Probing intracellular oxygen by quenched phosphorescence lifetimes of nanoparticles containing polyacrylamide-embedded [Ru(dpp(SO3Na)2)3]Cl2†‡
Michael P. Coogana, Jonathan B. Courtb, Victoria L. Grayb, Anthony J. Hayesc, Siôn H. Lloydb, Coralie O. Milletb, Simon J. A. Popea and David Lloyd*b
aSchool of Chemistry, Cardiff University, Cardiff, Wales, UK CF10 3AT
bCardiff School of Biosciences, Main Building, Museum Avenue, Cardiff, Wales, UK CF10 3AT. E-mail: lloydd@cf.ac.uk
cConfocal Microscopy Unit, Cardiff School of Biosciences, Life Sciences Building, Cardiff, Wales, UK CF10 3US
First published on 7th December 2009
Abstract
Methods for measuring O2 within living cells that rely on luminescent probes are hampered by several factors: local conditions of hydrophobicity, pH, ionic composition, dielectric constant, and photobleaching by free radical species. Use of a polymer-embedded luminophore should minimize these problems. Here we use a Ru(II) coordination complex embedded within 45 nm hydrodynamic diameter nanoparticles, and demonstrate that both phosphorescence intensity and lifetimes are O2-sensitive, both in aqueous suspensions and intracellularly (e.g. 4.06 versus 1.55 microseconds under anaerobic or aerobic conditions, respectively). Electroporation is necessary for incorporation of the nanoparticles into yeasts: it is more effective with the fission yeast, Schizosaccharomyces pombe, than for the budding yeast, Saccharomyces cerevisiae. However, electroporation was not required for particle uptake into a cultured human cell-line (mammary adenosarcoma MCF-7), although the intracellular distribution of the probe is more general to intracellular compartments when electroporation is employed. These procedures did not compromise vitality of cells over periods of 6 h, as judged by retention of structural characteristics evident in Nomarski interference or confocal microscopy images. Spatial resolution of intracellular structures defined by nanoparticle phosphorescence intensity imaging indicates potential usefulness of the application of lifetime imaging techniques for mapping of intracellular O2 distributions.
Introduction
Energy generation in all aerobic organisms and cells is almost entirely dependent on the delivery of O2 to the sites of its utilization.1 The major sites of O2 consumption occur at the dinuclear redox centre of the predominant terminal oxidase (cytochrome c oxidase) at the inner surface of the inner mitochondrial membrane.2 Rapid O2 depletion during cellular respiration at these sites leads to heterogeneity of its distribution within the organelles: a gradient in concentration between the mitochondrial matrix and the cytosol is also formed across the mitochondrial membranes.3 At the plasma membrane there is a similar discontinuity in O2 concentration between the internal milieu of the cell and the extracellular fluid surrounding it.4 In order to understand the dynamics of respiration in a cell (or unicellular organism) during its changing functional states, the magnitude of these gradients and the consequent heterogeneity of O2 concentration must be measured using a rapid, non-invasive5 (or at best a minimally-invasive) method.6A variety of methods have been employed to measure O2 concentrations in biological systems,7 but those suitable for intracellular monitoring are rather few.8“Oximetric” methods that rely on the paramagnetic properties of O2 have been described,9 but are not of the spatial or temporal resolution essential for detailed studies. More promising are the procedures that employ O2-quenchable luminescent probes, e.g. pyrenebutyric acid fluorescence.10 This approach enabled the mapping of intracellular O2 distributions11 inside cultured cells of mouse liver using quantitative fluorescence microscopy.12
Criteria for the ideal probe include: (a) penetration of the compound into the cell, (b) uniform distribution within the highly structured and compartmentalised cellular ultrastructure, (c) quenching of the luminophore by O2 strictly according to the Stern–Volmer relationship, (d) similarity of quenching constants irrespective of binding to hydrophobic membranes or in aqueous solution, and (e) the presence of probe should be without metabolic effects and (for long-term monitoring) not influence either survival or cell propagation.
