Synthesis and room temperature photo-induced electron transfer in biologically active bis(terpyridine)ruthenium(II)–cytochrome c bioconjugates and the effect of solvents on the bioconjugation of cytochrome c†
Joshua R. Petersonab, Trevor A. Smithc and Pall Thordarson*a
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: p.thordarson@unsw.edu.au; Fax: +61-2-9385-6141; Tel: +61-2-9385-4478
bSchool of Chemistry, The University of Sydney, NSW 2006, Australia
cSchool of Chemistry, The University of Melbourne, VIC 3010, Australia
First published on 16th November 2009
Abstract
Photo-active bis(terpyridine)ruthenium(II) chromophores were synthesised and attached to the redox enzyme iso-1 cytochrome c in a mixed solvent system to form photo-induced bioconjugates in greater than 40% yield after purification. The effects of up to 20% (v/v) of acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide at 4, 25 and 35 °C on the stability and biological activity of cytochrome c and its reactivity towards the model compound 4,4′-dithiodipyridine (DTDP) was measured. The second-order rate constant for the DTDP reaction was found to range between k = 2.5–4.3 M−1 s−1 for reactions with 5% organic solvent added compared to k = 5.6 M−1 s−1 in pure water at 25 °C. Use of 20% solvent generally results in significant protein oxidation, and 20% acetonitrile and tetrahydrofuran in particular result in significant protein dimerization, which competes with the bioconjugation reaction. Cyclic voltammetry studies indicated that the rate of electron transfer to the heme in solution was reduced in the bis(terpyridine)ruthenium(II) cytochrome c bioconjugates compared to unmodified cytochrome c. Steady-state fluorescence studies on these bioconjugates showed that energy or electron transfer is taking place between the bis(terpyridine)ruthenium(II) chromophores and cytochrome c. The bis(terpyridine)ruthenium(II) cytochrome c bioconjugates demonstrate room temperature photo-activated electron transfer from the bis(terpyridine)ruthenium(II) donor to the protein acceptor. Two sacrificial donors were used; in 50% glycerol, the bioconjugates were reduced in about 15 min while in 20 mM EDTA the bioconjugates were fully reduced in less than 5 min upon irradiation with a xenon lamp source. Under these conditions, the reduction of the non-covalent mixture of cytochrome c and bis(terpyridine)ruthenium(II) mixtures took over 30 min. Control experiments showed that the photo-induced reduction of cytochrome c only occurs in the absence of oxygen and presence of a sacrificial donor. These results are encouraging for future incorporation of these bioconjugates in light-responsive bioelectronic circuits, including photo-activated biosensors and biofuel cells.
Introduction
The chemical modification or conjugation of biomolecules defines the field of bioconjugate chemistry1,2 which has become an increasingly important area of research at the interface of chemistry, biology and nanotechnology making possible the preparation of biologically active complexes with unique properties in solution3 or on surfaces.3,4 Light-activated bioconjugates use light energy to drive biological processes in such potential applications as bio-solar cells or photo-activated biosensors. Our goal is to develop light responsive bioconjugates using bis(terpyridine)ruthenium(II) chromophores to harvest light energy and the redox enzyme yeast iso-1 cytochrome c from Saccharomyces cerevisiae to convert that energy to biologically relevant work under normal physiological conditions.While many techniques have been developed to functionalize proteins,1,2,5,6 the proteins are not always stable to the conditions required for the reaction, especially if organic solvents are required.6–9 When site-specific attachment is required, the reactivity of the target residue may also be less than optimal due to steric or electronic reasons. The stability of enzymes in mixed-solvent systems has been studied extensively.7–12 Cytochrome c, in particular, has been studied for its stability in mixed-solvent systems by monitoring the Soret band – an indicator of protein conformation easily measured by UV-Vis spectroscopy.13–16 Significant changes in Soret peak intensity have been observed outside the pH range of 5.0 to 7.5 and with increasing solvent concentration.13,17 Dimethylformamide and tetrahydrofuran were found to be solvents of relatively low and high “denaturing power”, respectively.17 In addition, cytochrome c has been reported to catalyse oxidation of organic substrates,18–22 leading some investigators to monitor stability by measuring catalytic activity of the protein in mixed-solvent systems.20,23,24
The bioconjugation of cytochrome c has also been studied extensively, although significant focus has been on horse heart cytochrome c and the conditions reported are quite varied. Native yeast iso-1 cytochrome c has been functionalised at CYS102 by attachment of a bromo-methyl functionalized ligand in approximately 5% dimethylformamide, buffered with Tris-HCl at pH 8 for 16 h at room temperature.25 Yeast iso-1 cytochrome c modified electrodes have been prepared by reaction of CYS102 with maleimide26 and thiol27 functionalized surfaces. For maleimide–cysteine reactions in particular, the use of a chelating agent such as ethylenediaminetetraacetic acid (EDTA) may be beneficial as reactions between free thiols and maleimide groups have been shown to be dependent on the presence of chelators in certain circumstances.28 Despite the significant amount of data reported on the stability of cytochrome c in organic solvent mixtures, and the functionalisation of cytochrome c, there is little data on the reactivity of yeast iso-1 cytochrome c CYS102 in mixed solvent systems and there are no comprehensive reports of how exposure to mixed-solvent systems affects the biological activity of cytochrome c.
Although bis(terpyridine)ruthenium(II) chromophores are widely studied complexes with a great deal of literature devoted to both synthesis and their photophysical properties,29–32 the room temperature photo-induced electron transfer from these complexes to a suitable redox protein (such as cytochrome c) has not been well studied. In contrast, room temperature photo-induced electron transfer between tris(bipyridine)ruthenium(II) type complexes and cytochrome c has been well established through a number of studies33–39 including the use of sacrificial electron donors such as aniline40 and ethylenediaminetetraacetic acid (EDTA).41 The lack of data using bis(terpyridine)ruthenium(II)-based complexes is likely due to the much shorter fluorescent lifetimes (120 ps) and lower quantum yields (10−6) at room temperature29,42 compared to the tris(bipyridine)ruthenium(II)-based complexes (lifetime ∼ 1 ms and quantum yield > 10−2).43 However, terpyridine based complexes are synthetically appealing as the use of 4′-functionalised terpyridine ligands does not introduce complications of chirality, which in turn allows for the synthesis of symmetric complexes and simplifies the synthesis of asymmetric complexes. In addition, terpyridine-based complexes have been shown to reduce heme groups upon photo-activation – studies have included Ru(bipyridine)(terpyridine) complexes coordinated to histidine residues in plastocyanin to measure transient room temperature photo-induced election transfer44 and in yeast iso-1 cytochrome c to measure room temperature fluorescence.45 Room temperature electron transfer has also been studied in apomyoglobin reconstituted with a synthetic bis(terpyridine)ruthenium(II)–heme which resulted in a protein with characteristics nearly identical to myoglobin with the exception of the added chromophore.46 Reduction of the heme group was found to be essentially complete after 5 h of photoexcitation in 20 mM EDTA, pH 6.3 at 25 °C and initial rates varied significantly with pH and EDTA concentration. In addition, no photoreduction was observed for a comparable non-covalent mixture of [Ru(tpy)2]2+ (3, Scheme 1) and myoglobin implying that proximity through a covalent bond is required for the photoreduction to occur.
