Neutral push-pull chromophores for nonlinear optical imaging of cell membranes†
Cyril Barsua, Rouba Cheaibbc, Stéphane Chambertbc, Yves Queneaubc, Olivier Maurya, Davy Cottetd, Hartmut Weged, Julien Douadyd, Yann Bretonnière*a and Chantal Andraud*a
aUniversité de Lyon, Laboratoire de Chimie de l'ENS Lyon, UMR 5182 CNRS-ENS Lyon, 46 allée d'Italie, 69364, Lyon, France. E-mail: Chantal.Andraud@ens-lyon.fr
bINSA Lyon, Laboratoire de Chimie Organique, Bâtiment J. Verne, 20 av A. Einstein, 69621, Villeurbanne, France
cCNRS, UMR 5246, ICBMS, Université Lyon 1, INSA-Lyon, CPE-Lyon, Bâtiment CPE, 43 bd du 11 novembre 1918, 69622, Villeurbanne, France
dLaboratoire de Spectrométrie Physique, UMR 5588 CNRS-Université Joseph Fourier (Grenoble I), 140 avenue de la physique, 38402, Saint Martin d'Hères, France
First published on 30th October 2009
Abstract
A new class of push-pull molecules was recently identified, based on pyridine dicarboxamide as an electron acceptor group and bearing a fluorenethynyl π-conjugated bridge. The molecules present good second and third order nonlinear properties, with a static quadratic hyperpolarisability μβ (at 1.907 μm) of 320 × 10−48 esu (μβ0 = 249 × 10−48 esu) and a maximum two-photon absorption cross-section of 1146 GM. Starting from this generic structure, we designed a series of eight amphiphilic nonlinear probes, varying the length of the lipophilic tail and the nature of the polar head, and tested their cell membrane affinity by nonlinear optical imaging. A good membrane affinity was observed with probes bearing short alkyl chains and carbohydrate moieties as the polar head, emphasizing the importance of the lipophilic–hydrophilic balance, as well as the role of the polar head. This original approach based on carbohydrate head groups is an important advancement in the design of membrane probes, since these highly versatile functional groups confer adequate hydrophilicity and yet conserve overall molecular neutrality.
Introduction
The description of the electrical activity of neuron networks in the brain necessitates simultaneous measurement of the activity in a great number of neurons in vivo,1 taking into account not only the action potential with a large amplitude of 100 mV, but also the small graduated potentials of lower amplitude (5 mV).2 Only optical imaging methods, and especially fluorescence, meet these requirements. A great deal of work has been dedicated to the design of efficient fluorescent fast-response voltage-sensitive optical probes showing good affinity for the cell membrane.3 This engineering was based quasi-exclusively on hemicyanine dyes featuring a charged pyridinium ring as an electron acceptor group, such as the now commercially available ANEPs (amino naphthyl ethynyl pyridinium), or the longer dialkylaminophenylpolyenylpyridinium RH dyes (Chart 1). These are amphiphilic dyes with hydrocarbon chains acting as membrane anchors, and a hydrophilic group attached to the pyridinium ring that aligns the probes perpendicular to the membrane–water interface. Systematic variations of the alkyl chain lengths and of the polar head enabled the identification of probes presenting the requisite water solubility and membrane affinity in neuronal cells,4 and the most recent works have successfully extended the class of available hemicyanine dyes to the near infrared.5 Styryl or naphthylstyryl dyes with short alkyl chains (such as di-4-ANEPPS) have been shown to be quickly internalized by the cell, whereas longer hydrocarbon tails tend to anchor the dyes into the outer leaflet, preventing flip-flop (i.e. the inversion of the probes from one leaflet to another resulting in the staining of both of the leaflets). The hydrophilic groups introduced consisted principally of ionic groups, such as sulfonium or polyammonium, giving overall zwitterionic or polycationic molecules. The molecular mechanisms resulting in voltage-dependent fluorescence properties have been discussed and, in the case of these fast-response probes, certainly arise from electrochromism—electric field-driven changes in the intramolecular charge distribution.6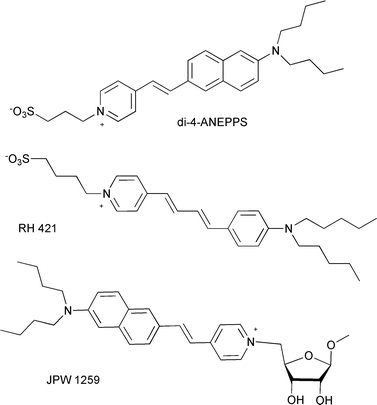 | ||
| Chart 1 Chemical structures of some voltage-sensitive fast response probes. | ||
The same molecules have large second order nonlinear coefficients,7 and have set the basis of the recent development of new membrane potential imaging techniques based on nonlinear optics: two-photon excited fluorescence microscopy (TPM) combined with second-harmonic imaging microscopy (SHIM). Both microscopy techniques originate from fundamentally different nonlinear optical phenomena, two-photon excited fluorescence (TPEF) and second harmonic generation (SHG), respectively. TPEF involves the simultaneous absorption of two photons (TPA) of lower frequency to trigger the fluorescence of the molecule, whereas in SHG, two incident photons are scattered into one coherent output photon with exactly twice the incident frequency. As the amplitude of both nonlinear phenomena are proportional to the square of the incident light intensity, they only occur at the focal point of a tightly focused laser, providing an intrinsic confocality, which results in a high z resolution. Furthermore, since the incident laser wavelengths are in the near infrared (NIR, typically in the tissue transparency region between 750–850 nm), lower tissue autofluorescence or self-absorption, increased penetration depth, reduced out-of-plane photobleaching and phototoxicity can be achieved.8,9 The SHG signal requires a non-centrosymmetric local environment, and only molecules asymmetrically distributed in the cell membrane will contribute to the observed signal,9–11 without any background signal originating from isotropic distribution in the surrounding medium. On the other hand, TPEF is an incoherent process and imposes no restriction on symmetry. The observation of TPEF will only depend on the presence of an emissive molecule, giving information about molecular distribution.