Luminescent transition metal complexes are increasingly used as molecular reporters in luminescent probes, designed to be selective for a variety of analytes. Those based around polypyridyl complexes of certain platinum group metals (Ru, Os, Re, Rh and Ir) have many desirable properties for such applications.13 These include:
– Long luminescence lifetimes (hundreds of nanoseconds to tens of microseconds) by comparison with most organic fluorophores (with low nanosecond to picosecond lifetimes);
– Luminescence quantum yields sometimes exceeding 0.5, although values of 0.04 and 0.2 are representative;
– Large Stokes shifts (often >5000 cm−1) eliminating self-quenching processes;
– Excitation wavelength-independent photoefficiencies;
– Intense absorption in the visible spectral range as well as thermal, chemical and photochemical stability and metabolic inertness.13–18
Recently, in addition to their application as in vitro sensors and probes, the application of late transition metal complexes, and especially polypyridyl complexes, in biological imaging has been the focus of a great deal of interest, with systems based on Re,14–16 Ru,17 and Ir18 in particular having been applied in cellular imaging studies. The photophysical attributes discussed above in the context of applications as molecular probes recommend these species as lumophores in imaging applications. Amongst these useful imaging agents, however, Ru2+ complexes19 show many advantages over other transition metal complexes for O2 sensing.20–21
The intense, broad 454 nm visible absorption band of [Ru(bpy)3]2+ and its derivatives is due to a metal-to-ligand charge transfer (MLCT) transition, involving electron transfer from ruthenium-based orbitals to a ligand-centred antibonding orbital (t52gπ*1 configuration).13 The allowed nature of this transition advantageously leads to much higher absorption coefficients than those associated with formally forbidden d–d transitions. The luminescent emission transition from the lowest vibrational level of the triplet MLCT excited state, then occurs via spin–orbit coupling mediated by the metal, which potentiates rapid and efficient intersystem crossing over to the triplet states: since emission then occurs from the triplet state, it is of a formally spin-forbidden phosphorescent nature. The photophysical and photochemical characteristics of these inorganic complexes with favourable properties include (a) a lowest excited state that is a charge transfer, thus having photochemical stability rendering them resistant to photobleaching processes, (b) d–d states well above the emissive states, thereby preventing thermal non-radiative deactivation, (c) high spin–orbit coupling constants associated with large Z values, allowing efficient population of emissive triplet states, and (d) an emitting state not too low in energy, as this would promote the rate of radiationless decay.13“Fine-tuning” of these characteristics has been described by substitution of 2,2′-bipyridine and 1,10-phenanthroline with groups of varying steric and electronic character.22
Most fluorophores are quenched by oxygen where the relationship between the resultant observed lifetime in oxygenated solution and the fundamental unquenched lifetime is proportional to the degree of quenching (Stern–Volmer). Ruthenium polypyridines and in particular those with extended aromatic substitutions on the coordinated diimine, which reduce solvent perturbations, are especially prone to luminescence quenching by dioxygen as there is an efficient, diffusion-controlled collisional interaction between the triplet MLCT excited state of the ruthenium polypyridyl and the triplet ground state of dioxygen, leading to the formation of the ground state of the ruthenium polypyridyl and singlet dioxygen (eqn (1)).21
| (3MLCT)[Ru(bipy)3]2+ + 3O2→ {3MLCT[Ru(bipy)3]2+⋯3O2} → (A1)[Ru(bipy)3]2+ + 1O2 | (1) |
Thus, the excited-state lifetime of a ruthenium polypyridyl species is exquisitely sensitive to oxygen concentration due to the efficiency associated with a diffusion-controlled quenching process leading to a very high rate of deactivation of the excited state species, and a concomitant drop in the average luminescence lifetime. Such behaviour can be exploited in FLIM of human bronchial epithelia cells where variations in the emission lifetime of embedded [Ru(bpy)3]2+ units were used to deduce the intracellular distribution of oxygen.23 Optical nanosensors (20–200 nm diameter) have been delivered into cells by liposomal delivery or gene-gun bombardment, and also by pico-injection into single cells:24–26 when incorporating a Ru(II) diphenylphenanthroline complex,25,26 these nanoparticles (photonic explorers for bioanalysis with biologically localised embedding, PEBBLES) successfully reported O2, and have been used for imaging O2 inside cells.26 Other O2-dependent phosphorescent quenching has also been utilised for biological monitoring. Examples include porphyrins (Zn protoporphyrins,27 Pd meso-tetrakis(4-carboxyphenyl)porphyrin, and tetrabenzoporphyrins.28 Endogenous protoporphyrin IX in hepatocyte mitochondria acts as an O2 sensor by virtue of its quenchable delayed fluorescence.29
Immobilization20,30 or encapsulation31,32 in polymeric materials provide possibilities for minimilization of toxicity and invasive perturbation of biological systems. Major disadvantages of Ru(II) coordination complexes used in aqueous solution, but without prior encapsulation include: minimal uptake into cells; toxic effects on metabolism, survival and proliferation of cells and organisms; and non-specific binding to proteins and membranes. In the present work we show that further extension of these fundamental principles permit the fabrication of nanoscale O2 sensor particles that when electroporated into yeast or animal cells report local O2 concentrations and rapidly sense changes during aerobic-anaerobic shifts in the extracellular environment and avoid at least some of the problems associated with non-encapsulated Ru coordination complex in biological imaging applications.