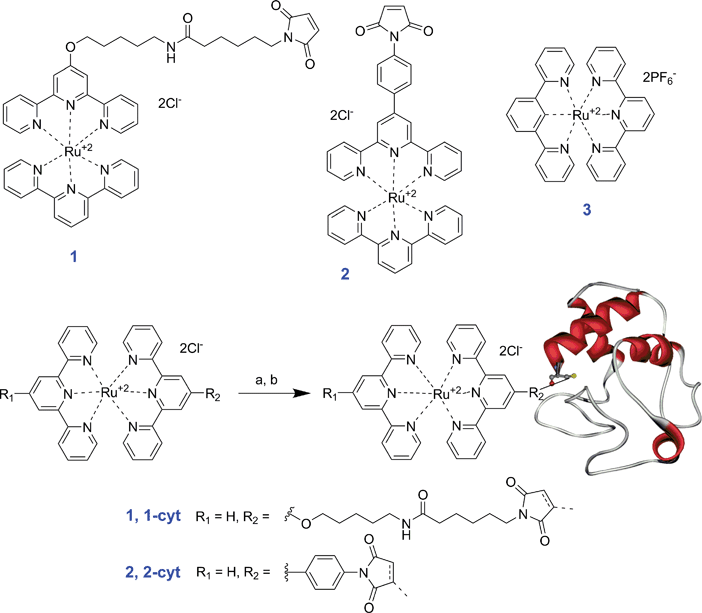 | ||
| Scheme 1 Bis(terpyridine)ruthenium(II) chromophores 1, 2 and 3 and synthesis of bioconjugates 1–cyt and 2–cyt. (a) 10 μM iso-1 cytochrome c, excess ligand (1) unknown concentration, 50 mM NaH2PO4, 15 mM EDTA, 5% CH3CN, pH 7.0, 35 °C, 22 h followed by purification on IMAC, 42%; (b) 8 μM iso-1 cytochrome c, excess ligand (2) unknown concentration, 50 mM NaH2PO4, 15 mM EDTA, 5% CH3CN, pH 7.0, 35 °C, 18 h followed by purification on IMAC, 47%. | ||
We have previously reported on the development of model biologically active light-activated bis(terpyridine)ruthenium(II)–cytochrome c bioconjugates 1–cyt and 2–cyt (Scheme 1)10 which were studied at low temperature (77 K) where increased luminescence quantum yields and lifetimes render measurements more feasible. Here we report additional low temperature luminescence data as well as results confirming room temperature photo-induced electron transfer in these bioconjugates resulting in the complete reduction of protein within 5 to 15 min in the presence of a sacrificial electron donor.
Despite the significant amount of data reported on the stability of cytochrome c in organic solvent mixtures, and the functionalisation of cytochrome c, there is little data on the reactivity of yeast iso-1 cytochrome c CYS102 in mixed solvent systems and there are no comprehensive reports of how exposure to mixed-solvent systems affects the biological activity of cytochrome c. In the course of this work, a study was carried out to determine conditions for the bioconjugation reaction which would lead to an increased reaction rate with high yield, yet maintain the protein in a native state, preserving its biological activity.
Results and discussion
Preparation of bioconjugates
Complexes 1, 2 and 3 were prepared according to literature procedures10,32,42 and the synthesis of bis(terpyridine)ruthenium(II)–cytochrome c bioconjugates 1–cyt and 2–cyt was carried out as previously reported (Scheme 1).10 It should be noted that while N-ethylmorpholine is used in some instances to increase the rate of ruthenium complex formation,47 its use in the synthesis of complexes 1 and 2 resulted in degradation of the maleimide moiety to maleamic acid (likely due to the reaction mixture pH effectively being increased to above 8.0 where maleimide groups have been reported to break down significantly).48 Additionally, it is noteworthy that while the bioconjugation reactions between cytochrome c and the chloride salts of complexes 1 and 2 proceeded to yield the desired bioconjugates in 40–50% yields, the limited solubility of the hexafluorophosphate salt complexes of 1 and 2 in water made it necessary to use acetonitrile as a co-solvent, prompting us to study further the stability and reactivity of cytochrome c in mixed solvent systems.Stability and reactivity of cytochrome c in mixed solvents
Two related studies were undertaken to address this question – the stability of yeast cytochrome c in mixed-solvent systems (to identify conditions in which the biological activity of the protein was retained), followed by reactivity studies using model cysteine selective compounds 4,4′-dithiodipyridine (DTDP) and N-(1-pyrenyl)-maleimide (NPM, Scheme 2). The popular cysteine selective alternative, Ellman's reagent, was not used due to overlapping of the characteristic UV-Vis peak with that of the main peak of cytochrome c. One of the motivations for these studies was our initial comparison of the reactivity of yeast iso-1 cytochrome c with that of the model protein bovine serum albumin (BSA) which indicated a significant inherent challenge to functionalising CYS102 (Fig. 1, note that 50% yield is considered to be complete reaction with BSA based on results published in the literature).49 | ||
| Scheme 2 Reactions of 4,4′-dithiodipyridine (DTDP) and N-(1-pyrenyl)-maleimide (NPM) with free sulfhydryls. | ||
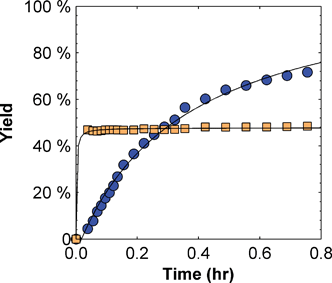 | ||
Fig. 1 Reactivity of iso-1 cytochrome c ( ) and bovine serum albumin (BSA) ( ) and bovine serum albumin (BSA) ( ) with 4,4′-dithiodipyridine (DTDP) at room temperature (8.5 μM protein, 200 μM DTDP, 50 mM NaH2PO4, 15 mM EDTA, pH 7.0). ) with 4,4′-dithiodipyridine (DTDP) at room temperature (8.5 μM protein, 200 μM DTDP, 50 mM NaH2PO4, 15 mM EDTA, pH 7.0). | ||
Biological stability was measured using the cytochrome c oxidase (CCOx) assay for water, 5% and 20% (v/v) solvent mixtures stored for 24 h at 4, 25 and 35 °C, and results for the initial activity with CCOx are shown in Table 1. Samples generally showed comparable activity (within experimental error) with samples stored at 35 °C often exhibiting lower initial activity than samples at 4 or 25 °C. The use of 20% (v/v) solvent, while often increasing protein oxidation (as discussed below), does not appear to have a significant impact on protein activity. All samples that were measured with CCOx showed some level of activity indicating that reactions under these conditions should not lead to significant loss of biological activity.
| Solvent (v/v) | 4 °C | 25 °C | 35 °C | |||
|---|---|---|---|---|---|---|
| 5% | 20% | 5% | 20% | 5% | 20% | |
| Water | 0.71 ± 0.19 | 1.00 ± 0.09 | 0.47 ± 0.16 | |||
| CH3CN | 0.46 ± 0.07 | 0.79 ± 0.52 | 0.63 ± 0.14 | 0.55 ± 0.08 | 0.74 ± 0.14 | 0.50 ± 0.07 |
| THF | 0.50 ± 0.07 | 0.58 ± 0.06 | 0.61 ± 0.14 | 0.92 ± 0.32 | 0.42 ± 0.01 | 0.54 ± 0.08 |
| DMF | 0.40 ± 0.07 | 0.74 ± 0.13 | 0.59 ± 0.02 | 0.91 ± 0.24 | 0.45 ± 0.01 | 0.70 ± 0.34 |
| DMSO | 0.55 ± 0.17 | 0.50 ± 0.09 | 1.15 ± 0.36 | 0.83 ± 0.09 | 0.55 ± 0.24 | 0.70 ± 0.24 |
Measurements of the UV-Vis spectrum indicate that the reduced protein is stable in water for up to 24 h at 4, 25 and 35 °C (Fig. 2, Panels A and C). Similar results were obtained for the 5% (v/v) solvent mixtures at 4, 25 and 35 °C and for the 20% (v/v) solvent mixtures at 4 °C (data not shown). However, in 20% (v/v) solvent mixtures protein oxidation is significant at 25 °C and even more pronounced at 35 °C as exemplified by the 20% dimethylformamide results (Fig. 2, Panels B and D). Protein oxidation was most pronounced in 20% tetrahydrofuran (total oxidation within 8 h at 25 °C and 1 h at 35 °C) followed by acetonitrile (24 h at 25 °C and 1 h at 35 °C), dimethylformamide (incomplete oxidation at 25 °C and total oxidation after 10 h at 35 °C), and dimethyl sulfoxide, which was comparable to water showing no significant protein oxidation after 24 h at 25 or 35 °C (see supplementary information†).
