Immunosuppressive ultraviolet-A radiation inhibits the development of skin memory CD8 T cells
Sabita Rana, Linda J. Rogers and Gary M. Halliday*
Discipline of Dermatology, Bosch Institute, Sydney Cancer Centre, The University of Sydney, NSW 2006, Australia. E-mail: garyh@med.usyd.edu.au
First published on 6th November 2009
Abstract
Ultraviolet A (UVA) radiation can have dual affects on the immune system depending on dose. At doses of approximately 1.8 J cm−2, UVA acts in an immunosuppressive manner, whilst at higher doses UVA can promote recovery and protection against UVB-induced immunosuppression in mice. We utilised a model of contact hypersensitivity (CHS) to investigate how different doses of UVA modulates CD8 T cell immunity against a hapten in vivo. Only 1.8 J cm−2 UVA decreased the CHS response compared to unirradiated mice, but this did not correlate with an inhibition of primary effector CD8 T cells. A similar expansion of effector CD8 T cells in skin-draining lymph nodes and accumulation of IFN-γ-producing CD8 T cells in the ear skin was observed between unirradiated and UVA-irradiated mice. However, dermal memory CD8 T cells examined 9 weeks post challenge showed decreased numbers in mice irradiated with 1.8 J cm−2 UVA compared with unirradiated, 1.3 J cm−2 and 3.4 J cm−2 UVA-irradiated mice. Therefore, UVA does not inhibit the expansion, migration or IFN-γ secretion of CD8 T cells during a primary immune response. However, exposure to immunosuppressive UVA causes a defect in CD8 T cell development that impairs the ability of cells to become long-term memory cells.
Introduction
The vast majority (95%) of the ultraviolet (UV) radiation in sunlight that reaches earth is of the UVA spectrum (320–400 nm). Although our understanding of how UV, in particular UVB, modulates the immune system is growing, there is very little understanding of how UVA regulates our immune system. There is increasing evidence to support the notion that UVA can indeed regulate the immune system, even at the low doses to which humans are frequently exposed to during routine daily activity. For example, nickel allergic volunteers irradiated with UVA at a dose approximating 6 min of exposure to the standard sun spectrum (European Cosmetic Toiletry and Perfumery Association Sun Protection Factor Test Method) demonstrate a reduced nickel patch test compared to unirradiated volunteers.1 Systemic (antigen and UVA applied at separate sites) models of contact hypersensitivity (CHS) and delayed hypersensitivity also exhibit decreased responses following exposure to UVA in C57BL/6 mice.2,3 In these instances, immunosuppression induced by UVA was limited to only short treatment regimes (1–3 days) as continued exposure did not result in immunosuppression, and to moderate doses (1.7–2.7 J cm−2) since higher doses (2.5–5.3 J cm−2) also do not cause immunosuppression. This is in contrast to the action of UVB, which is increasingly immunosuppressive upon increased dose and duration.2 Unlike the linear dose–response of UVB, UVA causes a bell-shaped curve relationship with immunosuppression. This indicates two important features of UVA-induced immunosuppression: that the mechanisms at play are different from those occurring after UVB, and that adaptive mechanisms at high doses of UVA may be acting to reverse the negative effects of UVA on the immune system. In support of the latter, a dose of UVA (10–30 J cm−2, at least 3-fold higher than what was used in this study) delivered to mice prior to Leishmania major infection at the local site of irradiation was found to enhance the immune response against the parasite.4 Moreover, an even greater dose of UVA (38.7 J cm−2) can protect against suppression initiated by UVB in mice.5 Hence, depending on dose, UVA can have both inhibiting and stimulating effects on the immune system.Due to its longer wavelength, UVA penetrates deeper into the skin compared to UVB.6 Depending on the dose and irradiation regime, UVA can modulate the migration of Langerhans cells (LC) from the epidermis. Studies utilising acute exposure (2–4 J cm−2) have not found UVA to perturb LC,7 whilst studies of chronic exposure (19.7 J cm−2 delivered 5 times per week for 4 weeks) demonstrate that UVA can cause the depletion of LC from the epidermis via a process mediated by nitric oxide.8 The formation of reactive nitrogen and oxygen species leads to both nitrosative and oxidative damage in the skin. Oxidative damage induced by UVA has been shown to be important for its ability to induce immunosuppression.9 The breakdown of lipids and proteins by nitration and peroxidation are thought to activate the alternative complement pathway, which has been shown by molecular studies to be upregulated by immunosuppressive doses of UVA.10
Elucidating the immunological processes modified by UVA during immunosuppression or higher dose recovery will facilitate our understanding of how UVA can have such widely disparate affects on the immune system. This knowledge will aid in the design of tailored sunscreens, which will prevent or promote UVA stimulated processes as required. Thus, this study utilised a systemic model of CHS to assess whether CD8 T cell immunity against a contact sensitiser (hapten) is regulated by UVA at 1.3 J cm−2 (inert), 1.8 J cm−2 (immunosuppressive) and 3.4 J cm−2 (recovery) doses. Primary effector CD8 T cells were not limited by the immunosuppressive dose of UVA, however, the development of dermal CD8 T cell memory was impaired by 1.8 J cm−2 UVA. 1.3 J cm−2 and 3.4 J cm−2 UVA did not alter the reactivity of CD8 T cells from what was observed in unirradiated mice.