Experimental
Maintenance and growth of cells and organisms yeasts
Organisms employed were brewer's yeast Saccharomyces cerevisiae IFO 0233 and the fission yeast Schizosaccharomyces pombe 972h-. Both were maintained and grown on Petri dishes containing Sabouraud Maltose Agar (Difco). Organisms were transferred to unbaffled Ehrlenmayer flasks (50 ml containing 10 ml YEPD liquid medium (0.3% yeast extract, 1% peptone and 1% glucose) and grown at 30 °C for two days on a rotary shaker at 150 rpm. Before electroporation, repeated centrifugation for 3 min at 2000 rpm (3000 g min) and resuspension in 1.0 M sorbitol solution removed all nutrients.Human cells
A cultured cell-line (human mammary adenocarcinoma, MCF-7) from the European Collection of Cell Cultures, Porton Down, Wiltshire, UK, was maintained in Eagles Minimal Essential Medium supplemented with 10% foetal bovine serum, penicillin and streptomycin. Detachment from the plastic culture flasks was carried out using trypsin/EDTA, and was followed by resuspension in growth medium.33Nanoparticle preparation
Reagents (from Sigma-Aldrich) were RuCl3 (57 mg, 0.0276 mmole) and 4,7-diphenyl-1,10-phenanthroline disulfonic acid disodium salt (518 mg, 0.0965 mmol) in 20 mL distilled water refluxed with stirring for two days. After filtration and rotary evaporation, the product, [Ru(dpp(SO3Na)2)3]Cl2·6H2O, was passed down a Sephadex G25 size exclusion column and eluted with water. The first brown and purple fractions were discarded and the red fraction dried.Polyacrylamide-coated nanoparticles (mean hydrodynamic diameter 45 nm) were prepared by encapsulation of the Ru complex using radical polymerization in inverse microemulsion.31,32
Polymer particles containing the Ru complex were passed through a suction filtration apparatus (Millipore Corporation, Bedford, MA) with a 0.025 μm nitrocellulose filter membrane and rinsed with 100 cm3 ethanol. Hydrodynamic particle diameter (45 nm) was determined by right-angle light scatter of 632.8 nm laser light (HeNe) using a BI-200SM Goniometer (Brookhaven Inst. NYC). Nanoparticles were stored at −18° C. Resuspension (18 mg mL−1) was in 1.0 M sorbitol and dispersion was by treatment with 20 kHz ultrasound for 30 s at 4 °C at an amplitude providing maximum cavitation intensity.
Electroporation of nanoparticles into yeasts or human cells
The Biorad Labs Gene Pulser Transfection Apparatus (Bio-Rad Laboratories, CA) was fitted with a Capacitor Extender set at 450 V for yeasts33 and 320 V for human cells (capacitance 960 μF, resistance 200 Ω). Electroporation cells were of 400 μl working volume; 3 high voltage pulses were employed.Confocal laser microscopy
After washing electroporated organisms or cells 3 times in their respective suspending buffers and harvesting (3 min at 1000 g for yeasts, 3 min 800 g for human cells) they were mounted on slides and viewed using a Leica TCS SP2 AOBS confocal scanning laser microscope using a 40× objective with 2.85× zoom, with excitation at 514 nm and detection at 520–600 nm. Where effects of changes in O2 concentrations were studied, the gas-phase composition flowing over a hanging-drop suspension was controlled (in a specially constructed flow cell)31 using mixtures of research grade N2 and air.34,35Phosphorescence lifetime measurements
Room temperature steady state emission spectra were obtained using aerated solutions and a Jobin-Yvon-Horiba Fluorolog spectrometer fitted with a JY TBX picosecond photodetection module. Aerated solution samples for luminescence lifetime decays were irradiated using a pulsed (15 Hz) Continuum Minilite Nd:YAG configured for either 355 or 532 nm output (low energy setting) and emission detected at 650 nm using a Hamamatsu R5509-73 detector (cooled to −80 °C using a C9940 housing). Data sets were obtained using the Jobin-Yvon-Horiba FluoroHub single photon counting module in multichannel scaler (MSC) mode, and lifetimes determined using the provided decay analysis software package, v6.1. Lifetime uncertainties are typically within 10%. De-oxygenated samples were obtained by gentle, cycled purging and evacuation of the degassing cuvette, but not freeze-pump-thaw due to the sensitivity of the biological samples.Results and discussion
Quenching of the triplet excited-state lifetime of the Ru complex within the acrylamide-based nanoparticles is strongly determined by collision with O2 molecules. Thus it is only those O2 molecules within the diameter/boundaries of the nanoparticles that are sensed and it is only local O2 levels that are reported.24–26 Consequently, increased O2 concentrations give increased rates of phosphorescence quenching, giving correspondingly decreased total phosphorescence intensities and shorter phosphorescence lifetimes.19 Both phosphorescence intensities and life-times are highly sensitive to solvent effects and/or binding to proteins/membranes due to restriction of vibrational freedom and consequently diminished quantum yields. However, embedding of the lumophore minimises its interactions with solvent and cellular constituents.24–26Although confocal images of mammalian macrophages containing Ru complex-containing osmosil nanoparticles have been published previously,25,26 we here demonstrate the applicability of polyacrylamide embedded O2 sensors to yeasts and human breast cancer cells. These organisms and cells do not show any signs of decreased vitality and, for the most part, retain viability after incorporation of the Ru(II) nanosensors.
When baker's yeast, the budding S. cerevisiae, was electroporated in the presence of the nanoparticles, only a few of the organisms showed extensive labelling with the luminophore when examined by confocal microscopy, as evident by comparison with the Nomarski differential interference contrast image of the identical observation field (Fig. 1).
 | ||
| Fig. 1 Images of Saccharomyces cerevisiae. Left: Luminescence. Right: Nomarski differential interference contrast. | ||
The basis for the heterogeneity of uptake was not investigated, but it seems that the cell walls of most of these organisms provide barriers to penetration. Where the nanoparticles had entered the organisms, luminescence labelling was intense and uniform throughout the cytoplasm. Nuclei were less intensely luminescent and cell walls were not visible.
Quite a different result was obtained after an identical electroporation treatment of the fission yeast Schiz. pombe, where samples were taken from a growing culture (Fig. 2). In this case almost every organism became labelled irrespective of the cell division cycle phase: hence short and longer rod-shaped stages showed intense luminescence, including those with transverse septa about to undergo cell division by fission. (Apparently spherical or elliptical profiles are a consequence of optical sectioning.) When non-growing Schiz. pombe were used, i.e. those taken from cultures where nutrients had become limiting to growth, electroporetic uptake of nanoparticles did not occur under the conditions employed here.
 | ||
| Fig. 2 Images of Schizosaccharomyces pombe. Left: Luminescence. Right: Nomarski differential interference contrast. | ||
Highly uniform uptake was observed throughout populations of human MCF-7 cells grown in culture; in this case electroporation was not necessary for uptake. Exposure for 15 min to nanoparticles produced cells which contain endosomal vacuoles that accumulated nanoparticles as dense clusters (Fig. 3a). Electroporation yields a much more uniform and diffusely-labelled luminophore distribution (Fig. 3b). Here it appears that, in addition to aggregated particles, a more diffuse background of cytoplasmic luminescence occurred.
 | ||
| Fig. 3 Images of MCF-7 Cells. A, Non-electroporated; B, electroporated. Left, Luminescence. Right, Nomarski differential interference contrast. | ||
Luminescence lifetime decays were obtained using solutions of nanoparticles and suspensions of cells (Table 1). Firstly, the free Ru-embedded acrylamide nanoparticles were photophysically assessed in aerated 1.0 M sorbitol. The luminescence properties were typical of triplet metal-to-ligand charge transfer (3MLCT) emitting species of the generic related type [Ru(phen)3]2+ with a broad structureless band observed at ca. 608 nm following excitation at 450 nm (i.e. selective irradiation of the 1MLCT transition). The luminescence lifetime of the 608 nm emission (using either 355 or 532 nm excitation) was satisfactorily fitted to a single exponential (τ = 1.81; 3.88 μs under a dinitrogen atmosphere), confirming the 3MLCT sensitivity to quenching via oxygen. When compared to free [Ru(dpp(SO3Na)2)3]Cl2 in water (lifetimes of 0.93 and 3.7 μs for aerated and argon atmospheres, respectively),36 these measurements suggest that the acrylamide shell enhances the photophysical properties of the embedded Ru(II) luminophores by extending the luminescence lifetime, most siginificantly in the case of an oxygenated environment.
Luminescence lifetime measurements were then conducted on suspensions of electroporated cells following different treatment regimes with an air atmosphere versus dinitrogen. Both lifetime profiles of the suspended cells fitted well enough to single exponential decays (see ESI‡), as determined by reduced χ2, to allow comparison, in air, of the electroporated cells with the nanoparticles, which revealed that the emission lifetime of the probe was dramatically lengthened reflecting the oxygen-depleted intracellular environment, due to respiratory activity. In non-electroporated control cells (Fig. 3A), the intensity of emission of the nanoparticles (presumably contained in endosomes) is comparable to that of those outside the cells in aqueous suspension at near air-saturated conditions. The intracellular probe was also found to be sensitive to changes in external atmospheric levels of oxygen; long-term depletion resulted in a luminescence lifetime comparable to that of the free nanoparticles in an oxygen-free atmosphere (ca. 4 μs). Interestingly, restoration of the aerobic atmosphere led to a dramatic shortening of the lifetime (1.86 μs) before returning to ca. 2.9 μs after approximately 5 min. This is difficult to rationalise as a purely chemical process, as the effect of increased oxygen concentration would be essentially instantaneous, and must reflect a change in the biochemistry of the cell compartment containing the nanoparticles. Possibly the initial shortening is indicative of rapid delivery of oxygen to the nanoparticle rich domains, which have shut down respiration due to the low oxygen concentration, leading to hyperoxic conditions and a short lifetime, followed by slower restoration of oxygen-consuming processes which restore the equilibrium values as oxygen concentration reverts to typical values.
Fig. 4 shows a series of images of MCF-7 cells showing changes of luminescence intensities at 20 s intervals, as obtained while changing the gaseous phase within the observation chamber of the confocal microscope. Initially the cells immersed in a thin layer of 1.0 M sorbitol were dinitrogen-equilibrated (frames at 220 and 240 s). Then the mobile gas phase was switched to air, and by 260 s emission intensities in most of the cells were greatly diminished. Restoration of dinitrogen (just after the 280 s scan) reversed these changes.
 | ||
| Fig. 4 Time series of images of MCF-7 cells labelled with nanoprobes and equilibrated in an atmosphere of dinitrogen (220, 240 s) then air (until 280 s) before switching back again to anaerobic conditions. | ||
Luminescence of selected areas within four cells (Fig. 5A) is plotted separately as time courses (Fig. 5B), and these indicate overall similarities between individuals, except that two cells showed an overshoot of emission intensity before re-attaining steady state anaerobic levels.
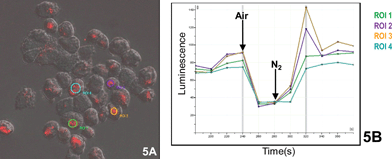 | ||
| Fig. 5 Luminescence of selected areas of anaerobic individual cells during an aerobic pulse. | ||
These data indicate the feasibility of using a Ru coordination complex embedded in polyacrylamide as an O2 nanoprobe for biological imaging. Advantages of this approach include minimisation of direct exposure of the sensing molecules to perturbative physical and chemical intracellular local environments or leakage,37 without restriction of access by gases. Use of fluorescence lifetime rather than intensity imaging promises further improved sensitivity and removal of ambiguities of interpretation due to heterogeneity of local conditions.38,39 Lifetime imaging of longer-lived emission processes is not straightforward with our commercially available laser scanning confocal microscope, but a more recent model with fluorescence lifetime imaging software also allows acquisition of images with microsecond emission lifetimes.40 Alternatively, a pinhole shifting method for collecting long-lifetime phosphorescence has recently been described that can be used with more generally available instrumentation, and could be used for the application described here.41
It becomes increasingly important that O2 distributions inside living cells be determined as a function of their respiratory activities and of external O2 partial pressures.42 This is especially so as the pivotal roles of O2˙−, HO˙, H2O2 and singlet oxygen in intracellular redox states,43 signalling pathways,44 mitochondrial functions,45 and oxidative damage46 become ever more evident. An intracellular O2 map has in fact now become “The Holy Grail of Cellular Physiology”.47
Acknowledgements
Thanks are due to Drs Allan Poulsen and Lars Folke Olsen for providing expert advice and Ru-complex-containing nanoparticles (preparation by Ms Anita Lunding) from the University of Southern Denmark, Odense. Mr Marc Isaacs gave expert advice and helped with confocal microscopy.References
- P. Rich, Nature, 2003, 421, 583 CrossRef CAS.
- B. Chance and G. R. Williams, J. Biol. Chem., 1955, 217, 429–438 CAS.
- D. F. Wilson, M. Erecinska, C. Drown and I. A. Silver, Arch. Biochem. Biophys., 1979, 195, 484–493.
- D. P. Jones and H. S. Mason, J. Biol. Chem., 1978, 253, 4874–4880 CAS.
- D. Lloyd, H. Mellor and J. L. Williams, Biochem. J., 1983, 214, 47–51 CAS.
- E. R. Gnaigner, G. Steinlechner-Maran, T. Mendez, T. Eberl and R. Margreiter, J. Bioenerg. Biomembr., 1995, 27, 583–596 CrossRef.
- D. Lloyd, in Bioinstrumentation Research, Developments and Applications, ed. D. L. Wise, Butterworths, Boston, MA, 1990, pp. 301–316 Search PubMed.
- R. Springett and H. M. Swartz, Antioxid. Redox Signaling, 2007, 9, 1295–1301 Search PubMed.
- H. M. Swartz and R. B. Clarkson, Phys. Med. Biol., 1998, 43, 1957–1975 CrossRef CAS.
- (a) J. Knopp and G. Weber, J. Biol. Chem., 1967, 242, 1353–1359 CAS; (b) W. M. Vaughan and G. Weber, Biochemistry, 1970, 9, 464–473 CrossRef CAS.
- J. A. Knopp and I. S. Longmuir, Biochim. Biophys. Acta, Gen. Subj., 1972, 279, 393–397 Search PubMed.
- D. H. Benson, J. A. Knopp and I. S. Longmuir, Biochim. Biophys. Acta, Bioenerg., 1980, 591, 187–197 CrossRef CAS.
- B. A. DeGraff and J. N. Demas, in Reviews in Fluorescence, ed. C. Geddes and J. R. Lakowicz, Springer Science, New York, vol. 2, 2005, pp. 125–151 Search PubMed.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066–3068 RSC.
- A. J. Amoroso, R. J. Arthur, M. P. Coogan, J. B. Court, V. Fernández-Moreira, A. J. Hayes, D. Lloyd, C. Millet and S. J. A. Pope, New J. Chem., 2008, 32, 1097–1102 RSC.
- K. K. Lo, M. Louie, K. Sze and J. Lau, Inorg. Chem., 2008, 47, 602–611 CrossRef CAS.
- K. K. Lo, T. K.-M. Lee, J. S.-Y. Lo, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef CAS.
- Mengxiao Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou and H. Yang, Chem. Commun., 2008, 2115–2117 RSC.
- R. J. Watts and G. A. Crosby, J. Am. Chem. Soc., 1971, 93, 3184–3188 CrossRef.
- J. R. Bacon and J. N. Demas, Anal. Chem., 1987, 59, 2780–2785 CrossRef CAS.
- G. Orellana and D. Garcia-Fresnadillo, in Optical Sensors: Industrial, Environmental and Diagnostic Applications, Springer Science, Berlin and Heidelberg, vol. 1, 2004, pp. 309–357 Search PubMed.
- J. M. Vanderkooi, G. Maniara, T. J. Green and D. F. Wilson, J. Biol. Chem., 1987, 262, 3476–3482.
- W Zhong, P. Urayama and M. A. Mycek, J. Phys. D: Appl. Phys., 2003, 36, 1689–1695 CrossRef CAS.
- H. A. Clark, M. Hoyer, M. A. Philbert and R. Kopelman, Anal. Chem., 1999, 71, 4831–4836 CrossRef CAS.
- Y.-E. Lee-Koo, Y. Cao, R. Kopelman, S. M. Koo, M. G. Brasuel and M. A. Philbert, Anal. Chem., 2004, 76, 2498–2505 CrossRef CAS.
- S. M. Buck, Y.-E. L. Koo, E. Park, H. Xu, M. A. Philbert, M. A. Brasuel and R. Kopelman, Curr. Opin. Chem. Biol., 2004, 8, 540–546 CrossRef CAS.
- M. Vanderkooi, W. W. Wright and M. Erecinska, Biochemistry, 1990, 29, 5332–5338 CrossRef.
- S. A. Vinogradov, L. W. I. W. T. Jenkins, S. M. Evans, C. Koch and D. F. Wilson, Biophys. J., 1996, 70, 1609–1617 CrossRef CAS.
- E. G. Mik, T. Johannes, C. Zuurbier, A. Heinen, J. H. Howben-Weerts, G. M. Balestra, J. Stap, J. F. Beek and C. Ince, Biophys. J., 2008, 95, 3977–3990 CrossRef CAS.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 337–342 CrossRef CAS.
- H. D. Soule, J. Vazquez, A. Long, S. Albert and M. Brennan, J. Natl. Cancer Inst., 1973, 51, 1409–1416 CAS.
- A. K. Poulsen, L. Arleth, K. Almdal and A. M. Scharff-Poulsen, J. Colloid Interface Sci., 2007, 306, 143–153 CrossRef CAS.
- A. K. Poulsen, A. M. Scharff-Poulsen and L. F. Olsen, Anal. Biochem., 2007, 366, 29–36 CrossRef CAS.
- D. Lloyd, Adv. Appl. Microbiol., 2002, 51, 155–183 Search PubMed.
- H. Degn and J. S. Lundsgaard, J. Biochem. Biophys. Methods, 1980, 3, 233–242 CrossRef CAS.
- F. N. Castellano and J. R. Lakowicz, Photochem. Photobiol., 1998, 67, 179–183 CrossRef CAS.
- M. Bradley, L. Alexander, K. Duncan, M. Cennaoui, A. C. Jones and R. M. Sanchez-Martin, Bioorg. Med. Chem. Lett., 2008, 18, 313–317 CrossRef CAS.
- D. M. Owen, P. M. Lanigan, C. Dunsby, I. Munro, D. Grant, M. A. A. Neil, P. M. W. French and A. I. Magee, Biophys. J., 2006, 90, L80–82 CrossRef CAS.
- J. A. Levitt, D. R. Matthews, S. M. Ameer-Beg and K. Suhling, Curr. Opin. Biotechnol., 2009, 20, 28–36 CrossRef CAS.
- F. Manjon, D. Garcia-Fresnadillo and G. Orellana, Photochem. Photobiol. Sci., 2009, 8, 926–932 RSC.
- V. K. Ramshesh and J. J. Lemasters, J. Biomed. Opt., 2008, 13(6), 64001 CrossRef.
- D. Lloyd, K. L. Thomas, J. Tamman and A. J. Williams, J. Microbiol. Methods, 2002, 48, 289–302 CrossRef CAS.
- D. Lloyd and D. B. Murray, BioEssays, 2007, 29, 465–473 CrossRef CAS.
- M. A. Aon, M. R. Roussel, S. Cortassa, B. O'Rourke, D. B. Murray, M. Beckmann and D. Lloyd, PLoS One, 2008, 3, e3624 CrossRef.
- K. M. Lemar, M. A. Aon, S. Cortassa, B. O'Rourke, C. T. Muller and D. Lloyd, Yeast, 2007, 24, 695–706 CrossRef CAS.
- B. Chance, Methods Enzymol., 2004, 385, 361–370 CAS.
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS.
Footnotes |
| † To the memory of Gregorio Weber (1916–1997), who made seminal advances in fluorescence techniques and taught one of us (DL) at the Biochemistry Department, University of Sheffield, 1958–1961. See http://www.cf.ac.uk/biosi/staffinfo/lloyd/weber/index.html |
| ‡ Electronic supplementary information (ESI) available: Data fits for lifetime decay measurements. See DOI: 10.1039/b9pp00071b |
| This journal is © The Royal Society of Chemistry and Owner Societies 2010 |