Some membrane-bound dyes of the ANEP family were found to engender a SHG signal highly sensitive to membrane potential,9,12 and others to present sub-millisecond response times to voltage change, which was shown to arise from electron redistribution or molecular tilt mechanisms.13 These assets make combined TPM/SHIM a unique and specific tool for the optical imaging of cellular membranes providing complementary information, and for direct time-resolved measurement of membrane potential in living cells.14 These studies have shown the proof of concept, but also pointed out the need for new chemical probes specifically designed for that purpose.15 In a recent report, Anderson, Clays and co-workers specifically designed a series of amphiphilic porphyrin-based second order nonlinear chromophores for SHG imaging of cell membranes, but still bearing a charged pyridinium polar head.16 Although a very strong SHG signal was observed in lipid vesicles for all compounds, the in vitro membrane localization strongly depends on the nature of the polar head. Therefore, despite the push in establishing pyridinium dyes as effective SHG sensors of membrane potential, there is room for improved molecules presenting largely different structures.
Results and discussion
We have recently identified a new class of push-pull molecules, based on pyridine dicarboxamide as an electron acceptor group and bearing a fluorenethynyl π-conjugated bridge (1, Chart 2), that displayed good second and third order nonlinear properties, with a static quadratic hyperpolarisability μβ (at 1.907 μm) of 320 × 10−48 esu (μβ0 = 249 × 10−48 esu) and a maximum TPA cross-section of 1146 GM, and whose intense fluorescence (with a quantum yield of 89% as reported in Table 1) is characterized by a strong positive solvatochromism, a key requirement for efficient voltage-dependent nonlinear probes.17 The absorption of this reference chromophore 1 is centered around 410 nm (Table 1), thus enabling efficient TPEF and resonance-enhanced SHG by working at 800 nm, and showed only little change with the solvent polarity. The present work focuses on the modification of this basic structure to tailor the dyes for cell membrane imaging, concentrating especially on the nature of the polar head.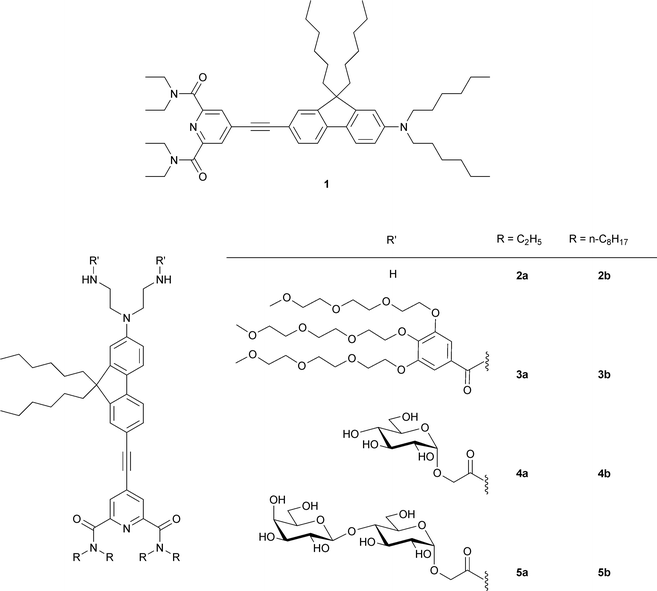 | ||
| Chart 2 Structure of new pyridine dicarboxamide nonlinear probes. | ||
| n | CH2Cl2a | Water | s/cm−1e | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λmax/nm | ε(λmax)/103 M−1 cm−1 | λem/nmb | Δṽ/cm−1 | ΦFc | λmax/nm | ε(λmax)/103 M−1 cm−1 | λem/nmb | Δṽ/cm−1 | ΦFc | ||
| a Except for 1 in CHCl3.b λexc = 410 nm.c λexc = 410 nm, reference Coumarin 153 in MeOH (ΦF = 0.45).26d Ref. 17.e Excluding water and methanol.f Excluding cyclohexane. | |||||||||||
| 1d | 411 | 37.0 | 525 | 5438 | 0.89 | — | — | — | — | — | 19385 |
