Mechanism of aerobic visible light formic acid oxidation catalyzed by poly(tri-s-triazine) modified titania†
Received 8th July 2009, Accepted 15th September 2009
First published on 12th November 2009
Abstract
Visible light aerial oxidation of formic acid catalyzed by N/C-modified titania (TiO2-N,C) is investigated by wavelength-dependent photocatalytic and photoelectrochemical experiments in the presence of oxygen, tetranitromethane, and methylviologen as electron acceptors. The title reaction is shown to proceed both through oxidative and reductive primary processes. Contrary to the urea-derived (TiO2-N,C), so-called “N-doped” titania (TiO2-N) as prepared from ammonia is inactive. In accord with photocurrent action spectra of corresponding powder electrodes, this different activity of the two photocatalysts is traced back to the different chemical nature of the reactive holes localized at the modifier. Hole stabilization by delocalization within an extended poly(tri-s-triazine) network of TiO2-N,C is proposed to render recombination with conduction band electrons less probable than in TiO2-N.
Introduction
The development of novel functional materials capable of solar-driven chemical transformations or production of electricity has attracted significant interest motivated by the need to develop technologies based on a clean and sustainable energy supply.1,2 In this context, particularly semiconductor photocatalysis is one of the promising approaches since semiconductors, due to their unique electronic and optical properties, enable efficient photon-induced generation and separation of charges that can be utilized for selective oxidation and reduction reactions at the semiconductor's surface. Due to its low cost, non-toxicity and excellent chemical stability, one of the most commonly employed photocatalysts is titanium dioxide. However, utilization of TiO2 is hampered by the fact that, due to the large bandgap of 3.2 eV (∼390 nm), it can make use of only a very small UV part (about 3%) of solar radiation. Therefore strong efforts are presently being made to shift its photocatalytic activity to the visible spectral region. The fundamental issue at stake here is that all viable strategies for large-scale applications should avoid expensive manufacturing procedures or using rare noble metals like platinum, iridium, palladium, etc. Accordingly, one of the most promising methods seems to be doping and/or surface modification of TiO2 with nonmetals, such as carbon, nitrogen and sulfur.3–13Recently we reported14 that calcining a mixture of urea and TiO2 at 400 °C produces poly(amino-tri-s-triazine) derivatives covalently attached to the semiconductor surface (Scheme 1). The resulting visible light-active photocatalysts TiO2-N,C consist of a titania core covered by a polytriazine shell symbolized as “C,N”. These condensed heteroaromatics were found to be responsible for the visible light activity, contrary to many other proposals made for so-called urea-derived “N-TiO2” in the literature.15–30
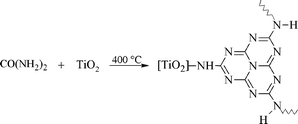 |
| | Scheme 1 Modification of titania with urea affording the core-shell particle TiO2-N,C. For simplicity only one tri-s-triazine unit (melem) is depicted.14 | |
TiO2-N,C exhibits a weak absorption shoulder in the visible region at 400–500 nm and photocatalyzes the aerial oxidation of formic acid to CO2 and H2O (λ > 455 nm). The quasi-Fermi level for electrons, which practically merges with the conduction band edge, was −0.48 V compared to −0.56 V for unmodified TiO2 (pH 7).
Different from TiO2-N,C, the catalyst TiO2-N prepared from ammonia by treating anatase powder in the NH3(67%)/Ar atmosphere at 600 °C for 3 h9 was found to be inactive in decomposition of formic acid under visible light irradiation.25,31 The nature of the nitrogen species responsible for visible light activity of TiO2-N is still a matter of discussion. Proposals range from N3−,9,32 NO, NO2, and NO2− to NHx-like species32,33 substituting oxide ions or occupying interstitial lattice positions. TiO2-N exhibits a similar absorption spectrum as the urea-derived catalyst. The distinct difference of TiO2-N,C and TiO2-N in the photocatalytic oxidation of formic acid very likely can be attributed to an efficient electron–hole recombination in the latter material although the location of relevant energy states are very similar (Fig. 1). We proposed that this might originate from the different electronic nature of the nitrogen-centered intra-bandgap states present in the two materials. Whereas in TiO2-N the nitrogen species do not allow for hole stabilization through charge delocalization, this should be possible in TiO2-N,C.34 This effect is expected to increase the hole lifetime and favor interfacial electron exchange over recombination. The assumption of an energy band-like structure of the intra-bandgap states of TiO2-N,C, instead of a manifold of discrete levels as drawn for TiO2-N in Fig. 1, is supported by the fact that pristine poly(amino-tri-s-triazine) derivatives themselves exhibit semiconductor photocatalytic properties.34,35
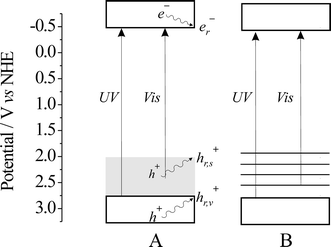 |
| | Fig. 1 Energy level schemes of TiO2-N,C (A) and TiO2-N (B) at pH = 7. The reactive, surface-trapped holes generated upon UV and Vis irradiation are denoted as hr,v+ and hr,s+, respectively, and the reactive, surface-trapped electron as er−.34 | |
Although rarely found in literature, detailed studies of the mechanism of the photocatalytic action are crucially important. Such mechanistic knowledge is indispensable for development of further photoactive materials with optimized properties. Accordingly, herein we report in detail our investigations of the mechanism of formic acid photocatalytic oxidation by TiO2-N,C. We are particularly interested in elucidation of the role of visible light generated holes in TiO2-N,C because the nature and fate of hr,s+ may have decisive bearings on the general photocatalytic activity. Similarly, the efficiency of dye-sensitized titania solar cells is also known to be dependent on the electronic structure of the visible-light generated dye radical cation.36 In this paper the mechanism of aerial formic acid oxidation is investigated by conducting a series of photochemical and photoelectrochemical experiments in which the electron scavenger oxygen is replaced by tetranitromethane or methylviologen. By combining these techniques we arrive at a conclusive mechanism of formic acid degradation catalyzed by TiO2-N,C.