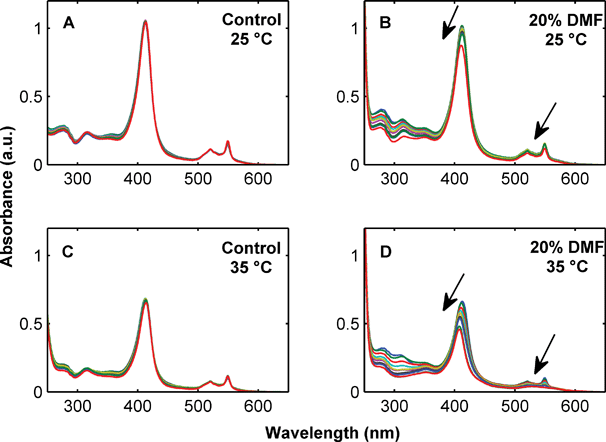 | ||
| Fig. 2 Stability of 10 μM cytochrome c over 24 h measured by UV-Vis absorbance in 20 mM NaH2PO4, 20 mM NaCl, 5 mM EDTA, pH 7.0 at 25 °C (Panel A) and 35 °C (Panel C) and 20 mM NaH2PO4, 20 mM NaCl, 5 mM EDTA, 20% DMF, pH 7.0 at 25 °C (Panel B) and 35 °C (Panel D). Arrows indicate decreasing UV absorbance over time. | ||
From the UV-Vis data alone, it is unclear whether the protein oxidation shown in Fig. 2 is limited to an Fe2+ to Fe3+ transition in the heme group, or if there is also an associated protein dimerization which would compete with the CYS102 bioconjugation reaction. To address this question, gel electrophoresis was performed to monitor dimer formation at 25 °C for up to 24 h in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 with or without 5% or 20% (v/v) solvents (results are shown in Fig. 3).
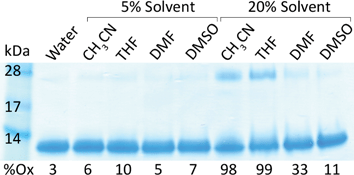 | ||
Fig. 3 Gel electrophoresis showing cytochrome c monomer (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 710 kDa), dimer (25420 kDa) and percent oxidation (%Ox) after 24 h at 25 °C in 20 mM NaH2PO4, 20 mM NaCl, 5 mM EDTA, pH 7.0 with or without 5% or 20% (v/v) solvent. 710 kDa), dimer (25420 kDa) and percent oxidation (%Ox) after 24 h at 25 °C in 20 mM NaH2PO4, 20 mM NaCl, 5 mM EDTA, pH 7.0 with or without 5% or 20% (v/v) solvent. | ||
It is clear from the results in Fig. 3 that 20% acetonitrile and 20% tetrahydrofuran exhibit the highest levels of protein oxidation and dimer formation, making these two conditions the least favourable for bioconjugation. However, it should be noted that while protein oxidation is an indicator of dimer formation, there is a far lesser degree of dimerization than oxidation. For example, in the case of 20% acetonitrile, the protein is nearly completely oxidized after 24 h at 25 °C, but it appears that only 20–25% of the protein is actually dimerized. Additional results indicate that 20% tetrahydrofuran (which leads to the most significant dimer formation) is ca. 5% and 10% dimerized after 2 and 8 h, respectively (data not shown). It should also be noted that the use of kosmotropes (such as glycine, sorbitol, etc.) may enhance the stability of the protein structure in these mixed solvent systems, although the use of such additives was beyond the scope of this study.
As expected, reactions between 4,4′-dithiodipyridine (DTDP) and cytochrome c in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 at 25 °C indicate that reagent concentration, ligand excess and temperature are all significant factors in determining the reaction rate and the extent of reaction (Table 2). Results indicate that using a 5-fold excess of ligand results in approximately 90% yield after 6.5 h while under the same conditions a stoichiometric amount of ligand results in a slower reaction that achieves only 70% yield after 13.5 h (entries 1 and 2, Table 2). Performing the reaction with 20-fold excess DTDP (entry 3) appears to provide a moderate advantage over the 5-fold excess reaction, reaching 90% yield in only 3 h. In addition, a reaction carried out at 4 °C (entry 5) results in a slower initial reaction that achieves only 50% yield after nearly 20 h. Neither removal of EDTA (entry 6), nor an increase to 20 mM (entry 7) appears to provide added benefit over the standard 5 mM EDTA reaction (entry 1).
| Entry | Solvent | [cyt]/μM | [DTDP]/μM | Second order rate constanta/M−1s−1 | Empirical initial rateb/10−9 M s−1 | Plateau yieldc(%) | Time to 90% of plateau yieldc/h |
|---|---|---|---|---|---|---|---|
| a Second order rate constant calculated from reaction data.b Average rate over first 2 h calculated from the empirical model.c Calculated from empirical model.d Results may be confounded by UV-Vis spectral shifts due to protein oxidation resulting in an artificially low plateau yield.e Results confounded by UV-Vis spectral shifts due to protein oxidation as described in the text. | |||||||
| 1 | Water | 10 | 50 | 5.60 | 1.16 | 108 | 6.6 |
| 2 | 10 | 10 | 9.23 | 0.65 | 80 | 13.6 | |
| 3 | 10 | 200 | 3.61 | 1.33 | 106 | 2.9 | |
| 4 | 100 | 500 | 3.87 | 10.6 | 90 | 1.0 | |
| 5 | 4 °C | 10 | 50 | 0.71 | 0.40 | 56 | 18.8 |
| 6 | 0 mM EDTA | 10 | 50 | 4.75 | 1.07 | 90 | 4.6 |
| 7 | 20 mM EDTA | 10 | 50 | 1.11 | 106 | 7.9 | |
| 8 | 5% CH3CN | 10 | 50 | 4.34 | 1.16 | 103 | 5.5 |
| 9 | 100 | 500 | 4.24 | 10.4 | 80d | 0.5d | |
| 10 | 5% THF | 10 | 50 | 2.54 | 1.01 | 87 | 5.3 |
| 11 | 5% DMF | 10 | 50 | 3.55 | 1.07 | 100 | 7.2 |
| 12 | 5% DMSO | 10 | 50 | 4.09 | 1.18 | 98 | 4.8 |
| 13 | 20% CH3CN | 100 | 500 | 1.22 | e | e | e |
Achievable ligand concentration will depend on the solubility of the ligand, so the results presented in Table 2 using water alone are not necessarily representative of bioconjugation reactions which require an organic co-solvent to solubilise the ligand. Therefore, a series of reactions were carried out at 25 °C by introducing DTDP dissolved in organic solvent (acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide) to a final concentration of 10 μM cytochrome c in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 with 50 μM DTDP and 5% (v/v) solvent (entries 8–12, Table 2). Surprisingly, there appears to be little difference between reactions with the water solubilised DTDP ligand and the ligand prepared in organic solvent. The calculated second-order rate constants range from k = 2.5 M−1 s−1 for 5% tetrahydrofuran to k = 4.3 M−1 s−1 for 5% acetonitrile, compared to k = 5.6 M−1 s−1 for DTDP in water alone under standard conditions at 25 °C (entry 1, Table 2). This indicates that the solvent mixture needs only to be able to solubilise the ligand to achieve bioconjugation reactions comparable to those of water alone. Again, this is more easily accomplished when the required ligand concentration is low, or it's solubility in water is high.
Given that reagent concentration has a significant effect on SN2 reactions, the reaction between cytochrome c and DTDP was studied at an elevated concentration of 100 μM protein, 500 μM DTDP in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 alone (entry 4, Table 2), with 5% acetonitrile (entry 9), or with 20% acetonitrile (entry 13). The results indicate that increasing the protein and ligand concentrations (to maintain 5-fold excess) has a significant effect on the reaction in both the phosphate buffer alone and in 5% (v/v) acetonitrile with the reactions achieving completion within 1 h, although yield may suffer (achieving a maximum of approximately 85%). The increased reaction rate is consistent with the fact that the second-order rate constant for these reactions is more or less the same. One complicating factor of analysing these mixed-solvent reactions is made apparent in the reaction with 20% (v/v) acetonitrile, which appears to proceed equivalently to the phosphate buffer and 5% reaction mixtures for the first 20 min, at which point yield decreases dramatically and eventually becomes negative (Fig. 4– Panel A).