| 2a | 389 | 12.2 | 543 | 7290 | 0.69 | 385 | 10.1 | 570 | 8368 | 0.05 | 19700 |
| 3a | 396 | 50.0 | 530 | 6348 | 0.75 | 402 | 30.0 | 575 | 7824 | 0.09 | 15552 |
| 4a | 391 | 24.8 | 529 | 6808 | 0.60 | 390 | 11.9 | 598 | 8720 | 0.07 | 24819f |
| 5a | 390 | 18.4 | 542 | 7190 | 0.49 | 397 | 10.1 | 584 | 8129 | 0.08 | 20718f |
| 2b | 390 | 35.7 | 537 | 7019 | 0.79 | 393 | 24.7 | 561 | 7620 | 0.16 | 20879 |
| 3b | 394 | 50.0 | 531 | 6477 | 0.78 | 397 | 27.9 | 552 | 7458 | 0.20 | 31497 |
| 4b | 388 | 32.9 | 525 | 5884 | 0.69 | 395 | 27.5 | 574 | 2299 | 0.17 | 24779f |
| 5b | 387 | 21.8 | 529 | 6936 | 0.50 | 388 | 18.8 | 561 | 7783 | 0.14 | 30588f |
Fluorescent probes with charged groups were found to occupy only a shallow location within the polar headgroup region of the bilayer, no matter how lipophilic the molecule to which they are linked.18 Therefore, in order to obtain a deeper membrane penetration, we incorporated two carbohydrate entities (glucosyl or lactosyl residues) into the probe, with the aim of providing an appropriate hydrophilicity while keeping overall neutrality. Carbohydrates are major constituents of cell membrane surfaces as glycoproteins or glycolipids. Carbohydrates mediate cell–cell contacts through sugar–protein specific interactions, and as such, are involved in many significant biological processes at the cell surface such as cell adhesion, cell recognition and differentiation, development of the neuronal network, inflammation, metastasis, and viral or bacterial infection.19 By grafting carbohydrate residues into the precedent nonlinear chromophores, we sought to mimic the overall structure of natural glycolipids and give access to neutral probes with good affinities for the cell membrane. To the best of our knowledge, no specially designed non-ionic carbohydrate-containing probes have been developed to date for nonlinear membrane imaging. The only example is a ribose-containing ANEP dye (JPW1259, Chart 1) in which the sugar cycle has been introduced as a chiral group to help assure a non-centrosymmetric environment.12 Carbohydrates are highly versatile scaffolds offering huge structural variation and can easily be introduced to primary amine groups through the use of carboxymethylglycoside lactones (CMGLs).20 In this article, we describe the synthesis and appraisal in nonlinear optical cell imaging of eight new amphiphilic compounds 2a/2b–5a/5b (Chart 2) bearing either neutral carbohydrate entities (two glucosyl or lactosyl residues) or simple amino and polyethyleneglycol (PEG) motifs for comparison. These different moieties enable considerable variation in hydrophilicity, whereas the length of the alkyl chains on the pyridinecarboxamide groups and the presence of two n-hexyl chains on the fluorenyl ring served to modulate the lipophilic character.