Before communicating our results, the mechanism of the photocatalytic oxidation of formic acid over unmodified titanium dioxide irradiated with UV light is briefly discussed. It is generally agreed to proceed by electron transfer to photogenerated valence band holes (eqn (1)) and/or reaction with OH˙ radicals (eqn (2)) produced through (i) water or hydroxyl group oxidation by valence band holes or (ii) dioxygen reduction (eqn (3)) via intermediate O2−, HO2 and H2O2.37–40
| | | HCOO− + hr,v+→ COO−˙ + H+ | (1) |
| | | HCOO− + OH˙→ COO−˙ + H2O | (2) |
Importantly, it is well known that the one-electron
oxidation product of
formic acid can inject a second electron into the conduction band of a semiconductor, which can be directly observed by enhancement of photocurrent and has become known as the so-called ‘photocurrent multiplication (photocurrent doubling) effect’.
41–42,43Experimental
The catalysts TiO2-N/C14 and TiO2-N9 were prepared by calcining TiO2 in the presence of urea and ammonia, respectively. In both cases Hombikat UV-100 (100% anatase, Sachtleben Chemie) was used as titania source. Methylviologen (1,1′-dimethyl-4,4′-bipyridinium dichloride; Acros Organics) and tetranitromethane (Aldrich) were used as received.Photomineralization experiments were carried out in a cylindrical 20 mL cuvette attached to an optical train. The reaction mixture was stirred magnetically. Irradiation was performed with an Osram XBO 150 W xenon arc lamp installed in a light-condensing lamp housing (PTI, A1010S). A water filter and an appropriate cut-off filter were placed in front of the cuvette. 20 mL of a 1 g L−1 powder suspension in 10−3 mol L−1 formic acid was sonicated for 15 min prior to illumination. When C(NO2)4 (10−2 mol L−1) was used as an electron scavenger, argon was bubbled for 40 min prior and during irradiation. Withdrawn samples were filtered through a syringe filter and subjected to ion chromatography analysis (Dionex DX120, Ion Pac 14 column, conductivity detector; NaHCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaCO3 = 0.001:0.0035 mol L−1 as eluent); no oxalate was detectable. Formation of C(NO2)3− anion was monitored by electronic spectrum (Varian Cary 50 UV-Vis spectrometer).
:NaCO3 = 0.001:0.0035 mol L−1 as eluent); no oxalate was detectable. Formation of C(NO2)3− anion was monitored by electronic spectrum (Varian Cary 50 UV-Vis spectrometer).
Quasi-Fermi potentials of electrons (nEF*) were measured according to the literature44 using methylviologen dichloride ((MV)Cl2, E°MV2+/+˙ = −0.445 V vs. NHE) as a pH-independent redox system. In a typical experiment 50 mg of catalyst and 10 mg of methylviologen dichloride were suspended and sonicated for 15 min prior to illumination in a 100 mL two-necked flask in 50 mL of 0.1 mol L−1 KNO3. Only a water filter was placed in front of the flask for UV-Vis tests, whereas a 420 nm cut-off filter was added for the Vis measurements. The tests were performed in the presence or absence of formic acid (10−3 mol L−1). A platinum flag and Ag/AgCl served as working and reference electrodes and a pH meter for recording the proton concentration. Initially the pH of the suspension was adjusted using HNO3 to pH ca. 2.5 before measurement. Stable photovoltages were recorded about 20 min after changing the pH value. The suspension was magnetically stirred and purged with nitrogen gas about 30 min prior and throughout the experiment. Titration was performed using NaOH (0.1, 0.01 and 0.001 mol L−1), also purged with nitrogen gas. The light source was the same as used in photodegradation experiments.
For the photostability test, 20 mL of a 1 g L−1TiO2-N,C suspension in 4.6 × 10−4 mol L−1 formic acid was sonicated for 15 min prior to illumination in a centrifuge glass. After centrifugation, 2 mL of liquid was withdrawn, filtered through a syringe filter and subjected to ion chromatography analysis (see above). Subsequently, the suspension was sonicated for 1 min and irradiated with visible light (λ≥ 420 nm). After 4 h of irradiation the suspension was again centrifuged, 2 mL of liquid were withdrawn, filtered through a syringe filter and analyzed by ion chromatography. The powder suspension was filled up with 4 mL of concentrated formic acid to achieve the initial concentration and volume of suspension. This procedure was repeated three times. The light source was the same as used in photomineralization.