Materials and methods
Mice
Female C57BL/6J mice were used at 8 weeks of age (Animal Resource Centre, Perth, Australia). All experiments were conducted under the approval of the University of Sydney Animal Ethics Committee.UVA source
A 1000 W xenon arc lamp solar simulator (Oriel, Stanford, CT) filtered with two 200–400 nm dichroic mirrors and a UVB blocking filter (CVL Laser, Albuquerque, NM) were used to produce a broadband UVA spectrum that had a wavelength range of 325 to 420 nm and a peak irradiance of 0.11 mW cm−2 at 370 nm. UVB (below 320 nm) and UVC (below 290 nm) contaminated the spectra by less than 0.01% and 0.001%, respectively. Spectral output and intensity was measured with a calibrated OL-754 spectroradiometer (Optronics Laboratories, Orlando, FL). A broadband radiometer (International Light Technologies, Inc., Peabody, MA) calibrated against the source with the spectroradiometer was used continuously to monitor fluctuations in output. Timing of UVA delivery was accurately maintained using an automated timing device.UVA irradiation protocol
Mice were removed of dorsum fur 24 h before irradiation with animal clippers (Oster, TN) and an electric razor (Remington, Austria). During irradiation, mice were restrained within black Perspex boxes with a quartz lid, and ears and heads were protected with black Perspex. The dorsums were exposed to 1.3, 1.8 or 3.4 J cm−2 of UVA daily for 3 consecutive days. UVA-irradiated but unsensitised mice were included in every experiment to control against any non-specific effects caused by irradiation.Contact hypersensitivity (CHS) response
Abdomens were sensitised epicutaneously 3 days following the last UVA-irradiation with 50 μl of 2% (w/v) oxazolone (4-ethoxymethylene-2-pheyl-2-oxazolin-5-one; Sigma, St. Louis, MN) in acetone. Ears were challenged topically 7 days later with 10 μl of 2% (w/v) oxazolone in acetone per ear. The increase in ear thickness was calculated based on pre- and 24 h post-challenge measurements using micrometer calipers (Interapid, Switzerland). Irritant control measurements from challenged but unsensitised mice were subtracted from sensitised mice to determine the CHS response.Flow cytometry
Single cell suspensions of draining lymph nodes (DLN; inguinal lymph node) and ear skin cells were prepared and processed by flow cytometry as previously described.11 Briefly, lymph nodes were disassociated through cell strainers before staining for flow cytometry. For ears, the skin was first split into dorsal and ventral sides and incubated in EDTA. The epidermis and dermis were then separated and chopped together, before being incubated with DNase and collagenase. Skin fragments were filtered before being processed for flow cytometry. Cells were blocked with anti-CD16/CD32 (anti-FcRγIII/II receptor; clone 2.4G2) antibody, before staining with T cell activation surface antibodies. The following monoclonal antibodies were used: βTCR (clone H57-597), CD8α (clone 53-6.7), CD127 (clone A7R34), CD44 (clone IM7.8.1) and CD62L (clone MEL-14). All antibodies were purchased from BD Pharmingen (San Diego, CA) and eBiosciences, Inc (San Diego, CA). Activated CD8 T cells were detected as being βTCR+CD8+CD127−CD44hiCD62L−, and analysis was performed on a FACSAria™ (BD Immunocytometry Systems, San Jose, CA). A minimum of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events was acquired for every sample. Data analysis was performed using FlowJo software v. 6.4 (Tree Star Inc, Ashland, OR).