Synthesis
As shown in Scheme 2, all the final compounds 3–5 were obtained from the key diamines 2a–b, the synthesis of which involved two successive Sonogashira cross-coupling reactions (Scheme 1). Direct alkylation of 6 by the commercially available N-(-2-bromoethyl)phthalimide yielded only sparse amounts of dialkylated product. Thus, alkylation of 2-amino-7-iodo-9,9-di-n-hexylfluorene (6)21 with 2-bromoethanol in DMF produced the diol 7, which was further transformed to the diphthalimide product 8 under Mitsonobu reaction conditions with phthalimide. For the synthesis of the acceptor side of diamines 2, chelidamic acid (9) was converted to 4-chloro-2,6-bis(dialkylcarbamoyl)pyridine (10a–b) by treatment with thionyl chloride followed by reaction of the acyl chloride in dichloromethane with excess diethylamine (to give 10a), or two equivalents of di-n-octylamine and excess triethylamine (to give 10b) . 10a–b were then converted to the 4-iodo analogues 11a–b as previously described,21 with careful control of the reaction conditions to avoid cleavage of the amide groups. The first Sonogashira coupling with trimethylsilylacetylene followed by classical trimethylsilyl deprotection (K2CO3 in methanol) afforded 4-ethynyl-2,6-bis(diakylcarbamoyl)pyridine (12a–b) in moderate to good yields (86% and 61% for the two steps for 12a and 12b, respectively). The second Sonogashira coupling between 8 and 12a or 12b gave 13a and 13b in 65% and 60% yield, respectively. Hydrazinolysis of phthalimides 13a–b was performed in THF with the addition of 15 equivalents of crotyl alcohol (2-buten-1-ol) to quench the diimide formed by the oxidation of hydrazine.22 Under these conditions, compounds 2a–b were obtained exclusively without reduction of the triple bond.23 Conversion to the PEG derivatives 3a–b was achieved by coupling with the carboxylic acid 14 using DiC/HOBt. The sugar moieties were introduced by the ring opening of lactones 1524 and 16,25 followed by removal of the acetyl groups using catalytic sodium methoxide in methanol, to give probes 4a–b and 5a–b, respectively (Scheme 2).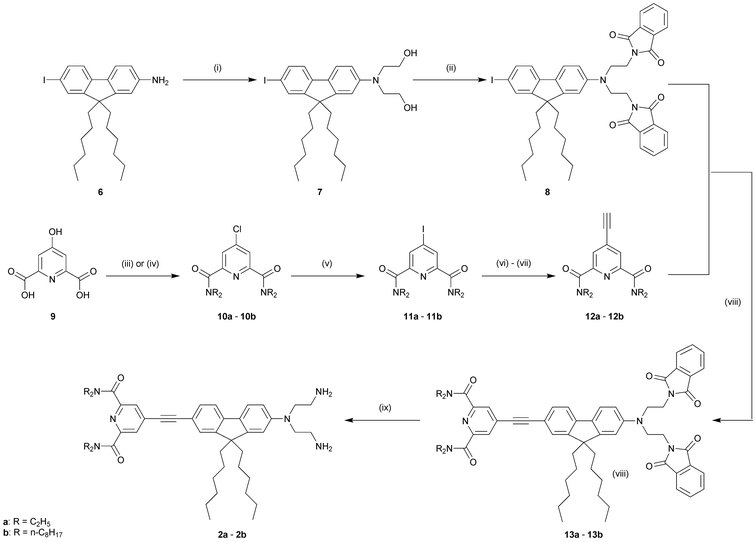 | ||
| Scheme 1 Synthesis of the diamino compounds (2a) and (2b). Reagents and conditions: (i) 1-bromoethanol, NaI, Na2CO3, DMF, 95 °C, 48 h, 66%; (ii) N-phthalimide, PPh3, DEAD, THF, rt, 18 h, 95%; (iii) R = ethyl: SOCl2, DMF cat., reflux, 2 h, then Et2NH, CH2Cl2, reflux, 2 h, 93% or (iv) R = n-C8H17: SOCl2, DMF cat., reflux, 2 h, then NH(n-C8H17)2, Et3N, CH2Cl2, reflux, 2 h, 90%; (v) 57% HI, H3PO3, 80 °C, 3.5 h, 72% (11a)/76% (11b); (vi) trimethylsilylacetylene, CuI, PdCl2(PPh3)2, THF, Et3N, rt, 4 d; (vii) K2CO3, MeOH, rt, 2 h, for the two steps 86% (12a)/61% (12b); (viii) CuI, PdCl2(PPh3)2, THF, Et3N, 50 °C, 48 h, 60% (13a)/65% (13b); (ix) NH2NH2·H2O, crotyl alcohol, THF, reflux, 18 h, 90% (2a)/95% (2b). | ||
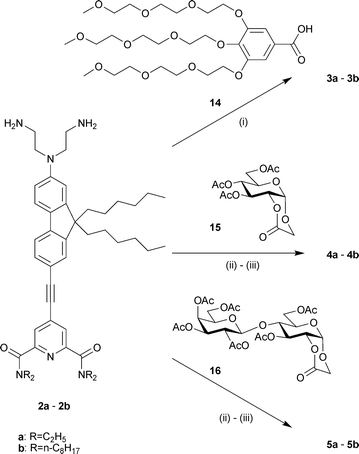 | ||
| Scheme 2 Synthesis of the target probes 3–5. Reagents and conditions: (i) DiC, HOBt, CH2Cl2, rt, 2 d, 51% (3a)/58% (3b); (ii) CH2Cl2, rt, 12 h; (iii) MeONa cat., MeOH, rt, 16 h, 45% (4a)/74% (4b), 30% (5a)/63% (5b) overall yield for the 2 steps. | ||
The molecules varied considerably in their water solubility. The six PEG groups in molecules 3a–b for instance provided very high solubility. Short chain 3a is soluble at 10−2 M and a clear solution was obtained, whereas the n-octyl analogue at 10−3 M in water gave a cloudy solution but no precipitate, suggesting the formation of micelles. On the other hand, the solubility in water of the sugar compounds 4a–b and 5a–b remains very low (a precipitate was observed in water at 10−6 M) even for the short-chain lactose compound 5a possessing four sugar residues.