For photocurrent measurements, electrodes consisting of a material film deposited on FTO–glass were prepared. The conducting FTO–glass substrate (Solaronix, sheet resistance of ∼10 Ω per square) was first cut into 2.5 × 1.5 cm pieces and then subsequently degreased by sonicating in acetone and boiling NaOH (0.1 mol L−1), rinsed with demineralized water, and blown dry in a nitrogen stream. A suspension of 200 mg of a catalyst in 1 mL of ethanol was sonicated for 20 min and then deposited onto the FTO glass by doctor blading using scotch tape as frame and spacer. The electrodes were then dried in air, covered with a glass plate, and pressed for 3 min at a pressure of 200 kg cm−2 using an IR pressing tool (Paul Weber, Stuttgart, Germany). Such a procedure yields an opaque layer of the investigated material having an excellent mechanical stability. Photocurrent experiments were performed with a tunable monochromatic light source provided with a 1000 W Xenon lamp and a universal grating monochromator Multimode 4 (AMKO, Germany) with a bandwidth of 10 nm. The electrochemical setup consisted of a BAS Epsilon Electrochemistry potentiostat and a three-electrode cell using a platinum counter electrode and a Ag/AgCl (3 mol L−1 KCl) reference electrode. During photoelectrochemical measurements the electrodes were pressed against an O-ring of an electrochemical cell leaving a working area of 0.636 cm2. The photocurrent experiments were carried out in a LiClO4 (0.1 mol L−1) containing formic acid (10−3 mol L−1) or potasium iodide (0.1 mol L−1) as hole scavengers. Nitrogen was passed through the electrolyte prior to the experiment, whereas it was supplied only to the gas phase above the electrolyte during the experiment. The wavelength dependence of photocurrent was measured at a constant potential of 0.5 V vs. Ag/AgCl. The electrodes were irradiated from the backside (through the FTO glass) with light and dark phases of 5 and 10 s, respectively. The value of photocurrent density was taken as the difference between current density under irradiation and in the dark. The incident photon-to-current efficiency (IPCE; the number of electrons generated in the external circuit divided by the number of incident photons) for each wavelength was calculated according to equation IPCE (%) = (iphhc)/(λPq)×100, where iph is the photocurrent density, h is Planck's constant, c velocity of light, P the light power density, λ is the irradiation wavelength, and q is the elementary charge. The spectral dependence of lamp power density was measured by the optical power meter Oriel 70260 (Oriel, Stratford, USA).
Results and discussion
To address the problem of photostability and catalytic function of TiO2-N,C in the mineralization of formic acid, one unique sample was subjected to a twelve hour irradiation experiment (λ≥ 420 nm). After every fourth hour the irradiation was interrupted for formic acid measurement followed by addition of a new portion of formic acid solution (Fig. 2, see Experimental). Complete photomineralization occurred even after the third irradiation cycle evidencing the photocatalytic function of TiO2-N,C. This finding corroborates also the recently reported photostability of polymeric melon modified with Pt for photocatalytic (λ≥ 420 nm) H2 production in the presence of a reducing agent.35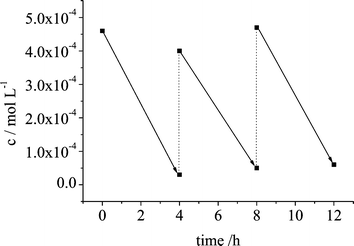 |
| | Fig. 2 Decrease of concentration upon addition of formic acid after every fourth hour (dashed lines) during irradiation of TiO2-N,C (λ≥ 420 nm). See text. | |
As already mentioned, contrary to TiO2-N, the suspension of TiO2-N,C is highly active in the mineralization of formic acid under visible (λ≥ 420 nm) light irradiation in the presence of dissolved oxygen (Fig. 3, curves a and c). It is reasonable to assume that in the presence of oxygen this will be reduced by er−according to eqn (3). Subsequent protonation of superoxide affords HO2 that disproportionates to O2 and H2O2. Photoreduction of the latter finally produces OH− and OH˙ radicals.45 These are able (E°OH˙/H2O≈ 2.8 V)46,47 to oxidize formic acid (E°CO2−˙/HCO2−≈ 1.90 V).31 The initial pH in the photoactivity tests was ca. 3–4 and increased in the course of the photocatalytic reaction to values of ca. 7. The resulting COO−˙ can either reduce oxygen to superoxide48–50 (eqn (4)) or increase the number of reactive electrons through electron injection into the conduction band of TiO2-N,C (E°CO2/CO2−˙≈−1.90 V,51,52eqn (5)). Alternatively and less likely, it can react with H2O2 and give rise to OH˙ radicals (eqn (6)).48 No experimental evidence for oxalic acid formation could be found, which is in line with the fact that the COO−˙ radical is known to dimerize only in neutral and alkaline solution.53
| | | COO−˙ + O2→ CO2 + O2−˙ | (4) |
| | | COO−˙ + H2O2→ CO2 + OH− + OH˙ | (6) |
 |
| | Fig. 3 Photomineralization of formic acid (c = 1 × 10−3 mol L−1); c0, ct are concentrations at times 0 and t; (a) TiO2-N,C in the presence of oxygen, (b) TiO2-N,C with C(NO2)4 in deoxygenated system, (c) TiO2-N, in the presence of oxygen; λ≥ 420 nm. | |
In order to differentiate the role of visible light generated holes and electrons the reaction was carried out in oxygen-free catalyst suspension containing tetranitromethane as electron acceptor. This hinders the formation of OH˙ radicals through oxygen reduction and subsequent reactions, hence the reductive photodegradation pathway is effectively “switched-off”. Under such conditions the mineralization of formic acid is still observed (Fig. 3, curve b). The flattening of the degradation curve observed after two hours is most likely due to depletion of tetranitromethane and increasing competitive light absorption and photodegradation of the C(NO2)3− product. Clearly, under the Vis irradiation the reactive holes hr,s+ are able to oxidize formic acid either directly (eqn (7)) or indirectly through OH˙ radical formed by hole oxidation of surface-bound OH groups. The latter possibility can be excluded since TiO2-N,C in pristine water does not enable visible light reduction of methylviologen.42 This becomes possible only in the presence of formic acid. In other words, the surface-bound OH groups do not scavenge holes induced by visible light. Degradation is slower by a factor of ∼2–3, suggesting that the reductive pathway via intermediate superoxide plays an essential role in formic acid degradation. In the presence of tetranitromethane the photogenerated electrons er− reduce the nitroalkane to C(NO2)3− and NO2 (eqn (8)). The COO−˙ radical, in addition to the reaction described by eqn (5), may also reduce tetranitromethane according to eqn (9).