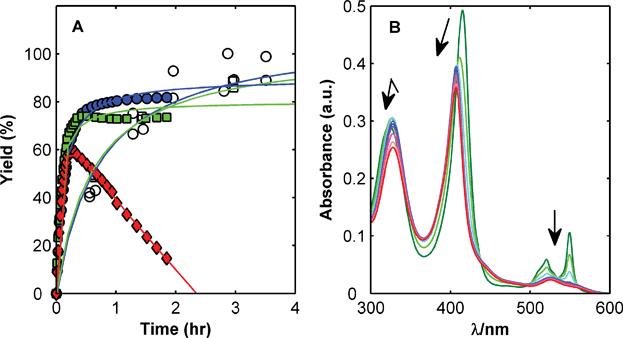 | ||
Fig. 4 Panel A – Effect of increased protein and ligand concentration on reactivity of CYS102 with DTDP. Reactions in 20 mM NaH2PO4, 20 mM NaCl, 5 mM EDTA, pH 7.0 with 10 μM cytochrome c, 50 μM DTDP (open markers) in water (○) and 5% CH3CN (□) and 100 μM cytochrome c, 500 μM DTDP (closed markers) in water ( ), 5% CH3CN ( ), 5% CH3CN ( ), and 20% CH3CN ( ), and 20% CH3CN ( ). Panel B – Confounding effect of protein oxidation on interpreting UV-Vis spectral changes resulting from reaction of cytochrome c and DTDP in 20% CH3CN at 25 °C. Arrows indicate changes in UV absorbance over time. ). Panel B – Confounding effect of protein oxidation on interpreting UV-Vis spectral changes resulting from reaction of cytochrome c and DTDP in 20% CH3CN at 25 °C. Arrows indicate changes in UV absorbance over time. | ||
This dramatic decrease in calculated yield is due to protein oxidation during the reaction which is characterised by changes in the UV-Vis spectrum (Fig. 4– Panel B). The DTDP reaction is monitored at 324 nm, which overlaps a region in the cytochrome c spectra that decreases substantially when the protein is oxidized. The end result is a reaction that appears to proceed normally, only to be confounded when the oxidation process becomes significant.
Similar studies were carried out for the bioconjugation of the N-(1-pyrenyl)-maleimide (NPM) fluorescent dye with cytochrome c. Although absolute rate constants for the reactivity of NPM could not be determined accurately, similar general trends were seen as for the reactivity of DTDP (see supplementary material†). To demonstrate successful formation of bioconjugate, a reaction was performed using 10 μM crude reduced cytochrome c, 50 μM NPM in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 with 20% (v/v) dimethylformamide at 25 °C for 24 h and the resulting bioconjugate characterized by UV-Vis, fluorescence and MALDI-TOF (see supplementary material†).
Characterisation of bis(terpyridine)ruthenium(II) bioconjugates
The photophysical and electrochemical characterisation of bioconjugates 1–cyt and 2–cyt was carried out using [Ru(tpy)2](PF6)2 (6) and iso-1 cytochrome c as control compounds. These studies focused on the photophysical and electrochemical properties of the bioconjugates to determine the oxidation and reduction potentials and to determine the photo-induced electron transfer properties as ‘proof of principle’ for the eventual attachment of ruthenium terpyridine bioconjugates to electrode surfaces. In addition to UV-Vis and MALDI-TOF characterisation of bioconjugates 1–cyt and 2–cyt,10 gel electrophoresis and cyclic voltammetry were performed to further characterise the purified bioconjugates. Gel electrophoresis results for bioconjugates 1–cyt and 2–cyt are shown in Fig. 5.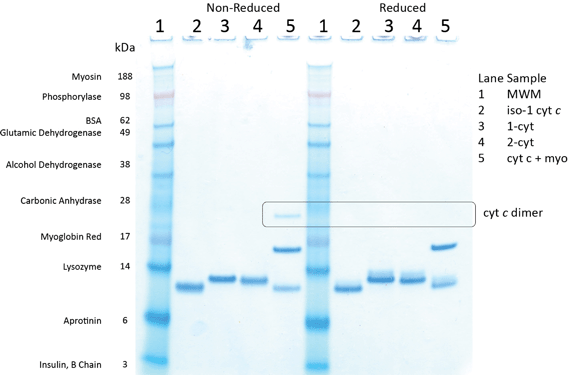 | ||
| Fig. 5 Gel electrophoresis of SeeBlue® Plus2 molecular weight marker (lane 1) and iso-1 cytochrome c (lane 2, calculated 12707), 1–cyt (lane 3, calculated 13568), 2–cyt (lane 4, calculated 13462) and a mixture of iso-1 cytochrome c and myoglobin (lane 5, calculated 16948). Samples on the left are not reduced while samples on the right are reduced with dithiothreitol (DTT). | ||
As expected, bioconjugates 1–cyt and 2–cyt (lanes 3 and 4, respectively, in Fig. 5) migrate as slightly larger species than unmodified iso-1 cytochrome c (lane 2 in Fig. 5). It is also noteworthy that while reduction with dithiothreitol (DTT) is capable of reducing cytochrome c disulfide dimer (indicated by the box in Fig. 5 and present in the non-reduced cytochrome c and myoglobin sample but absent in the reduced sample), it does not have an effect on the bioconjugates. This indicates that the maleimide–cysteine thioether bond cannot be reduced by a strong reducing agent such as DTT. The biological activity of these bioconjugates, assayed using cytochrome c oxidase, was also confirmed as described in our earlier communication.10
Cyclic voltammetry has been used to study not only the oxidation and reduction of cytochrome c over the last several decades, but also conformation, binding and electron transfer.50 Based on the literature, cyclic voltammetry was performed on [Ru(tpy)2](PF6)2 (3), iso-1 cytochrome c, a non-covalent mixture of [Ru(tpy)2](PF6)2 and cytochrome c (3:cyt), and bioconjugate 2–cyt. The results are shown in Fig. 6.
2 (3, black), a non-covalent mixture of [Ru(tpy)2](PF6)2 and iso-1 cytochrome c (3–cyt, green), and bioconjugate 2–cyt (red). Samples at 34 μM in 20 mM NaH2PO4, 0.1 M NaCl, pH 6.8. Glassy carbon electrode, Ag/AgCl reference electrode, Pt counter electrode, at 50 mV s−1.](/image/article/2010/OB/b919289a/b919289a-f6.gif) | ||
| Fig. 6 Cyclic voltammetry of iso-1 cytochrome c (blue), [Ru(tpy)2](PF6)2 (3, black), a non-covalent mixture of [Ru(tpy)2](PF6)2 and iso-1 cytochrome c (3–cyt, green), and bioconjugate 2–cyt (red). Samples at 34 μM in 20 mM NaH2PO4, 0.1 M NaCl, pH 6.8. Glassy carbon electrode, Ag/AgCl reference electrode, Pt counter electrode, at 50 mV s−1. | ||
The Faradaic oxidation and reduction peaks of iso-1 cytochrome c are visible in the protein alone and the non-covalent mixture at ca. 0.2 V (Fig. 6, blue and green traces respectively), but not in bioconjugate 2–cyt. This result is characteristic of bioconjugate 1–cyt as well (data not shown) and indicates possible impedance to electron transfer in bioconjugates 1–cyt and 2–cyt that is not present in the pure protein or the mixture.