Spectroscopic properties
Table 1 summarizes the data obtained for all compounds in dichloromethane and water. Spectroscopic data in chloroform for the reference molecule 1 are also included for comparison, but no data were obtained in water due to the very low solubility in this solvent.The absorption spectra of all compounds displayed an intense broad band in the range 350–450 nm with maxima at around 390 nm in dichloromethane. It is worth noting that the position of the absorption band allows an enhancement of the SHG signal by resonance at around 800 nm. The 20 nm blue shift of the linear absorption of 2–5 with respect to that of 1 could be related to the decreased donor strength due to the existence of hydrogen-bonding between the aniline nitrogen and the primary amine protons (compounds 2a–2b) or the amide hydrogen (3–5), as suggested for push-pull chromophores where only two or three carbon atoms separate the nitrogen donor atom from a hydrogen donor functionality such as a hydroxyl or an amide group.27 The molar extinction coefficients, at the wavelength of maximum absorption (εmax) ranged from 12.2 × 103 M−1 cm−1 for the diamine 2a to 50.0 × 103 M−1 cm−1 for the PEG compounds 3a and 3b in dichloromethane. A substantial decrease is observed in water for all systems with values decreasing from 50.0 × 103 M−1 cm−1 in CH2Cl2 to 30.0 and 27.9 × 103 M−1 cm−1 in water for 3a and 3b, respectively, for instance (Table 1).
All the studied compounds exhibited an intense emission with a structureless band centered at 525–545 nm in dichloromethane (Fig. 1). No residual emission was observed at 400 nm for each compound, suggesting an unambiguous separation of TPEF and SHG signals, which is a relevant condition for nonlinear imaging. The quantum yields measured in dichloromethane were high (ΦF = 0.50 for 5a and 5b to ΦF = 0.79 for 3b and 2b) and similar to that measured for the reference 1 (ΦF = 0.89), independent of the nature of the polar head. No differences were observed between short chain and long chain amides. Only the presence of four sugar groups (lactose compounds) resulted in a decrease of the quantum yield. In water, however, long chain compounds displayed a much higher quantum yield than the short chain analogs, suggesting a different organization in solution for both cases.
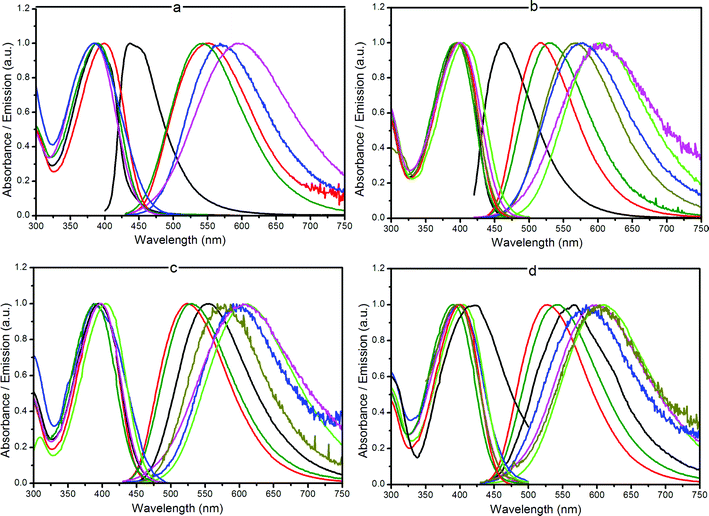 | ||
Fig. 1 Normalized absorption and emission spectra for compounds 2a (a), 3a (b), 4a (c) and 5a (d) in different solvents: (—) cyclohexane, ( ) THF, ( ) THF, ( ) CH2Cl2, ( ) CH2Cl2, ( ) acetone, ( ) acetone, ( ) DMSO, ( ) DMSO, ( ) methanol, ( ) methanol, ( ) water. ) water. | ||
The influence of the functional changes on the chromophores’ optical properties was studied by recording the absorption and fluorescence spectra in solvents of different polarity and proton donating ability at room temperature. Fig. 1 shows the absorption as well as the fluorescence spectra of the short chain compounds 2a–5a in the various solvents. Data for other probes are given in detail in the ESI.† An increase in solvent polarity induced a marked red shift of the emission band. The positive solvatochromic properties were analyzed following the Lippert–Mataga model.28 The solvent polarity parameter is thereby defined by the factor 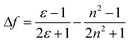 , where ε and n are the dielectric constant and the refractive index of the solvent, respectively. The Stokes shift (
, where ε and n are the dielectric constant and the refractive index of the solvent, respectively. The Stokes shift (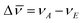 in cm−1) is correlated to Δf according to eqn (1), where K is a constant, c and h are the light speed and the Planck constant, and a is the radius of the spherical cavity of the solute.
in cm−1) is correlated to Δf according to eqn (1), where K is a constant, c and h are the light speed and the Planck constant, and a is the radius of the spherical cavity of the solute.