| | | hr,s+ + HCOO−→ COO−˙ + H+ | (7) |
| | | er− + C(NO2)4→ C(NO2)3− + NO2 | (8) |
| | | COO−˙ + C(NO2)4→ CO2 + C(NO2)3− + NO2 | (9) |
Formation of relatively stable C(NO2)3− is confirmed by enhancement of its typical absorbance at 350 nm (Fig. 4).54 The peak at time t = 0 min (at λ = 350 nm) is caused by contamination of commercial tetranitromethane with its reduced form. On the contrary, in the case of unmodified TiO2 no degradation of formic acid and no increasing of absorbance at 350 nm were observed (Fig. 5). The decomposition of C(NO2)3− observed after prolonged irradiation in both cases is caused by its photolysis taking place with and without TiO2.54,55 Both the formation of C(NO2)3− and consumption of formic acid provide clear evidence for interfacial electron transfer reactions occurring at the TiO2-N,C surface irradiated with visible light. The overall reaction describing processes occurring during oxidation of formic acid in an oxygen free TiO2-N,C suspension in the presence of tetranitromethane can be summarized according to the eqn (10) (the sum of eqn (7)–(9)).
| | | er− + hr,s+ + HCOO− + 2C(NO2)4→ CO2 + 2C(NO2)3− + 2NO2 + H+ | (10) |
 |
| | Fig. 4 Concentration of C(NO2)3− during irradiation (λ≥ 420 nm) of deoxygenated system containing C(NO2)4 (10−2 mol L−1), formic acid (1 × 10−3 mol L−1), and TiO2-N,C (1 g L−1). | |
 |
| | Fig. 5 Concentration of C(NO2)3− during irradiation (λ≥ 420 nm) of deoxygenated system containing C(NO2)4 (10−2 mol L−1), formic acid (1 × 10−3 mol L−1), and TiO2 (1 g L−1). | |
Upon UV-Vis (λ≥ 320 nm) light irradiation, as expected, the holes hr,v+ generated in a TiO2-N,C suspension containing tetranitromethane instead of oxygen can easily oxidize formic acid. As shown in Fig. 6, already within 15 min 75% mineralization is achieved. At the same time increase of absorbance of C(NO2)3− is observed (Fig. 7) indicating tetranitromethane reduction by er−. The faster mineralization is due to a stronger light absorption of TiO2-N,C resulting in a higher concentration of hr,v+. Interestingly, when the same oxygen-free experiment was carried out in the absence of tetranitromethane, 20% mineralization of formic acid was still detected (Fig. 6). In the course of this reaction the color of suspension changed from yellow to dark blue which is caused by reduction of titania lattice ions by photogenerated er-, since any other electron acceptor is absent.56,57
 |
| | Fig. 6 Photomineralization of formic acid (c = 1 × 10−3 mol L−1) TiO2-N,C in a deoxygenated system in the presence (a) and absence (b) of C(NO2)4. λ≥ 320 nm. | |
 |
| | Fig. 7 Concentration of C(NO2)3− anion during irradiation (λ≥ 320 nm) of deoxygenated system containing C(NO2)4 (10−2 mol L−1), formic acid (1 × 10−3 mol L−1), and TiO2-N,C. | |
Further experiments were conducted using methylviologen MV2+ as an electron acceptor. In this case the pH dependence of the Fermi potential induces also a pH-dependent formation of MV+˙. It can be followed easily by recording the open-circuit photopotential of a platinum electrode immersed into the solution. This experimental set-up is typically used for determination of the quasi-Fermi level of photogenerated electrons.44,58 In solutions without any added hole scavengers the reduction of methylviologen is observed when TiO2-N,C is excited with UV-Vis (λ≥ 320 nm) light but is absent within the full pH range under visible light irradiation (λ > 420 nm) (Fig. 8). It means that in the latter case recombination processes overcome the interfacial electron transfer reactions even at pH values enabling the methylviologen reduction. The reason for the inefficiency of MV2+ reduction is the absence of a suitable, i.e. more easily oxidizable, hole scavenger than water and surface OH groups. However, when formic acid was added the suspension formed a blue color at pH values higher than ca. 6.0 (Fig. 8), whereby the initially transient green color resulted from superimposition of the colors of yellow photocatalyst with blue MV+˙. The titration curve does not have the typical sigmoidal shape, probably due to slow kinetics of formic acid oxidation. Notice that without addition of MV2+ to the TiO2-N,C suspension in formic acid no blue color and no photovoltage change were observed. Therefore, upon visible light irradiation Ti4+ reduction is negligible and the blue color of the suspension is solely due to MV+˙. All these results clearly reveal that under the Vis excitation experiment MV2+ is reduced by e−r (eqn (11)) whereas formic acid is oxidized by hr,s+ according to eqn (7). The resulting COO−˙ can convert to CO2 in oxygen free conditions by reducing MV2+ directly (eqn (12)) or through intermediate involvement of conduction band electrons (eqn (5)). Thus, the overall reaction occurring during potentiometric titration of oxygen free TiO2-N,C suspension under visible light irradiation in the presence of methylviologen and formic acid can be summarized according to the eqn (13) (the sum of eqn (7), (11), (12)). Importantly, when TiO2-N,C was replaced by TiO2-N in this experiment, no inflection point and no methylviologen reduction were observed.34