000 events was acquired for every sample. Data analysis was performed using FlowJo software v. 6.4 (Tree Star Inc, Ashland, OR).Immunohistochemistry
Mouse ears were snap-frozen, sectioned and stained for CD8 and IFN-γ as previously described.11 Briefly, 7 μm sections were fixed in 4% paraformaldehyde, and were then blocked for endogenous biotin activity (Dako, Glostrup, Denmark) and non-specific antibody labelling with TNB blocking buffer (Perkin Elmer Life Sciences, Wellesley, MA) supplemented with 5% normal rabbit serum. Sections were first labelled with biotinylated anti-CD8 (clone 53-6.7, BD Pharmingen), followed by streptavidin-Alexa Fluor®488 (Molecular Probes™, Eugene, OR). Blocking was then applied again, before staining with anti-IFN-γ (clone AF-585-NA, R&D Systems, MN), biotinylated donkey anti-goat F(ab′)2 (Jackson Immunoresearch Laboratories, West Grove, PA), streptavidin-Alexa Fluor®594 (Molecular Probes™) and finally SlowFade® Gold antifade reagent with DAPI (Molecular Probes™). A BX51 fluorescent microscope with a DP70 camera attachment (Olympus, Tokyo, Japan) was used to visualise and photograph the sections.Data analysis and statistics
One-way ANOVA analyses with Tukey post-hoc tests were used for CHS responses. The effect of UVA on oxazolone-induced T cell activation and skin infiltration was determined by subtracting unsensitised (baseline activation) from sensitised groups to enable the direct comparison of sensitisation in unirradiated and UVA-irradiated groups by an unpaired Student's t tests. SPSS v. 11 software (SPSS Inc, Chicago, IL) was used to determine significance, where P < 0.05 was considered to be significant.Results
Suppression of CHS by UVA depends on dose
Fig. 1 shows final normalised data pooled from 3 replicate experiments. Mice that were irradiated with 1.3 J cm−2 UVA exhibited a CHS response that was not significantly different to unirradiated mice. When this was increased to 1.8 J cm−2 UVA, the CHS response was significantly reduced by 28% compared to unirradiated mice (P = 0.011). However, double this dose, 3.4 J cm−2 UVA, did not significantly suppress the response. To put the normalised values into context, absolute values for a representative experiment were 0.12, 0.13, 0.03 and 0.10 mm increase in ear thickness for mice exposed to 0, 1.3, 1.8 and 3.4 J cm−2 UVA, respectively. This is consistent with previous data.2,3,12 | ||
| Fig. 1 1.8 J cm−2 UVA reduces the CHS response in mice. Mice were exposed to 0, 1.3, 1.8 or 3.4 J cm−2 UVA for 3 consecutive days on the dorsum, before they were sensitised with oxazolone on the abdomen. The CHS response was elicited in ears 7 days after sensitisation and was determined 24 h later. The increase in the ear thickness was normalised to the unirradiated control to enable the multiple experiments to be pooled. UV effects can therefore be observed independent of variations in control reactions between experiments. The mean + SEM are shown. Mice exposed to 1.8 J cm−2 UVA had a significantly reduced CHS response (P = 0.011) compared to unirradiated mice. N = 24 mice pooled from 8 individual experiments. | ||
Immunosuppressive UVA does not inhibit the primary response of CD8 T cells
We first examined whether UVA alters the total number of cells in the DLN in sensitised mice compared to unirradiated sensitised controls. We found no significant difference between unirradiated and irradiated mice. In a representative experiment, the total number of cells per DLN of sensitised mice were found to be 5.3, 4.6, 5.0 and 5.4 × 106 from mice exposed to 0, 1.3, 1.8 and 3.4 J cm−2, respectively. As hapten-specific CD8 T cells are the principal effector cells known to mediate CHS reactions, we examined whether UVA differentially modulates the number of effector CD8 T cells in skin DLN activated by oxazolone sensitisation. Activated CD8 T cells were identified by flow cytometry based on the phenotype of being βTCR+CD8+CD127−CD44hiCD62L−.13 In unirradiated mice, oxazolone sensitisation significantly (P = 0.014) increased the number of effector CD8 T cells in DLN compared to unsensitised mice. We found that exposure to either 1.3, 1.8 and 3.4 J cm−2 UVA did not significantly alter the oxazolone-induced expansion of effector CD8 T cells compared to unirradiated mice based on normalised data pooled from 3 independent experiments (Fig. 2A). From a representative experiment, the actual values were 4.85, 4.71, 3.96 and 4.05 × 103 effector CD8 T cells per DLN in mice exposed to 0, 1.3, 1.8 and 3.4 J cm−2 UVA, respectively. Ear skin was then investigated to determine whether UVA modifies the migration of effector CD8 T cells during elicitation. The skin of unirradiated sensitised mice was infiltrated with a significantly increased number of activated CD8 T cells compared to unsensitised mice (P = 0.0001). However, again we did not detect a significant difference in the number of infiltrated activated CD8 T cells between any of the UVA doses and unirradiated mice (Fig. 2B). Actual values from a representative experiment, to put the numbers into biological context, were 3.45, 2.99, 3.90 and 2.95 × 102 CD8 T cells per ear from mice exposed to 0, 1.3, 1.8 and 3.4 J cm−2 UVA, respectively. Once CD8 T cells are in the skin, they respond to keratinocytes presenting hapten by producing cytokines like IFN-γ.14 Therefore, we assessed the functional activity of the skin infiltrating CD8 T cells for IFN-γ production during a CHS reaction (Fig. 3). In unirradiated mice, most CD8 T cells in the skin 48 h after CHS elicitation are positive for IFN-γ. Mice that were irradiated with 1.8 J cm−2 UVA also demonstrated IFN-γ production by most dermal CD8 T cells. Taken together, these findings suggest that immunosuppressive 1.8 J cm−2 UVA does not inhibit the capacity of CD8 T cells to become activated, proliferate, infiltrate skin and produce IFN-γ. | ||
| Fig. 2 UVA does not inhibit the oxazolone-induced increase in DLN effector T cell number and infiltration of CD8 T cells into elicited skin. Mice were exposed to 0, 1.3, 1.8 or 3.4 J cm−2 UVA for 3 consecutive days on the dorsum, before they were sensitised with oxazolone on the abdomen. The CHS response was elicited in the ears 7 days after sensitisation, and the draining lymph nodes (DLN) (A) and ear skin (B) were assessed at 48 h after elicitation during the peak period of infiltration for activated effector CD8 T cells. The increase in the number of activated CD8 T cells due to oxazolone sensitisation was determined by subtracting the background number of effector T cells in unsensitised but oxazolone challenged mice. The number of activated CD8 T cells was normalised to the unirradiated control to enable multiple experiments to be pooled. UV effects can therefore be observed independently of variations in control reactions between experiments. In either the DLN or skin, there was no significant difference between unirradiated and UVA-irradiated mice for any UVA dose. N = 9–12 mice per group, pooled from 3–4 individual experiments. Means + SEM are shown. | ||