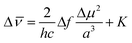 | (1) |
The solvatochromic slopes s derived from the Lippert–Mataga model are reported in Table 1. Even if this model is limited to a point dipole approximation, which is not the case here, a good linear correlation with relationship (1) was found if hydrogen bonding solvents (methanol and water) and, in the case of sugar-based molecules, cyclohexane are excluded (Fig. 2, and S2–S10 in the ESI†). This points out that the emission process occurs from a dipolar excited state, and is indicative of a rather high variation of the dipole moment between the excited and the ground state (Δμ). It is interesting to note that the emission wavelength is more sensitive to solvent than the absorbance, which indicates that the relaxed excited state may have a significantly different structure to that of the ground state. The solvatochromism was not analyzed further, as the shape and the dimension of the solute cavity a can hardly be simply estimated for such chromophores. More importantly, very close values have been found for all molecules 2–5, which are similar to that of the reference compound 1. This led us to think that the substitution does not significantly modify the chromophore’s electrochromic properties, and by extension, the nonlinear properties. The ETN scale29 emphasized that the points for protic solvents are outside the linear regression zone (see the ESI, Fig. S11–S12†). This was observed for all compounds including the reference 1, showing the existence of a hydrogen bond involving the electron acceptor group (pyridine dicarboxamide) of the chromophores and the solvent.
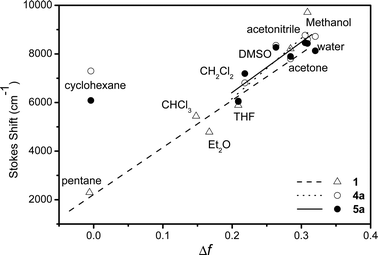 | ||
| Fig. 2 Variations of the Stokes shift (cm−1) Δṽ with Δf for compounds 4a–5a and reference compound 1. | ||
Nonlinear optical imaging
Cellular staining was investigated using Chinese hamster ovarian cancer cells (CHO) and F98 glial cells grown on borosilicate microscope cover slips, which were clamped for microscopy imaging into a custom-made circular Teflon/aluminium observation chamber. A few microlitres of concentrated (1–10 mM) aqueous (compounds 3a–b) or DMSO (compounds 2a–b, 4a–b and 5a–b) dye solutions were injected into the culture medium-filled chamber to yield final dye concentrations of ca. 10−7 M. At higher concentrations, the low water solubility of most compounds resulted in the formation of fluorescent aggregates after final dilution in the aqueous culture media.Cell staining was observed by TPEF imaging immediately after dye injection, without washing. Experiments were run on a modified upright microscope adapted for two-photon imaging with an external scan head and pulsed laser illumination, with direct immersion of the objective into the cell culture medium. Irradiation was carried out at 810 nm in the TPA band17 with a mode-locked Ti:sapphire laser, characterized by an output pulse width of ∼100 femtoseconds and a repetition rate of 78 MHz.
Surprisingly, the most lipophilic long chain compounds (2b–5b) did not stain the cell membranes. No intracellular fluorescence could be observed at any concentration, but a high background signal arising from the surrounding liquids suggested that the dyes stayed in the extracellular media, even after long incubation times (overnight). For the short chain compounds 2a–5a, marked differences were observed depending on the nature of the polar head, as shown in Fig. 3 and 4. The diamino compound 2a (protonated at physiological pH) and compound 3a bearing six PEG groups quickly entered the cell and accumulated in the cell cytoplasm where fluorescence was observed, as shown in Fig. 3. The cell nucleus remained unstained. The exact localization of the dyes, which did not evolve with time, has not been further investigated.