| | | COO−˙ + MV2+→ CO2 + MV+˙ | (12) |
| | | e−r + h+r,s + HCOO- + 2MV2+→ CO2 + 2MV+˙ + H+ | (13) |
 |
| | Fig. 8 Variation of photovoltage with pH value for a suspension of the catalyst in the presence of (MV)Cl2; (a) TiO2-N,C, no cut-off filter, no formic acid, (b) TiO2-N,C, 420 nm cut-off filter, formic acid, (c) TiO2, 420 nm cut-off filter, formic acid. | |
The reactivity of photogenerated holes was also examined using wavelength-resolved photocurrent measurements on photocatalyst powders deposited on FTO–glass electrodes. Here it should be noted that a photocurrent is observed only if two processes occur simultaneously—the photogenerated electron is transferred to the FTO electrode and the hole oxidizes a donor species. Since the first process can be assumed to proceed readily at anodically-biased FTO electrodes, it is the latter process which exerts control over the photocurrent response. In other words, under such experimental conditions photocurrents are observed only when the holes can oxidize a suitable reducing agent. Therefore the photocurrent action spectra were recorded with and without addition of potassium iodide and formic acid as different hole scavengers.
As expected, an unmodified TiO2 photoelectrode oxidizes neither iodide nor formic acid in the visible, as shown in Fig. 9. This is because titania does not absorb visible light and is in agreement with its inactivity in visible light photodegradation of formic acid. However, when TiO2-N,C is used, both formic acid and iodide can be oxidized in the visible spectral range (Fig. 10). As expected, the incident photon-to-current efficiency is higher for iodide than for formic acid since the standard reduction potential (vs. NHE) is more negative for iodide (ca. 1.3 V) than for formic acid (1.90 V).31,51 It is noted that the quite small IPCE values at 400–450 nm observed in pristine water are significantly increased upon addition of formic acid (Fig. 10, curves b and c). This is not observed when TiO2-N is employed as electrode. In this case even a very small decrease is found (Fig. 11, curves b and c). However, a significant visible light response is observed in the presence of iodide (Fig. 11, curve d). This finding is in line with the fact that this material does not exhibit photocatalytic activity towards formic acid in visible light.14 The rather low IPCE values observed in the range of 400–450 nm in the experiments in the absence of formic acid (curves b in Fig. 10 and 11) may arise from oxidation of water or organic impurities present in TiO2-N,C.
 |
| | Fig. 9 Photocurrent action spectra of TiO2 (Hombikat) measured at 0.5 V vs. Ag/AgCl (3 mol L−1) in LiClO4 (0.1 mol L−1) electrolyte containing various hole-scavengers: (a) no added hole scavenger, (b) HCOOH (10−3 mol L−1), (c) KI (0.1 mol L−1). | |
 |
| | Fig. 10 Photocurrent action spectra of TiO2 (Hombikat) and TiO2-N,C measured at 0.5 V vs. Ag/AgCl (3 mol L−1) in LiClO4 (0.1 mol L−1) electrolyte containing various hole-scavengers: (a) TiO2, no added hole scavenger; TiO2-N,C, (b) no added hole scavenger, (c) HCOOH (10−3 mol L−1), (d) KI (0.1 mol L−1). | |
 |
| | Fig. 11 Photocurrent action spectra of TiO2 (Hombikat) and TiO2-N measured at 0.5 V vs. Ag/AgCl (3 mol L−1) in LiClO4 (0.1 mol L−1) electrolyte containing various hole-scavengers: (a) TiO2, no added hole scavenger; TiO2-N, (b) no added hole scavenger, (c) HCOOH (10−3 mol L−1), (d) KI (0.1 mol L−1). | |
Notably, the visible light induced mineralization of formic acid shows maximum reaction rates at pH 3–4. This corresponds with the results reported for the reaction conducted in the presence of UV light and unmodified titania, started at about pH 3.4 and was completely inhibited above pH 6.59 It can be rationalized by electrostatic repulsion between formate and the titania surface as indicated by the formic acid pKa value of 3.75 and by the point of zero charge of ∼5.8 for anatase.60 Moreover, it should be noted that the oxidation potential of the OH˙ radical decreases from 2.6 V to 1.9 V upon going from pH 0 to pH 6, and therefore the driving force of formate oxidation is also decreased.46,61,62 At pH 3.5 the visible light generated holes have enough oxidative force (∼2.2 V) to oxidize formic acid (E°HCO2−/CO2−˙≈ 1.90 V).31 Additionally, protonation of superoxide radical (pKa = 4.88),63 a reaction necessary for OH˙ radical production on the reductive pathway, is also more effective in acidic solution.