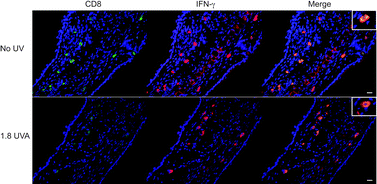 | ||
| Fig. 3 Skin-infiltrating CD8 T cells do not have inhibited IFN-γ production in 1.8 J cm−2 UVA-irradiated mice. Mice were exposed to 1.8 J cm−2 UVA, sensitised and challenged with oxazolone. Forty-eight hours after elicitation, the ear skin was assessed for CD8 (green) and IFN-γ (red). All CD8+ cells were found in the dermis and were co-localised with IFN-γ (orange, merge) in both unirradiated (no UV) and 1.8 J cm−2 UVA treated mice (1.8 UVA). The scale bar indicates 20μm. The inserts show higher magnifications of single CD8+IFN-γ+ cells. | ||
Immunosuppressive UVA inhibits the development of memory dermal CD8 T cells
Following the resolution of a CHS reaction, the skin maintains a subpopulation of CD8 T cells in the dermis in the form of memory CD8 T cells. As we previously showed that UVB irradiation limits the ability of CD8 T cells to become peripheral memory CD8 T cells in the skin,11 we investigated what effect UVA has on the development of memory CD8 T cells. In the absence of UVA and following sensitisation, previously elicited skin was able to support the localisation of CD8 T cells in the dermis 9 weeks after the initiation of a CHS reaction (Fig. 4). The number of these cells was significantly higher than in unirradiated unsensitised control mice (P = 0.02). Mice that had been irradiated with 1.3 J cm−2 UVA before sensitisation showed a similar number of oxazolone activated CD8 T cells in the dermis. However, mice exposed to 1.8 J cm−2 immunosuppressive UVA had a significantly reduced number of oxazolone activated CD8 T cells compared to unirradiated mice (>6-fold reduction, P = 0.0009). When mice were treated with 3.4 J cm−2 UVA, the number of dermal CD8 T cells recovered were not significantly different from unirradiated mice. Actual values from a representative experiment, to put the numbers into biological context, were 6.09, 5.43, 0.49 and 4.95 × 102 CD8 T cells per ear from mice exposed to 0, 1.3, 1.8 and 3.4 J cm−2 UVA, respectively. | ||
| Fig. 4 Dermal memory CD8 T cells are decreased in mice exposed to 1.8 J cm−2 UVA. After mice were irradiated, sensitised and challenged, they were rested for 9 weeks. The ear skin was then examined in the resting memory mice for activated CD8 T cells. There was a significant reduction in the number of CD8 T cells in mice irradiated with 1.8 J cm−2 UVA compared with unirradiated mice (P = 0.0009). N = 9–12 mice per group pooled from 3–4 individual experiments. Means + SEM are shown. | ||
To confirm the ability of these cells to rapidly respond to oxazolone like a memory CD8 T cell, we rechallenged resting memory mice (9 weeks from the time of first challenge) with oxazolone without further resensitisation, and examined CD8 T cells 24 h later for IFN-γ production. The short time frame between rechallenge and IFN-γ assessment was necessary to ensure that only resident memory CD8 T cells and not newly recruited CD8 T cells were the source of IFN-γ. Dermal CD8 T cells in the ears of mice not exposed to UVA showed reactivity to oxazolone by producing IFN-γ (Fig. 5). However, only few to no CD8 T cells were found in mice exposed to 1.8 J cm−2 UVA. These findings indicate that 1.8 J cm−2 immunosuppressive UVA can impair the ability of CD8 T cells to develop into memory CD8 T cells in the dermis.
 | ||
| Fig. 5 Dermal memory CD8 T cells can produce IFN-γ upon rechallenge. Mice were exposed to 1.8 J cm−2 UVA, sensitised, challenged and were then rested. Ears were rechallenged 9 weeks later, and 24 h after rechallenge, the ears were assessed for CD8 (green) and IFN-γ (red). In unirradiated (no UV) mice, all the CD8+ cells were located in the dermis and were co-localised with IFN-γ (orange, merge). No CD8+ or IFN-γ+ labelling were detected in mice irradiated with 1.8 J cm−2 UVA (1.8 UVA). The scale bar indicates 20μm. The insert shows higher magnification of a single CD8+IFN-γ+ cell. | ||
Discussion
UVA and the immune system share a complex relationship, which is highlighted by the dose-dependent bell-shaped affect UVA has on CHS. This study has shown that systemic suppression of CHS by 1.8 J cm−2 UVA occurs independent of primary effector CD8 T cell number and IFN-γ production. However, the development of dermal memory CD8 T cells is impeded by this immunosuppressive dose of UVA. Normal sunlight largely consists of UVA and the doses used here are below the amounts of UVA found within sunlight needed to cause barely detectable sunburn. Therefore, this study indicates for the first time that daily exposure to UVA at doses achievable during normal daily activities may have a long-term deleterious affect on the development of long-term T cell memory.The doses of UVA used here did not perturb the CD8 T cell response in relation to the expansion of effector CD8 T cells, migration into skin and capacity to produce IFN-γin situ at the elicitation site. Therefore, the lessened CHS reaction in mice exposed to 1.8 J cm−2 UVA cannot be due to a reduced number of IFN-γ-producing CD8 T cells infiltrating the skin. CHS magnitude is determined by infiltrate size and function. In the case of UVB-induced immunosuppression, a reduction in the generation of effector CD8 T cells in skin DLN and decreased accumulation in skin was correlated with immunosuppression.11 The absence of these size-related factors during 1.8 J cm−2 UVA-induced immunosuppression suggests that different mechanisms must be involved than those prevailing in UVB-irradiated mice. Instead, 1.8 J cm−2 UVA may have impacted a functional phenotype independent of IFN-γ that was not investigated in this study. Although IFN-γ has a role in supporting and sustaining CHS inflammation, absence of the IFN-γ receptor and neutralisation of IFN-γ does not abrogate CHS reactivity.15,16 Other cytokines produced by CD8 T cells can also act as inflammatory mediators including IL-17, which has been shown to be necessary for CHS reactivity.16,17 Another critical function of CD8 T cells is cytotoxic activity. Mice that are double deficient in perforin and FasL do not mount a CHS reaction akin to wild-type mice despite an intact ability to generate hapten-specific CD8 T cells.14 Thus, a combination of dysfunctional IL-17 secretion and poor cytotoxic activity by CD8 T cells may be responsible for the dampened CHS response observed in mice irradiated with 1.8 J cm−2 UVA.
1.8 J cm−2 UVA inhibited the maintenance of dermal CD8 T cells in resting mice for several weeks following ear challenge compared to unirradiated, 1.3 or 3.4 J cm−2 UVA-irradiated mice. This indicates that the immunosuppressive dose of UVA did have a functional effect on activation of CD8 T cells as they had impaired ability to survive as memory cells when they had migrated into challenged skin. Retention of CD8 T cells in the form of tissue-resident peripheral memory cells in the dermis helps to protect the skin against repeat challenges. Skin memory CD8 T cells have also been reported in mice following 2,4-dinitrofluorobenzene challenge or herpes simplex virus-1 infection.18,19 Inadequate survival support via IL-7 and IL-15 cytokine production in the skin or improper signalling early during T cell activation in sensitisation could have influenced this outcome.20,21 As both effector and memory CD8 T cells were affected by 1.8 J cm−2 UVA, the latter would be more plausible given that antigen-specific T cells receive their instructions for development very early during activation.22,23
Absorption of UVA energy by unknown chromophores in the irradiated skin may have initiated immunosuppressive pathways leading to defects in activation of CD8 T cells in DLN. Activation of the alternative complement pathway in response to immunosuppressive UVA at a similar dose to the amount used here is known to be involved in UVA-immunosuppression.10 At higher non-immunosuppressive doses (38.7 J cm−2), the skin upregulates a separate profile of processes including haem-oxygenase I, IL-12 and IFN-γ that prevent this UVA dose from being immunosuppressive.24 Therefore, it would seem that UVA differentially modulates processes in the skin with the result that they either favour or disfavour the reactivity of CD8 T cells. The delay-accelerating factor (DAF) is a key inhibitor of complement pathways. Local binding of DAF to the cell surface of UVA-induced migrating LC or possibly the circulation of increased DAF levels after UVA irradiation may have directly interfered with the development of hapten-specific CD8 T cells. DAF bound to antigen-presenting cells acts as an inhibitor of T cell immunity as shown by studies in mice deficient in DAF, wherein T cells exhibit an enhanced immune response or autoimmunity.25,26 Thus, activation of the alternative complement pathway by immunosuppressive doses of UVA may lead to this defect in peripheral memory CD8 T cell development while haem-oxygenase I, IL-12 and IFN-γ produced in response to higher doses of UVA may enable normal memory cell development.
This study has shown for the first time that immunosuppressive doses of UVA can prevent the functional differentiation of newly activated hapten-specific CD8 T cells into long-lasting memory dermal CD8 T cells. In light of the fact that UVA is also known to inhibit existing immune responses,1,27 these findings impress the need to protect against the UV spectrum of sunlight that primarily consists of UVA radiation.
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council of Australia and Epiderm to GH.References
- D. L. Damian, R. S. Barnetson and G. M. Halliday, Low-dose UVA and UVB have different time courses for suppression of contact hypersensitivity to a recall antigen in humans, J. Invest. Dermatol., 1999, 112, 939–44 CrossRef CAS.
- S. N. Byrne, N. Spinks and G. M. Halliday, Ultraviolet A irradiation of C57BL/6 mice suppresses systemic contact hypersensitivity or enhances secondary immunity depending on dose, J. Invest. Dermatol., 2002, 119, 858–864 CrossRef CAS.
- S. N. Byrne, N. Spinks and G. M. Halliday, The induction of immunity to a protein antigen using an adjuvant is significantly compromised by ultraviolet A radiation, J. Photochem. Photobiol., B, 2006, 84, 128–134 CrossRef CAS.
- N. M. Khaskhely, M. Maruno, A. Takamiyagi, H. Uezato, K. M. A. Kasem, A. Hosokawa, K. Kariya, Y. Hashiguchi, E. A. G. Landires and S. Nonaka, Pre-exposure with low-dose UVA suppresses lesion development and enhances Th1 response in BALB/c mice infected with Leishmania (Leishmania) amazonensis, J. Dermatol. Sci., 2001, 26, 217–232 CrossRef CAS.
- V. E. Reeve, M. Bosnic, C. Boehm-Wilcox, N. Nishimura and R. D. Ley, Ultraviolet A radiation (320-400 nm) protects hairless mice from immunosuppression induced by ultraviolet B radiation (280–320 nm) or cis-urocanic acid, Int. Arch. Allergy Immunol., 1998, 115, 316–322 CrossRef CAS.
- G. M. Halliday, S. N. Byrne, J. M. Kuchel, T. S. Poon and R. S. Barnetson, The suppression of immunity by ultraviolet radiation: UVA, nitric oxide and DNA damage, Photochem. Photobiol. Sci., 2004, 3, 736–740 RSC.
- A. A. El-Ghorr, F. Pierik and M. Norval, Comparative potency of different UV sources in reducing the density and antigen-presenting capacity of Langerhans cells in C3H mice, Photochem. Photobiol., 1994, 60, 256–261 CrossRef CAS.
- K. S. Yuen, M. R. Nearn and G. M. Halliday, Nitric oxide-mediated depletion of Langerhans cells from the epidermis may be involved in UVA radiation-induced immunosuppression, Nitric Oxide, 2002, 6, 313–318 CrossRef CAS.
- I. Iwai, M. Hatao, M. Naganuma, Y. Kumano and M. Ichihashi, UVA-induced immune suppression through an oxidative pathway, J. Invest. Dermatol., 1999, 112, 19–24 CrossRef CAS.
- M. P. Stapelberg, R. B. Williams, S. N. Byrne and G. M. Halliday, The alternative complement pathway seems to be a UVA sensor that leads to systemic immunosuppression, J. Invest. Dermatol., 2009, 129, 2694–2701 CrossRef CAS.
- S. Rana, S. N. Byrne, L. J. MacDonald, C. Y. Chan and G. M. Halliday, Ultraviolet B suppresses immunity by inhibiting effector and memory T cells, Am. J. Pathol., 2008, 172, 993–1004 Search PubMed.
- S. N. Byrne, J. Ahmed and G. M. Halliday, Ultraviolet B but not A radiation activates suppressor B cells in draining lymph nodes, Photochem. Photobiol., 2005, 81, 1366–170 CrossRef CAS.
- S. M. Kaech, J. T. Tan, E. J. Wherry, B. T. Konieczny, C. D. Surh and R. Ahmed, Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells, Nat. Immunol., 2003, 4, 1191–1198 CrossRef CAS.
- J. Kehren, C. Desvignes, M. Krasteva, M. Ducluzeau, O. Assossou, F. Horand, M. Hahne, D. Kagi, D. Kaiserlian and J. Nicolas, Cytotoxicity is mandatory for CD8+ T cell-mediated contact hypersensitivity, J. Exp. Med., 1999, 189, 779–786 CrossRef CAS.
- M. Saulnier, S. Huang, M. Aguet and B. Ryffel, Role of interferon-γ in contact hypersensitivity assessed in interferon-γ receptor-deficient mice, Toxicology, 1995, 102, 301–312 CrossRef CAS.
- D. He, L. Wu, H. K. Kim, H. Li, C. A. Elmets and H. Xu, CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses, J. Immunol., 2006, 177, 6852–6858 CAS.
- D. D. Kish, X. Li and R. L. Fairchild, CD8 T cells producing IL-17 and IFN-γ initiate the innate immune response required for responses to antigen skin challenge, J. Immunol., 2009, 182, 5949–5959 CrossRef CAS.
- F. M. Mbitikon-Kobo, M. Vocanson, M. C. Michallet, M. Tomkowiak, A. Cottalorda, G. S. Angelov, C. A. Coupet, S. Djebali, A. Marçais, B. Dubois, N. Bonnefoy-Bérard, J. F. Nicolas, C. Arpin and J. Marvel, Characterization of a CD44/CD122int memory CD8 T cell subset generated under sterile inflammatory conditions, J. Immunol., 2009, 182, 3846–3854 CrossRef CAS.
- T. Gebhardt, L. M. Wakim, L. Eidsmo, P. C. Reading, W. R. Heath and F. R. Carbone, Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus, Nat. Immunol., 2009, 10, 524–530 CrossRef CAS.
- H. Matsue, P. R. Bergstresser and A. Takashima, Keratinocyte-derived IL-7 serves as a growth factor for dendritic epidermal T cells in mice, J. Immunol., 1993, 151, 6012–6019 CAS.
- R. A. Clark and T. S. Kupper, IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin, Blood, 2007, 109, 194–202 CrossRef CAS.
- E. Teixeiro, M. A. Daniels, S. E. Hamilton, A. G. Schrum, R. Bragado, S. C. Jameson and E. Palmer, Different T cell receptor signals determine CD8+ memory versus effector development, Science, 2009, 323, 502–505 CrossRef CAS.
- J. T. Chang, V. R. Palanivel, I. Kinjyo, F. Schambach, A. M. Intlekofer, A. Banerjee, S. A. Longworth, K. E. Vinup, P. Mrass, J. Oliaro, N. Killeen, J. S. Orange, S. M. Russell, W. Weninger and S. L. Reiner, Asymmetric T lymphocyte division in the initiation of adaptive immune responses, Science, 2007, 315, 1687–1691 CrossRef CAS.
- J. Shen, S. Bao and V. E. Reeve, Modulation of IL-10, IL-12, and IFN-γ in the epidermis of hairless mice by UVA (320–400 nm) and UVB (280–320 nm) radiation, J. Invest. Dermatol., 1999, 113, 1059–1064 CrossRef CAS.
- J. Liu, T. Miwa, B. Hilliard, Y. Chen, J. D. Lambris, A. D. Wells and W. C. Song, The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo, J. Exp. Med., 2005, 201, 567–577 CrossRef CAS.
- P. N. Lalli, M. G. Strainic, F. Lin, M. E. Medof and P. S. Heeger, Decay accelerating factor can control T cell differentiation into IFN-γ-producing effector cells via regulating local C5a-induced IL-12 production, J. Immunol., 2007, 179, 5793–5802 CAS.
- D. X. Nghiem, N. Kazimi, G. Clydesdale, H. N. Ananthaswamy, M. L. Kripke and S. E. Ullrich, Ultraviolet A radiation suppresses an established immune response: implications for sunscreen design, J. Invest. Dermatol., 2001, 117, 1193–1199 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2010 |