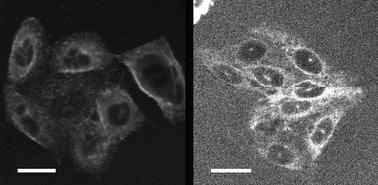 | ||
| Fig. 3 TPEF images of CHO cells stained externally (0.1 μM) by 2a (left) and 3a (right). The average excitation power was 10 mW at the sample and the total acquisition time was 0.9 s. Scale bars are 20 μm. | ||
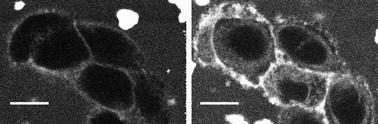 | ||
| Fig. 4 TPEF images of CHO cells stained externally by 4a (10 μM) showing the progressive internalization of the dye: immediately after dye injection (left) and after 10 min (right). The average excitation power was 10 mW at the sample and the total acquisition time was 0.9 s. Scale bars are 10 μm. | ||
Conversely, showing the role of the peripheral substituents and of the balance between hydrophilicity and lipophilicity, a good membrane staining was achieved with dyes 4a and 5a bearing two and four pyranoside structures, respectively. It was observed immediately after dye injection and remained as long as the extracellular medium was not washed. The cytoplasm, where no fluorescence was observed immediately after injection, was progressively stained, as shown in Fig. 4. This indicates that the dyes were able to cross the membrane bilayer and diffuse into the cytoplasm. As for the PEG compound, the cell nucleus was not stained. When stained cells were washed by fresh culture medium after a few minutes of incubation, fluorescence from the membrane vanished as the dyes progressively internalized. Focusing the laser beam on a small area of the cell resulted in too strong absorption and rapid cell death. These encouraging results prompted us to try to simultaneously record TPEF and second harmonic generation signals. In SHG, being a coherent process, the signal propagates roughly in the same direction as the incident beam9,10 and was therefore collected in a transmitted light configuration. With incident light at 810 nm in the TPA band, the SHG was considerably enhanced by resonance, and the signal was also spectrally separated from the fluorescence which allowed for its selective detection by means of a 405 nm narrow band-pass filter. The TPEF signal was collected in epi-configuration. We also used commercially available di-4-ANEPPS as a reference membrane-staining compound to assess instrument performance and to adjust the recording configuration.
Fig. 5 shows the compared TPEF (left)–SHG (right) images recorded with compound 5a on F98 glial cells. The TPEF signal of the stained membrane was very strong and a single scan was sufficient for recording a good quality image, while the SHG signal observed at 405 nm, arising only from molecules in the cell membrane, was obtained after averaging of 3 to 10 successive scans, due to the weak intensity of the signal. It is worth noting that similar results were obtained with CHO cells. Although the SHG signal strength depends quadratically on incident intensity, it could not be improved by increasing the laser power, due to a simultaneous enhancement of the fluorophore TPA, resulting in increased photo-induced damage in the cells, and limiting the experiment to a laser power of 5–15 mW under the objective. Focusing and scanning a reduced area of the cell to assess the kinetics of internalization also resulted in cell death for the same reasons.
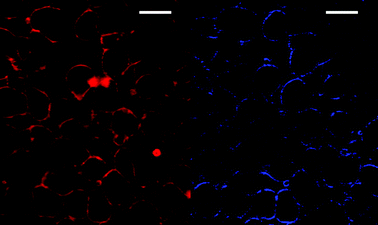 | ||
| Fig. 5 Simultaneous TPEF (left) and SHG (right) images of F98 cells stained externally with 5a (2 μM) and excited at 810 nm. The SHG image has been contrasted for better visualization. The average excitation power was 15 mW at the sample, and the total acquisition time was 0.9 s for the TPEF image and 3.6 s for the SHG image. Scale bars are 20 μm. | ||
Similar results were obtained for both compounds 4a and 5a, bearing a glucosyl and a lactosyl structure, respectively. On the other hand, although quantification could not be obtained, membrane staining with compounds 4a and 5a was long-lasting and the signal was observed for a long time span by comparison with di-4-ANEPPS. In the same imaging experiment, the latter showed a stronger signal, almost immediately seen, but it vanished quickly as the dye was internalized.
Conclusion
In order to develop efficient probes for cell membrane imaging and to find adequate polar end groups, as well as the right balance between hydrophilicity and lipophilicity, we synthesized a series of novel push-pull probe molecules based on pyridine dicarboxamide bearing lipophilic alkyl chains of varying length as an electron acceptor group, a fluorenethynyl π-conjugated bridge and a dialkylamino with different hydrophilic attachments as a donor moiety. We studied their optical properties and their efficiency for TPEF and SHG imaging using two different cell lines, CHO and F98 glial cells, and showed that membrane imaging depends strongly on the peripheral substitution pattern, and that a small variation in the structure of the probe can induce dramatic changes in the cell uptake or efficiency of the membrane staining. The most hydrophilic probes diffuse with ease through the medium, but show no affinity for the membranes, resulting in fast cellular internalization. On the other hand, the molecules with the longest lipophilic acyl tail groups are difficult to deliver to the membranes, at least if delivery is based on molecular dissolution in a biocompatible solvent. Different alternative methods, based on liposomes, poloxamers and/or cyclodextrins as stabilizing agents or delivery vehicles are currently under research.Good membrane staining was observed on CHO and F98 glial cells for the dyes 4a and 5a bearing short alkyl chains and containing two and four pyranoside structures, respectively, while no significant difference was found between glucose- and lactose-based groups. The approach based on carbohydrate head groups is an important advancement in the design of membrane probes, since these highly versatile functional groups confer adequate hydrophilicity and yet conserve overall molecular neutrality. Membrane specificity of the most promising dyes is currently being tested in neuronal cells and brain slices in order to test the voltage dependence in physiological conditions. With the huge structural variety offered by carbohydrate scaffolds, more selective staining, as well as nonlinear imaging of specific sugar receptors on the cell membrane surface can be envisaged.