The results reported above can be summarized as follows. Clearly, the separation of photogenerated charges is more efficient in TiO2-N,C than in TiO2-N, which makes it manifest both in photocatalytic and photocurrent experiments. This can be rationalized in terms of stabilization of photogenerated holes by delocalization within an extended poly(amino-tri-s-triazine) heterocycle at the surface of TiO2-N,C, which, in turn, renders electron–hole recombination less favorable. The tri-s-triazine core is based on a cyclic system of twelve C–N bonds which surround the central sp2 hybridized N atom. The 14 π-electrons form a doubly cross-conjugated aromatic planar system.64 Importantly, melamine rings and related materials are known to be of low flammability. Reasons for this property are the relatively high bond dissociation energy of C–N single and multiple bonds, as well as the relatively high electronegativity of nitrogen atoms, since it causes a partial oxidation of the carbon atoms. Oxidation reactions are therefore less likely for nitrogen rich C/N/(H) compounds as compared to other organic compounds such as hydrocarbons.65 It follows that the visible light-generated hole which is delocalized in the tri-s-triazine rings is not expected to induce self-oxidation processes of the TiO2-N,C shell (i.e. visible light photocorrosion) but more likely causes one-electron oxidation of compounds present in the catalyst suspension. Such hole stabilization is not possible in the case of TiO2-N since this contains non-aromatic small amidic or oxidic nitrogen species. As a consequence thereof, electron–hole recombination becomes more efficient than interfacial electron exchange and formic acid oxidation is not possible.
Conclusion
The oxidation of formic acid by TiO2-N,C irradiated with visible light in the presence of oxygen involves both reductive and oxidative primary process. Photocatalytic and photoelectrochemical measurements in the presence of different electron acceptors confirm that the visible light photogenerated holes in TiO2-N,C are able to oxidize formic acid. This is in contrast to the behavior of conventional TiO2-N. The reason for this difference most likely is an enhanced stabilization of the photogenerated hole in TiO2-N,C against recombination with conduction band electrons.
References
- V. Balzani, A. Credi and M. Venturi, Photochemical conversion of solar energy, ChemSusChem, 2008, 1, 26–58 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Powering the planet: chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- X. Qiu and C. Burda, Chemically synthesized nitrogen-doped metal oxide nanoparticles, Chem. Phys., 2007, 339, 1–10 CrossRef CAS.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, N-doped TiO2: Theory and experiment, Chem. Phys., 2007, 339, 44–56 CrossRef CAS.
- A. V. Emeline, V. N. Kuznetsov, V. K. Rybchuk and N. Serpone, Visible-light-active titania photocatalysts: the case of N-doped TiO2s-properties and some fundamental issues, Int. J. Photoenergy, 2008, 2008, 258394.
- S. Sakthivel and H. Kisch, Daylight photocatalysis by carbon-modified titanium dioxide, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef CAS.
- C. S. Enache, J. Schoonman and R. van de Krol, Addition of carbon to anatase TiO2 by n-hexane treatment - surface or bulk doping?, Appl. Surf. Sci., 2006, 252, 6342–6347 CrossRef CAS.
- S. Sato, Photocatalytic activity of nitrogen oxide (NOx)-doped titanium dioxide in the visible light region, Chem. Phys. Lett., 1986, 123, 126–128 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Visible-light photocatalysis in nitrogen-doped titanium oxides, Science, 2001, 293, 269–271 CrossRef CAS.
- S. Sakthivel, M. Janczarek and H. Kisch, Visible Light Activity and Photoelectrochemical Properties of Nitrogen-Doped TiO2, J. Phys. Chem. B, 2004, 108, 19384–19387 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, Nitrogen-Concentration Dependence on Photocatalytic Activity of TiO2-xNx Powders, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Enhanced Nitrogen Doping in TiO2 Nanoparticles, Nano Lett., 2003, 3, 1049–1051 CrossRef CAS.
- T. Ohno, Preparation of visible light active S-doped TiO2 photocatalysts and their photocatalytic activities, Water Sci. Technol., 2004, 49, 159–163 Search PubMed.
- D. Mitoraj and H. Kisch, The nature of nitrogen-modified titanium dioxide photocatalysts active in visible light, Angew. Chem., Int. Ed., 2008, 47, 9975–9978 CrossRef CAS.
- Y. Nosaka, M. Matsushita, J. Nishino and A. Y. Nosaka, Nitrogen-doped titanium dioxide photocatalysts for visible response prepared by using organic compounds, Sci. Technol. Adv. Mater., 2005, 6, 143–148 CrossRef CAS.
- K. Kobayakawa, Y. Murakami and Y. Sato, Visible-light active N-doped TiO2 prepared by heating of titanium hydroxide and urea, J. Photochem. Photobiol., A, 2005, 170, 177–179 CrossRef CAS.
- S. Yin, K. Ihara, Y. Aita, M. Komatsu and T. Sato, Visible-light induced photocatalytic activity of TiO2-xAy (A = N, S) prepared by precipitation route, J. Photochem. Photobiol., A, 2006, 179, 105–114 CrossRef CAS.
- S. Yin, K. Ihara, M. Komatsu, Q. Zhang, F. Saito, T. Kyotani and T. Sato, Low temperature synthesis of TiO2-xNy powders and films with visible light responsive photocatalytic activity, Solid State Commun., 2006, 137, 132–137 CAS.