Low temperature luminescence of bioconjugates
We have previously reported on luminescent lifetimes of bioconjugates 1–cyt and 2–cyt at 77 K in 50% glycerol (v/v).10 In addition to low temperature lifetime measurements, steady state luminescence emission was measured at 77 K (to visualize the quenching effects) for [Ru(tpy)2](PF6)2 (3) and bioconjugates 1–cyt and 2–cyt. Samples were prepared at similar concentrations (as measured by UV-Vis and shown in Fig. 3), then freeze–thaw degassed and measured over a range of excitation wavelengths from 250 to 550 nm, monitoring emission from 360 to 850 nm to generate a 3-dimensional luminescence map (Fig. 7).2 (3, black), non-covalent mixture of [Ru(tpy)2](PF6)2 and iso-1 cytochrome c (3–cyt, green) and bioconjugate 2–cyt (red) in 50% glycerol at 77 K. The diagonal series of peaks from λex = 350, λem = 350 to λex = 550, λem = 550 are due to scattering of the incident excitation light. The diagonal series of peaks from λex = 250, λem = 500 to λex = 425, λem = 850 and the peaks at ca.λex = 275, λem = 825 (3:cyt and 2–cyt) are due to 2nd and 3rd order harmonics of the residual incident excitation light. Fluorescence intensity (arbitrary units) as indicated by the color map.](/image/article/2010/OB/b919289a/b919289a-f7.gif) | ||
| Fig. 7 UV-Vis and steady state luminescence emission of [Ru(tpy)2](PF6)2 (3, black), non-covalent mixture of [Ru(tpy)2](PF6)2 and iso-1 cytochrome c (3–cyt, green) and bioconjugate 2–cyt (red) in 50% glycerol at 77 K. The diagonal series of peaks from λex = 350, λem = 350 to λex = 550, λem = 550 are due to scattering of the incident excitation light. The diagonal series of peaks from λex = 250, λem = 500 to λex = 425, λem = 850 and the peaks at ca.λex = 275, λem = 825 (3:cyt and 2–cyt) are due to 2nd and 3rd order harmonics of the residual incident excitation light. Fluorescence intensity (arbitrary units) as indicated by the color map. | ||
From the data shown in Fig. 7, it is apparent that not only are there multiple effective excitation wavelengths for the [Ru(tpy)2]2+ chromophore, but that the luminescence intensity of the free chromophore 3 (maximum intensity of 550 at λex = 440 nm, λem = 600 nm) is greater than that in the non-covalent mixture 3:cyt (400 at λex = 480 nm, λem = 600 nm, 27% quenching compared to 3) and even greater than in the bioconjugate 2–cyt (ca. 100 at λex = 480 nm, λem = 625 nm, 80% quenching compared to 3). This quenching effect in the bioconjugate is indicative of electron or energy transfer between the excited chromophore and the protein, in line with our reported luminescence lifetime studies and the observed reduction of 1–cyt in 50% glycerol after excitation with 480 nm light.10
Room temperature photo-induced electron transfer
Studies on the room temperature photo-induced electron transfer in bioconjugates 1–cyt and 2–cyt were performed by irradiation of bioconjugate solutions with a high powered xenon lamp in the presence of glycerol and EDTA.46 Samples were initially prepared in 50% glycerol (v/v) and include iso-1 cytochrome c alone, a non-covalent mixture of iso-1 cytochrome c and [Ru(tpy)2](PF6)2 (3:cyt), and bioconjugate 1–cyt. Samples were degassed in a cuvette modified for use under high vacuum using the freeze–thaw method then exposed to light from a high powered xenon lamp (450 W) fitted with a 2 mm iris to restrict the amount of outgoing light (thereby reducing power input to the sample and limiting heating effects). The photoreduction of bioconjugate 1–cyt and the control samples (iso-1 cytochrome c and 3:cyt) was followed by monitoring UV-Vis absorbance from 250 to 650 nm after a timed period of exposure to the xenon lamp. The spectra were then deconvoluted to estimate the molar percentage of reduced protein using representative spectra for oxidized and reduced iso-1 cytochrome c and [Ru(tpy)2](PF6)2 (3). The results are shown in Fig. 8.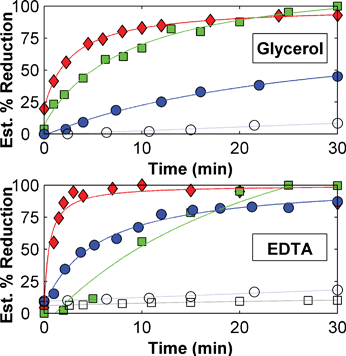 | ||
Fig. 8 Room temperature photoreduction of iso-1 cytochrome c ( ), 3:cyt ( ), 3:cyt ( ) and 1–cyt ( ) and 1–cyt ( ) in degassed 50% glycerol (v/v) or phosphate buffer with 20 mM EDTA. Iso-1 cytochrome c (○) and 3:cyt (□) in phosphate buffer alone shown for comparison. Samples exposed to light from xenon lamp with 2 mm iris. ) in degassed 50% glycerol (v/v) or phosphate buffer with 20 mM EDTA. Iso-1 cytochrome c (○) and 3:cyt (□) in phosphate buffer alone shown for comparison. Samples exposed to light from xenon lamp with 2 mm iris. | ||
As shown in Fig. 8, bioconjugate 1–cyt appears to be reduced over a period of 10 to 15 min in 50% (v/v) glycerol and within 5 min in 20 mM EDTA, while the non-covalent mixture 3:cyt appears to be fully reduced after 30 min. The iso-1 cytochrome c protein itself is also significantly photoreduced in the presence of glycerol and EDTA. This indicates that a significant contribution to the reduction of the protein does not come from the ruthenium terpyridine chromophore. It is possible that radicals generated by the photoexcitation of amino acids such as tyrosine or tryptophan may be responsible for the reduction of the protein heme group. Glycerol and EDTA appear to act as the sacrificial donors as protein in phosphate buffer shows only limited, if any, photoreduction. Also, while the electron transfer rate was on the order of 105 s−1 at 77 K,10 room temperature reduction of the entire sample takes several minutes, indicative of limited metal-to-ligand charge transfer at room temperature in bis(terpyridine)ruthenium(II) complexes and dependence on the nature of the light source. Studies with bioconjugate 2–cyt indicate behaviour similar to that of 1–cyt (data not shown).
Further studies investigated the effect of EDTA and oxygen removal (by freeze–thaw degassing or by alternating between a mild vacuum of ca. 200 mbar and a nitrogen overlay). Three samples of 3:cyt were prepared – (1) in degassed water with EDTA, (2) in degassed water without EDTA, and (3) in water with EDTA but not degassed. The samples were exposed to light from the xenon lamp and after 10 min, sample (2) was adjusted to 20 mM EDTA and degassed prior to additional irradiation. After 30 min, sample (3) was degassed prior to additional xenon lamp exposure. The results (shown in Fig. 9– Panel A) indicate that the presence of EDTA and degassing are both critical to the photoreduction of 3:cyt. Similar behaviour has been observed for 1–cyt (data not shown).
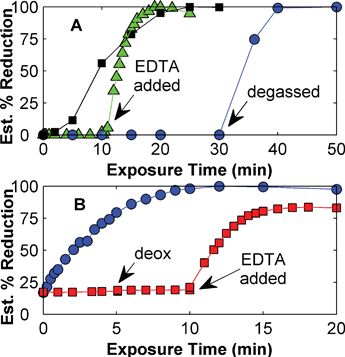 | ||
Fig. 9 Panel A – room temperature photoreduction of 3:cyt in 20 mM EDTA, pH 7.0 degassed (■) and not-degassed ( ) or in degassed 20 mM NaH2PO4, 20 mM NaCl, pH 7.0 ( ) or in degassed 20 mM NaH2PO4, 20 mM NaCl, pH 7.0 ( ). Samples exposed to light from xenon lamp. Panel B – room temperature photoreduction of 1–cyt in deoxygenated 20 mM EDTA ( ). Samples exposed to light from xenon lamp. Panel B – room temperature photoreduction of 1–cyt in deoxygenated 20 mM EDTA ( ), or in water alone ( ), or in water alone ( ) with deoxygenation and addition of 20 mM EDTA performed at the indicated time points. Samples exposed to light from mercury lamp. ) with deoxygenation and addition of 20 mM EDTA performed at the indicated time points. Samples exposed to light from mercury lamp. | ||
Additional measurements were made using bioconjugate 1–cyt to determine how the light source (using a 100 W mercury lamp instead of the 450 W xenon lamp) and modest oxygen removal (alternating vacuum and nitrogen overlay) may affect photoreduction. Two samples of 1–cyt were prepared – (1) in water with 20 mM EDTA, pH 7.0 deoxygenated, and (2) in water without EDTA and not deoxygenated. Both samples were exposed to unfiltered light from the mercury lamp. After 5 min, sample (2) was deoxygenated, then after an additional 5 min of irradiation EDTA was added to 20 mM and the sample was again deoxygenated and further irradiated. As shown in Fig. 9– Panel B, the removal of oxygen and the presence of EDTA are both necessary for the photoreduction of bioconjugate 1–cyt, in agreement with the results discussed above. This behaviour has been confirmed for the xenon lamp and for bioconjugate 2–cyt (data not shown).