Experimental
Cell-cultures
Cells were cultured as adherent monolayers in 30 mL polystyrene cultures dishes (Nunc) and on 28 mm circular borosilicate glass microscope cover slips (Mentzel) in DMEM/high glucose Dulbecco's modified Eagle's medium (HyQ), containing an added 10% v/v standard fetal bovine serum (Perbio), 1% v/v MEM/non-essential amino acids (HyQ) (for CHO only) and 1% v/v of Penicillin and Streptomycin solution (DB Difco) in a humidified incubator (Sanyo IncuSafe) at 37 °C and 5% CO2. Once the monolayer reached 80–90% confluence (every 3–4 d), the cells were resuspended as follows. The old growth medium was discarded and the dishes were washed 3 times with PBS (PAA) to remove from the cell monolayer all of the trypsin inhibitors contained in the growth medium. After that, the dishes were incubated at 37 °C for 3–5 min with a solution of trypsin/EDTA (Invitrogen Gibco), which detaches the cells from the surface via proteolysis. The resulting suspension was gently agitated with a pipette and 10% of it was transferred into duplicate wells of fresh, warmed media.Combined SHG-TPEF microscopy
Biphotonic laser scanning microscopy was performed with a confocal set-up consisting of a Biorad MRC 1024 scanhead and an Olympus BX50WI microscope. The fluorescence signal was directly epicollected, as shown in Fig. 6, whereas collection of SHG was performed on the other side of the sample because the SHG signal is directional and more efficient in transmission. An excitation beam at 810 nm (Tsunami femtosecond Ti:Sa laser pumped by a 5 W Spectra-Physics Millenia V) was focused on the sample using a 60× water immersion objective with a 0.9 numerical aperture (LumPlanFl/IR Olympus). The back aperture of the objective was slightly underfilled (∼70%), giving rise to a voxel size of 0.5 μm × 0.5 μm in the horizontal plane and 3 μm in the vertical dimension. The pulse length at the entrance of the microscope is 100 fs but the light is dispersed, mainly by the lenses of the objective. Therefore, we estimate a 500 fs length at the focal plane. The beam was scanned in the x–y plane to acquire 512 × 512 pixels images in 0.9 s. To vary the observation depth, z-scan was performed by vertical motion of a motorized objective. The incident laser intensity was adjusted by a half-wave plate and a polarizer placed before the microscope so that the total average power delivered at the surface ranged from 1–100 mW. Two channels could be simultaneously recorded using two added external photomultiplier tubes and appropriate filters. Images were acquired using the Biorad exploitation system and displayed using ImageJ.30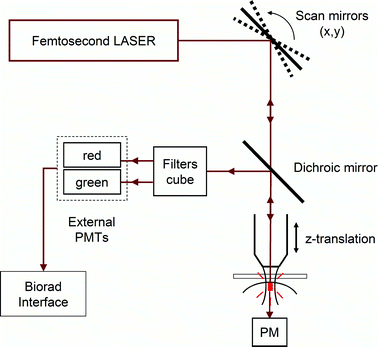 | ||
| Fig. 6 Schematic representation of the microscopy set-up. | ||
Acknowledgements
This work was supported by funding by région Rhône-Alpes through the CPER “Nouvelles approches physiques des sciences du vivant”, from “Cluster de recherche Chimie de la région Rhône-Alpes” for a PhD grant (R.C.), and from the RTRA “Fondation nanosciences: aux limites de la nanoélectronique”.Notes and references
- R. Cossart, D. Aronov and R. Yuste, Nature, 2003, 423, 283–288 CrossRef CAS.
- (a) J. H. Goldberg, R. Yuste and G. Tamas, J. Physiol., 2003, 551, 67–78 CAS; (b) S. Charpak, J. Mertz, E. Beaurepaire, L. Moreaux and K. Delaney, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1230–1234 CrossRef CAS.
- (a) A. S. Waggoner, Annu. Rev. Biophys. Bioeng., 1979, 8, 47–68 Search PubMed; (b) L. M. Loew, Pure Appl. Chem., 1996, 68, 1405–1409 CAS; (c) M. Zochowski, M. Wachowiak, C. X. Falk, L. B. Cohen, