 647 CrossRef CAS.
- L. Zhang, J. B. Thomas, P. D. C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123 CrossRef CAS.
- L. Zhang, W. Liu, L. Lin, D. Chen and M. H. Stenzel, Biomacromolecules, 2008, 9, 3321 CrossRef CAS.
- L. Zhang, K. Katapodi, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2177 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, D. B. Thomas, A. J. Convertine, M. S. Donovan and C. L. Mc Cormick, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1724 CrossRef CAS.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064 CrossRef CAS.
- Y. Nagasaki, K. Yasugi, Y. Yamamoto, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 1067 CrossRef CAS.
- Z. Hu, F. Luo, Y. Pan, C. Hou, L. Ren, J. Chen, J. Wang and Y. Zhang, J. Biomed. Mater. Res., Part A, 2008, 85a, 797 CrossRef CAS.
- X. B. Xiong, A. Mahmud, H. Uludag and A. Lavasanifar, Biomacromolecules, 2007, 8, 874 CrossRef CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385 CrossRef CAS.
- H. Lee, E. Lee, D. K. Kim, N. K. Jang, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2006, 128, 7383 CrossRef CAS.
- L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- L. Barner, C. Barner-Kowollik and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1064 CrossRef CAS.
- H. S. Wu, M. H. Chuang and J. W. Hwang, J. Appl. Polym. Sci., 1999, 73, 2763 CrossRef CAS.
- L. Barner, S. Pereira, S. Sandanayake and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 857 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS.
- T. S. C. Pai, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Polymer, 2004, 45, 4383 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923 CrossRef CAS.
- I. Hamley, Block copolymers in solution: Fundamentals and Applications, John Wiley and Sons, Ltd, Chichester, West Sussex, England, 2005 Search PubMed.
- O. Terreau, L. B. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601 CrossRef CAS.
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691 CrossRef CAS.
- H. Ni, Heather A. Nash, James G. Worden and Mark D. Soucek, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1677 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. Wang, Renliang Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033 CrossRef CAS.
- M. Oba, K. Aoyagi, K. Miyata, Y. Matsumoto, K. Itaka, N. Nishiyama, Y. Yarnasaki, H. Koyama and K. Kataoka, Mol. Pharmaceutics, 2008, 5, 1080 CrossRef CAS.
- X. B. Xiong, A. Mahmud, H. Uludag and A. Lavasanifar, Pharm. Res., 2008, 25, 2555 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: NMR of copolymerization, detailed results of styrene/TMIpolymerization, protocol for cell assay . See DOI: 10.1039/b9py00210c |
| This journal is © The Royal Society of Chemistry 2010 |