Conclusions
Photo-active bis(terpyridine)ruthenium(II) chromophores were successfully synthesised and covalently bound to yeast iso-1 cytochrome c to form light-activated donor–acceptor bioconjugates. In the course of this work yeast cytochrome c was found to be biologically stable in water with up to 20% (v/v) acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide for 24 h at temperatures up to 35 °C. However, protein oxidation is likely in the 20% (v/v) mixtures at 25 and 35 °C and the accompanying dimerization in 20% acetonitrile and 20% tetrahydrofuran may result in an unacceptable loss in yield. Using DTDP as a probe, the reactivity of cytochrome c in mixed solvent systems was also optimised. Under standard reaction conditions (20 mM sodium dihydrogen phosphate, 20 mM sodium chloride, 5 mM EDTA, 10 μM cytochrome c, 50 μM reagent (DTDP or NPM), 5% (v/v) solvent), pH 7.0 at 25 °C, the reaction generally reached completion in good yield after 6–8 h. It may be of benefit to decrease the phosphate buffer pH to 6.5 to 7.0 prior to addition of organic solvent to decrease the reaction time. Also, if ligand solubility is poor and 20% (v/v) solvent is required, dimethylformamide and dimethyl sulfoxide are preferred to avoid protein dimerization. These results might be of general relevance for other protein bioconjugation studies, especially where the reactivity of the target residue is compromised due to steric or electronic reasons and organic solvents are required to solubilise the ligands, as is the case in our current work on light-activated bis(terpyridine)ruthenium(II) cytochrome c bioconjugates.Cyclic voltammetry studies on indicated that the electron transfer rates to the heme are reduced in the bis(terpyridine)ruthenium(II) cytochrome c bioconjugates compared to cytochrome c itself. Low temperature luminescence measurements of the bioconjugates at 77 K in 50% glycerol (v/v) show significant quenching which indicates energy or electron transfer from the ruthenium terpyridine chromophores to the protein. Furthermore, irradiation at room temperature results in photo-induced electron transfer to the cytochrome c acceptor, although a portion of the reduction appears to be unrelated to the bis(terpyridine)ruthenium(II) chromophore – possibly due to radical generation and propagation from amino acid residues such as tyrosine or tryptophan. However, it is clear that electron transfer from the bis(terpyridine)ruthenium(II) chromophore excited state to the cytochrome c heme group is much more effective and is the major contributor to photo-induced electron transfer. Based on these results, bis(terpyridine)ruthenium(II) cytochrome c bioconjugates are a promising candidate as building block for novel light-activated devices including photo-activated biosensors and hybrid solar biofuel cells.51
Experimental
Chemicals, solvents and materials
Chemicals were purchased from Sigma Aldrich with the exception of sodium dihydrogen phosphate (Ajax Finchem Pty. Ltd.). Acetonitrile (CH3CN) and tetrahydrofuran (THF) were either distilled over appropriate drying agents or obtained from a Pure Solv dry solvent system (Innovative Technology, Inc. model #PS-MD-7) to remove additives. Dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich and used as received. Yeast cytochrome c from Saccharomyces cerevisiae and bovine serum albumin (BSA) were purchased from Sigma Aldrich (catalog numbers C-2436 and A-0281, respectively) and the former purified prior to use by strong cation exchange chromatography to yield pure iso-1 cytochrome c.52 Cytochrome c oxidase (CCOx) was purchased from Sigma as part of the cytochrome oxidase assay kit (catalog number CYTOCOX1) and buffers for the CCOx activity assay were prepared from those included in the kit. Phosphate buffers were prepared with concentrated stock solutions of sodium dihydrogen phosphate (1 M, adjusted to pH 7.0 with aqueous sodium hydroxide (5 M) to eliminate the need for adjusting the pH of small volume solutions), sodium chloride (1 M), ethylenediaminetetraacetic acid (EDTA, 200 mM) and diluted with deionised water (Milli-Q Ultrapure Water System, Millipore) as necessary (typically 50-fold). All aqueous stock solutions were pH adjusted (if necessary) using a Scholar 425 pH meter (Corning) and filtered 0.2 μm (Millipore, 47 mm regenerated cellulose) prior to use.UV-Vis spectra were measured using either a Varian Cary 1E UV-Vis, or a Cary 5E UV-Vis-NIR. Room temperature fluorescence measurements were made using a Varian Cary Eclipse fluorescence spectrophotometer with emission and excitation slits at 5 nm, excitation filter “auto”, emission filter “open”, and PMT Voltage set to “medium”, unless otherwise stated. MALDI-TOF mass spectra were recorded on either a Micromass Tof Spec 2E or an Applied Biosystems Voyager DE STR MALDI reflectron TOFMS (protein and bioconjugate measurements were made in linear mode).
Protein and bioconjugate purification was performed using a GE Healthcare Akta Purifier. Cation exchange chromatography (CEX) was performed using either strong cation exchange column (Sigmachrom IEX-S or TSKgel SP-5PW, Supelco) or a weak cation exchange column (HiPrep 16/10 CM FF, GE Healthcare). Immobilized metal affinity chromatography (IMAC) was performed using either a HisTrap HP (1 mL) or a HisPrep FF 16/10 (20 mL, GE Healthcare, charged with Ni2+). Cyclic voltammetry was performed on a BAS 100B Electrochemical Analyser (BASi). Solution cyclic voltammetry of bioconjugates 1–cyt and 2–cyt, iso-1 cytochrome c and [Ru(tpy)2](PF6)2 (3) was performed in 20 mM phosphate buffer, 0.1 M sodium chloride, pH 6.8 with a glassy carbon working electrode, Ag/AgCl reference electrode and platinum counter electrode.