- J. Yuan, M. Chen, J. Shi and W. Shangguan, Preparations and photocatalytic hydrogen evolution of N-doped TiO2 from urea and titanium tetrachloride, Int. J. Hydrogen Energy, 2006, 31, 1326–1331 CrossRef CAS.
- D. Chen, Z. Jiang, J. Geng, Q. Wang and D. Yang, Carbon and Nitrogen Co-doped TiO2 with Enhanced Visible-Light Photocatalytic Activity, Ind. Eng. Chem. Res., 2007, 46, 2741–2746 CrossRef CAS.
- Y. Cong, J. Zhang, F. Chen and M. Anpo, Synthesis and Characterization of Nitrogen-Doped TiO2 Nanophotocatalyst with High Visible Light Activity, J. Phys. Chem. C, 2007, 111, 6976–6982 CrossRef CAS.
- D. Huang, S. Liao, S. Quan, L. Liu, Z. He, J. Wan and W. Zhou, Synthesis and characterization of visible light responsive N-TiO2 mixed crystal by a modified hydrothermal process, J. Non-Cryst. Solids, 2008, 354, 3965–3972 CrossRef CAS.
- G.-S. Shao, X.-J. Zhang and Z.-Y. Yuan, Preparation and photocatalytic activity of hierarchically mesoporous-macroporous TiO2-xNx, Appl. Catal., B, 2008, 82, 208–218 CrossRef CAS.
- R. Bacsa, J. Kiwi, T. Ohno, P. Albers and V. Nadtochenko, Preparation, Testing and Characterization of Doped TiO2 Active in the Peroxidation of Biomolecules under Visible Light, J. Phys. Chem. B, 2005, 109, 5994–6003 CrossRef CAS.
- H. Kisch, S. Sakthivel, M. Janczarek and D. Mitoraj, A Low-Band Gap, Nitrogen-Modified Titania Visible-Light Photocatalyst, J. Phys. Chem. C, 2007, 111, 11445–11449 CrossRef CAS.
- R. Beranek and H. Kisch, Tuning the optical and photoelectrochemical properties of surface-modified TiO2, Photochem. Photobiol. Sci., 2008, 7, 40–48 RSC.
- S. Yin, Y. Aita, M. Komatsu and T. Sato, Visible-light-induced photocatalytic activity of TiO2-xNy prepared by solvothermal process in urea-alcohol system, J. Eur. Ceram. Soc., 2006, 26, 2735–2742 CrossRef CAS.
- Y. Yamamoto, S. Moribe, T. Ikoma, K. Akiyama, Q. Zhang, F. Saito and S. Tero-Kubota, Visible light induced paramagnetic sites in nitrogen-doped TiO2 prepared by a mechanochemical method, Mol. Phys., 2006, 104, 1733–1737 CrossRef CAS.
- E. A. Reyes-Garcia, Y. Sun, K. Reyes-Gil and D. Raftery, 15N Solid State NMR and EPR Characterization of N-Doped TiO2 Photocatalysts, J. Phys. Chem. C, 2007, 111, 2738–2748 CrossRef CAS.
- M. Alvaro, E. Carbonell, V. Fornes and H. Garcia, Enhanced photocatalytic activity of zeolite-encapsulated TiO2 clusters by complexation with organic additives and N-doping, ChemPhysChem, 2006, 7, 200–205 CrossRef CAS.
- M. Mrowetz, W. Balcerski, A. J. Colussi and M. R. Hoffmann, Oxidative Power of Nitrogen-Doped TiO2 Photocatalysts under Visible Illumination, J. Phys. Chem. B, 2004, 108, 17269–17273 CrossRef CAS.
- H. Chen, A. Nambu, W. Wen, J. Graciani, Z. Zhong, J. C. Hanson, E. Fujita and J. A. Rodriguez, Reaction of NH3 with Titania: N-Doping of the Oxide and TiN Formation, J. Phys. Chem. C, 2007, 111, 1366–1372 CrossRef CAS.
- S.-K. Joung, T. Amemiya, M. Murabayashi and K. Itoh, Relation between photocatalytic activity and preparation conditions for nitrogen-doped visible light-driven TiO2 photocatalysts, Appl. Catal., A, 2006, 312, 20–26 CrossRef CAS.
- D. Mitoraj and H. Kisch, J. Phys. Chem. C Search PubMed , in print.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- J. R. Durrant, S. A. Haque and E. Palomares, Towards optimisation of electron transfer processes in dye sensitised solar cells, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef.
- M. F. J. Dijkstra, H. J. Panneman, J. G. M. Winkelman, J. J. Kelly and A. A. C. M. Beenackers, Modeling the photocatalytic degradation of formic acid in a reactor with immobilized catalyst, Chem. Eng. Sci., 2002, 57, 4895–4907 CrossRef CAS.
- F. Shiraishi, T. Nakasako and Z. Hua, Formation of Hydrogen Peroxide in Photocatalytic Reactions, J. Phys. Chem. A, 2003, 107, 11072–11081 CrossRef CAS.
- S.-H. Yoon, S.-E. Oh, J. E. Yang, J. H. Lee, M. Lee, S. Yu and D. Pak, TiO2 Photocatalytic Oxidation Mechanism of As(III), Environ. Sci. Technol., 2009, 43, 864–869 CrossRef CAS.
- E. Pelizzetti and C. Minero, Mechanism of the photooxidative degradation of organic pollutants over titanium dioxide particles, Electrochim. Acta, 1993, 38, 47–55 CrossRef CAS.
- W. P. Gomes, T. Freund and S. R. Morrison, Chemical reactions involving holes at the zinc oxide single-crystal anode, J. Electrochem. Soc., 1968, 115, 818–823 CAS.
- M. Mrowetz and E. Selli, Photocatalytic degradation of formic and benzoic acids and hydrogen peroxide evolution in TiO2 and ZnO water suspensions, J. Photochem. Photobiol., A, 2006, 180, 15–22 CrossRef CAS.
- T. L. Villarreal, R. Gomez, M. Neumann-Spallart, N. Alonso-Vante and P. Salvador, Semiconductor Photooxidation of Pollutants Dissolved in Water: A Kinetic Model for Distinguishing between Direct and Indirect Interfacial Hole Transfer. I. Photoelectrochemical, Experiments with Polycrystalline Anatase Electrodes under Current Doubling and Absence of Recombination, J. Phys. Chem. B, 2004, 108, 15172–15181 CrossRef CAS.
- A. M. Roy, G. C. De, N. Sasmal and S. S. Bhattacharyya, Determination of the flatband potential of semiconductor particles in suspension by photovoltage measurement, Int. J. Hydrogen Energy, 1995, 20, 627–630 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Photoinduced reactivity of titanium dioxide, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- U. K. Klaning, K. Sehested and J. Holcman, Standard Gibbs energy of formation of the hydroxyl radical in aqueous solution. Rate constants for the reaction chlorite (ClO2-) + ozone.dblarw. ozone(1-) + chlorine dioxide, J. Phys. Chem., 1985, 89, 760–763 CrossRef CAS.
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671 CrossRef CAS.
- E. J. Hart, Mechanism of the g-ray induced oxidation of formic acid in aqueous solution, J. Am. Chem. Soc., 1951, 73, 68–73 CrossRef CAS.
- E. J. Hart, g-Ray-induced oxidation of aqueous formic acid-oxygen solutions.sbd.effect of pH, J. Am. Chem. Soc., 1954, 76, 4198–4201 CrossRef CAS.
- E. J. Hart and A. Henglein, Free radical and free atom reactions in the sonolysis of aqueous iodide and formate solutions, J. Phys. Chem., 1985, 89, 4342–4347 CrossRef CAS.
- P. Wardman, Reduction potentials of one-electron couples involving free radicals in aqueous solution, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CAS.
- H. A. Schwarz and R. W. Dodson, Reduction potentials of CO2- and the alcohol radicals, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- H. Fricke and E. J. Hart, J. Chem. Phys., 1934, 2, 824 CrossRef CAS.
- A. Henglein, Colloidal titanium dioxide-catalyzed photo- and radiation chemical processes in aqueous solution, Ber. Bunsen-Ges., 1982, 86, 241–246 CAS.
- A. J. Frank, M. Graetzel and A. Henglein, The 347.2 nm laser photolysis of tetranitromethane in polar, nonpolar, and micellar solutions, Ber. Bunsen-Ges., 1976, 80, 593–602 CAS.
- R. F. Howe and M. Grätzel, EPR observation of trapped electrons in colloidal titanium dioxide, J. Phys. Chem., 1985, 89, 4495 CrossRef CAS.
- P. V. Kamat, I. Bedja and S. Hotchandani, Photoinduced Charge Transfer between Carbon and Semiconductor Clusters. One-Electron Reduction of C60 in Colloidal TiO2 Semiconductor Suspensions, J. Phys. Chem., 1994, 98, 9137–9142 CrossRef CAS.
- H. Kisch, G. Burgeth and W. Macyk, Visible light photocatalysis by a titania transition metal complex, Adv. Inorg. Chem., 2004, 56, 241–259 CAS.
- D. H. Kim and M. A. Anderson, Solution factors affecting the photocatalytic and photoelectrocatalytic degradation of formic acid using supported TiO2 thin films, J. Photochem. Photobiol., A, 1996, 94, 221–229 CrossRef CAS.
- S. R. Morrison, The Chemical Physics of Surfaces of Solids, Plenum Press, New York, 1980 Search PubMed.
- W. H. Koppenol and J. F. Liebman, The oxidizing nature of the hydroxyl radical. A comparison with the ferryl ion (FeO2+), J. Phys. Chem., 1984, 88, 99–101 CrossRef CAS.
- H. A. Schwarz and R. W. Dodson, Equilibrium between hydroxyl radicals and thallium(II) and the oxidation potential of hydroxyl(aq), J. Phys. Chem., 1984, 88, 3643–3647 CrossRef CAS.
- D. Behar, G. Czapski, J. Rabani, L. M. Dorfman and H. A. Schwarz, Acid dissociation constant and decay kinetics of the perhydroxyl radical, J. Phys. Chem., 1970, 74, 3209–3213 CrossRef CAS.
- M. Cyranski and T. M. Krygowski, Separation of the energetic and geometric contributions to the aromaticity of pi-electron carbocyclics. Part V. Analysis, of the aromatic character of aza-analogs of benzenoid hydrocarbons, Tetrahedron, 1996, 52, 13795–13802 CrossRef CAS.
- E. Kroke and M. Schwarz, Novel group 14 nitrides, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photocurrent measured under intermittent irradiation as a function of irradiated wavelength (Fig. S1). See DOI: 10.1039/b9pp00052f |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.