Synthesis of 1–cyt
To a solution of 500 mM sodium dihydrogen phosphate, 150 mM ethylenediaminetetraacetic acid, pH 7.0 (4 mL) in water (33.2 mL) at 35 °C was added [Ru(tpy)(4′-(maleimide-alkyloxy)-2,2′:6′,2″-terpyridine)]Cl2 (1) (crude, estimated to be in at least 10-fold excess based on UV-Vis absorbance) in acetonitrile (2 mL). Purified, reduced iso-1 cytochrome c (4.8 mg, 0.38 μmol) in water (0.8 mL) was then added and the resulting solution stirred in darkness at room temperature for 22 h. The reaction mixture was then concentrated, dialysed into water and purified by immobilized metal affinity chromatography (IMAC, HisPrep FF 16/10, GE Healthcare) using a gradient from 0 to 75 mM imidazole in 20 mM sodium dihydrogen phosphate, 0.5 M sodium chloride, pH 7.0 in 100 mL at 3 mL min−1. The product fraction (eluting from 35 to 85 mL) was pooled, concentrated, and dialysed into water to yield bioconjugate 1–cyt. (0.16 μmol, 42%). MS (MALDI) m/z 13566 ([M − 2Cl]+ requires 13568). MS (ESI) m/z 13567 ([M − 2Cl]+ requires 13568).Synthesis of 2–cyt
A solution of [Ru(tpy)(4′-(maleimide-phenyl)-2,2′:6′,2″-terpyridine)]Cl2 (2) (crude, estimated to be in at least 10-fold excess based on UV-Vis absorbance) in acetonitrile (1 mL) was added to a solution of 500 mM sodium dihydrogen phosphate, pH 7.0, 150 mM ethylenediaminetetraacetic acid (2 mL) in water (16 mL) at 35 °C. Purified, reduced iso-1 cytochrome c (2.0 mg, 0.16 μmol) in water (0.909 mL) was then added and the resulting solution stirred in darkness at 35 °C for 18 h. The reaction mixture was then concentrated, dialysed into water and purified by immobilized metal affinity chromatography (IMAC, HisPrep FF 16/10, GE Healthcare) using a gradient from 0 to 75 mM imidazole in 20 mM sodium dihydrogen phosphate, 0.5 M sodium chloride, pH 7.0 in 100 mL at 3 mL min−1. The product fraction (eluting from 35 to 85 mL) was pooled, concentrated, and dialysed into water to yield bioconjugate 2–cyt. (0.075 μmol, 47%). MS (MALDI) m/z 13465 ([M − 2Cl + OH]+ requires 13462). MS (ESI) m/z 13464 ([M − 2Cl + OH]+ requires 13462).Stability in mixed solvents measured by cytochrome c oxidase assay
Crude yeast cytochrome c was prepared at 66.7 μM in water or mixed solvent systems (up to 20% (v/v) of acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide). The samples were incubated at 4, 25, or 35 °C for up to 24 h during which samples were removed, reduced with dithiothreitol (DTT, 5 μL of 0.5 mM DTT added to 15 μL protein solution) and diluted to 12.5 μM cytochrome c with 17.3 mM Tris-HCl, 15.1 mM sucrose, 200 mM potassium chloride, pH 7.0 for analysis by UV-Vis spectroscopy (monitored between 250 and 650 nm). Cytochrome c oxidase solution was then injected (final conditions 10 μM cytochrome c, 12.2 mM Tris-HCl, 54.1 mM sucrose, 120 mM potassium chloride, pH 7.0 with a catalytic amount of cytochrome c oxidase) and decreasing UV-Vis absorbance at 550 nm was followed to measure the initial rate of cytochrome c oxidation. The data were fit with exponential decay functions using MATLAB and the initial rates of reaction (−dA550/dt) were calculated from the model equations and normalized to that of water at 25 °C.Oxidation and dimerization of cytochrome c in mixed solvents
Reduced, crude yeast cytochrome c was prepared at 12.5 μM in 20 mM sodium phosphate, 20 mM sodium chloride, 5 mM EDTA, pH 7.0 and stored for up to 24 h at 25 °C. Samples were removed at 0, 2, 8 and 24 h and analysed by UV-Vis spectroscopy (monitored between 250 and 650 nm) to determine the extent of protein oxidation. Protein dimerization was monitored using gel electrophoresis by mixing 15 μL of protein sample with 5 μL of sample buffer (4×) and loading 17 μL of the mixture onto the gel (2 μg protein/well).Reactivity of CYS102 in mixed solvents
Standard conditions for the reactions were 20 mM phosphate buffer, 20 mM sodium chloride, 5 mM ethylenediaminetetraacetic acid (EDTA), 10 μM cytochrome c, 50 μM 4-4′-dithiodipyridine (DTDP) or N-(1-pyrenyl)-maleimide (NPM), 5–20% (v/v) solvent. Reaction mixtures were prepared from stock solutions of sodium dihydrogen phosphate (1 M, pH 7), sodium chloride (1 M), ethylenediaminetetraacetic acid (EDTA, 200 mM, pH 7), and cytochrome c (200 μM) in water. A stock solution of 4 mM DTDP in water was prepared by stirring a mixture of DTDP (11.2 mg) in water (12.7 mL) and concentrated hydrochloric acid (12.7 μL) until all was dissolved.49 The 4 mM stock was stored at −20 °C until needed, at which point an aliquot was diluted 4-fold to 1 mM. Stock solutions of DTDP in organic solvent (10 or 1 mM) were prepared on the day of use by dissolving DTDP in the appropriate solvent (acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide). Stock solutions of NPM (10 mM) were prepared on the day of use and diluted to 2.5, 1 or 0.25 mM as needed using the appropriate solvent (acetonitrile, tetrahydrofuran, dimethylformamide, or dimethyl sulfoxide). The stock solutions were then added to 5 or 20% of the final volume to initiate the reaction. Detailed procedures for the preparation of reaction mixtures from stock solutions are provided in the supplementary information.† DTDP reactions were monitored by increasing absorbance at 324 nm using the previously reported extinction coefficient ε324 = 21400 M−1cm−1.49 DTDP reactions at 100 μM cytochrome c were measured neat in a sealed 1 mm path length cuvette. NPM reactions were monitored by increasing luminescence emission at 395 nm (λex = 342 nm, slit widths 5 nm, PMT Voltage “medium”). Reaction data were fit with rational functions using MATLAB and results were calculated from the model equations (described further in the supplementary information).Gel electrophoresis of bioconjugates
Gel electrophoresis was performed using Invitrogen Novex® NuPage® 12% Bis-Tris, 1 mm, 10-well gels, SeeBlue® Plus2 molecular weight marker, NuPage® LDS Sample Buffer (4×), NuPage® Sample Reducing Agent (10×), NuPage® MES SDS Running Buffer, SimplyBlue™ Safestain and the gels run using a Zoom Dual Power supply (model ZP10002, Invitrogen). Samples for gel electrophoresis were prepared by dilution in Novex® NuPage® LDS Sample Buffer (Invitrogen). If the samples were to be reduced (to eliminate disulfide dimers), NuPage® Sample Reducing Agent (active ingredient dithiothreitol (DTT)) was added. Samples were then heated at 70 °C for 10 min to denature the protein. Novex® NuPage® gels (12% Bis-Tris, 12-wells) were then loaded with 1 μg of protein per well, run at constant 200 V and stained according to the procedure included with SimplyBlue™ Safestain.Low temperature fluorescence measurements
Low temperature fluorescence measurements were made using an Oxford Instruments Optistat DN with an Intelligent Temperature Controller ITC601. Steady-state low temperature measurements were made using a Varian Cary Eclipse fluorescence spectrophotometer and Varian Cary 50 Bio UV spectrometer. Samples were prepared in glycerol/water (50:50 (v/v)) solutions and degassed using a freeze–thaw technique (liquid nitrogen) with a specialized cuvette and vacuum pump system (pressure less than 10−5 mbar).Room temperature photo-induced electron transfer measurements
Samples for room temperature photo-induced electron transfer were prepared by dissolution in either glycerol/water (50:50 (v/v)) or in water with ethylenediaminetetraacetic acid (EDTA). Specialized degassing cuvettes were designed and built with either 5 or 10 mm path length quartz fluorescence cuvettes to allow UV-Vis measurements to be made under oxygen free conditions. Cytochrome c and bioconjugate samples were prepared at 5 to 20 μM depending on the path length of the cuvette such that absorbance at 410 nm would be greater than or equal to 0.5 absorbance units. Samples with non-covalent mixtures of cytochrome c and [Ru(tpy)2](PF6)2 (3) were prepared by dilution of a stock protein solution to 5 to 20 μM and injection of a stock solution of [Ru(tpy)2](PF6)2 (3) in acetonitrile such that the total volume would be less than 20% acetonitrile (v/v, typically 5%). Samples were either degassed by freeze–thaw (the sample was frozen in liquid nitrogen, placed under high vacuum (10−3 mbar) until a stable pressure was achieved, then sealed and thawed and the procedure repeated until gases liberated during thawing were minimal) or deoxygenated using house vacuum (100 mbar) and nitrogen overlay. Once prepared, the samples were exposed to light from an Oriel Basic Power Supply (model 68806, 50–200 watts) fitted with either a xenon (450 W) or mercury (100 W) lamp and UV-Vis measurements were taken using a Varian Cary 5 at various time intervals.
Acknowledgements
We would like to thank Dr. Allan G. Larsen for assistance with cyclic voltammetry measurements and Dr. Timothy W. Schmidt at the University of Sydney and Dr. Graham E. Ball at the University of New South Wales for access to high-powered xenon and mercury light sources. This work was supported by the Australian Research Council (ARC DP0666325) and an ARC Australian Research Fellowship to PT and an ARC/NHMRC FABLS Network grant to PT and TAS. JRP was funded by the EIPRS and IPA scholarships from The University of Sydney.References
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, Inc., San Diego, California, 1996 Search PubMed.
- C. M. Niemeyer, ed., Bioconjugation Protocols: Strategies and Methods, Humana Press, Totowa, New Jersey, 2004 Search PubMed.
- I. Willner and E. Katz, in Bioelectronics: From Theory to Applications, eds. I. Willner and E. Katz, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, pp. xiii–xiv Search PubMed.
- D. C. Goldstein, P. Thordarson and J. R. Peterson, Aust. J. Chem., 2009, 62, 1320–1327 CrossRef CAS.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243–254 CrossRef CAS.
- M. Brinkley, Bioconjugate Chem., 1992, 3, 2–13 CrossRef CAS.
- L. Yang, J. S. Dordick and S. Garde, Biophys. J., 2004, 87, 812–821 CrossRef CAS.
- G. R. Castro and T. Knubovets, Crit. Rev. Biotechnol., 2003, 23, 195–231 CrossRef CAS.
- B. Castillo, Y. Pacheco, W. Al-Azzam, K. Griebenow, M. Devi, A. Ferrer and G. Barletta, J. Mol. Catal. B: Enzym., 2005, 35, 147–153 CrossRef CAS.
- J. R. Peterson, T. A. Smith and P. Thordarson, Chem. Commun., 2007, 1899–1901 RSC.
- R. M. Guinn, H. W. Blanch and D. S. Clark, Enzyme Microb. Technol., 1991, 13, 320–326 CrossRef CAS.
- L. M. Simon, M. Kotormán, A. Szabó, J. Nemcsók and I. Laczkó, Process Biochem., 2007, 42, 909–912 CrossRef CAS.
- L. S. Kaminsky and A. J. Davison, Biochemistry, 1969, 8, 4631–4637 CrossRef CAS.
- F. Sinibaldi, B. D. Howes, M. C. Piro, P. Caroppi, G. Mei, F. Ascoli, G. Smulevich and R. Santucci, J. Biol. Inorg. Chem., 2006, 11, 52–62 CrossRef CAS.
- C.-H. Lee, J. Lang, C.-W. Yen, P.-C. Shih, T.-S. Lin and C.-Y. Mou, J. Phys. Chem. B, 2005, 109, 12277–12286 CrossRef CAS.
- C. Bongiovanni, F. Sinibaldi, T. Ferri and R. Santucci, J. Protein Chem., 2002, 21, 35–41 CrossRef CAS.
- L. S. Kaminsky and A. J. Davison, FEBS Lett., 1969, 3, 338–340 CrossRef CAS.
- R. Radi, L. Thomson, H. Rubbo and E. Prodanov, Arch. Biochem. Biophys., 1991, 288, 112–117 CAS.
- N. H. Kim, M. S. Jeong, S. Y. Choi and J. H. Kang, Bull. Korean Chem. Soc., 2004, 25, 1889–1892 CAS.
- R. Vazquez-Duhalt, D. W. S. Westlake and P. M. Fedorak, Enzyme Microb. Technol., 1993, 15, 494–499 CrossRef CAS.
- L. Gębicka, Res. Chem. Intermed., 2001, 27, 717–723 CrossRef CAS.
- T. Yamada, K. Kikawa, S. Shinoda and H. Tsukube, Tetrahedron Lett., 1999, 40, 6967–6970 CrossRef CAS.
- E. Torres, J. V. Sandoval, F. I. Rosell, A. G. Mauk and R. Vazquez-Duhalt, Enzyme Microb. Technol., 1995, 17, 1014–1020 CrossRef CAS.
- R. Vazquez-Duhalt, K. M. Semple, D. W. S. Westlake and P. M. Fedorak, Enzyme Microb. Technol., 1993, 15, 936–943 CrossRef CAS.
- L. Geren, S. Hahm, B. Durham and F. Millett, Biochemistry, 1991, 30, 9450–9457 CrossRef CAS.
- E. Katz and I. Willner, J. Am. Chem. Soc., 2003, 125, 6803–6813 CrossRef CAS.
- M. Gerunda, C. A. Bortolotti, A. Alessandrini, M. Sola, G. Battistuzzi and P. Facci, Langmuir, 2004, 20, 8812–8816 CrossRef CAS.
- S. Papp, M. Rutzke and A. Martonosi, Arch. Biochem. Biophys., 1985, 243, 254–263 CrossRef CAS.
- J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez and C. Coudret, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147–155 RSC.
- U. S. Schubert, H. Hofmeier and G. R. Newkome, Modern Terpyridine Chemistry, WILEY-VCH, Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006 Search PubMed.
- H. Hofmeier, P. R. Andres, R. Hoogenboom, E. Herdtweck and U. S. Schubert, Aust. J. Chem., 2004, 57, 419–426 CrossRef CAS.
- B. Durham, L. P. Pan, J. E. Long and F. Millett, Biochemistry, 1989, 28, 8659–8665 CrossRef CAS.
- G. Engstrom, R. Rajagukguk, A. J. Saunders, C. N. Patel, S. Rajagukguk, T. Merbitz-Zahradnik, K. Xiao, G. J. Pielak, B. Trumpower, C.-A. Yu, L. Yu, B. Durham and F. Millett, Biochemistry, 2003, 42, 2816–2824 CrossRef CAS.
- L. P. Pan, M. Frame, B. Durham, D. Davis and F. Millett, Biochemistry, 1990, 29, 3231–3236 CrossRef CAS.
- K. Wang, H. Mei, L. Geren, M. A. Miller, A. J. Saunders, X. Wang, J. L. Waldner, G. J. Pielak, B. Durham and F. Millett, Biochemistry, 1996, 35, 15107–15119 CrossRef CAS.
- J. R. Winkler and H. B. Gray, Chem. Rev., 1992, 92, 369–379 CrossRef.
- M. J. Bjerrum, D. R. Casimiro, I.-J. Chang, A. J. D. Bilio, H. B. Gray, M. G. Hill, R. Langen, G. A. Mines, L. K. Skov, J. R. Winkler and D. S. Wuttke, J. Bioenerg. Biomembr., 1995, 27, 295–302 CAS.
- J. R. Winkler, H. B. Gray, T. R. Prytkova, I. V. Kurnikov and D. N. Beratan, in Bioelectronics: From Theory to Applications, eds. I. Willner and E. Katz, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, pp. 15–33 Search PubMed.
- T. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6497–6501 CrossRef CAS.
- T. Mizushima, A. Yoshida, A. Harada, Y. Yoneda, T. Minatani and S. Murata, Org. Biomol. Chem., 2006, 4, 4336–4344 RSC.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- A. J. Di Bilio, C. Dennison, H. B. Gray, B. E. Ramirez, A. G. Sykes and J. R. Winkler, J. Am. Chem. Soc., 1998, 120, 7551–7556 CrossRef CAS.
- X.-J. Yang, F. Drepper, B. Wu, W.-H. Sun, W. Haehnel and C. Janiak, Dalton Trans., 2005, 256–267 RSC.
- I. Hamachi, T. Matsugi, S. Tanaka and S. Shinkai, Bull. Chem. Soc. Jpn., 1996, 69, 1657–1661 CAS.
- H. Hofmeier, J. Pahnke, C. H. Weidl and U. S. Schubert, Biomacromolecules, 2004, 5, 2055–2064 CrossRef CAS.
- J. D. Gregory, J. Am. Chem. Soc., 1955, 77, 3922–3923 CrossRef CAS.
- C. K. Riener, G. Kada and H. J. Gruber, Anal. Bioanal. Chem., 2002, 373, 266–276 CrossRef CAS.
- H. A. O. Hill, L. H. Guo and G. McLendon, in Cytochrome c: A Multidisciplinary Approach, eds. R. A. Scott and A. G. Mauk, University Science Books, Sausalito, California, 1996, pp. 317–333 Search PubMed.
- M. Ihara, H. Nisihara, K.-S. Yoon, O. Lenz, B. Friedrich, H. Nakamoto, K. Kojima, D. Honma, T. Kamachi and I. Okura, Photochem. Photobiol., 2006, 82, 676–682 CrossRef CAS.
- J. R. Peterson and P. Thordarson, Chiang Mai J. Sci., 2009, 26, 236–246 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting data for protein stability and reactivity (UV-Vis, fluorescence emission) and further details on data analysis for DTDP and NPM reactions. See DOI: 10.1039/b919289a |
| This journal is © The Royal Society of Chemistry 2010 |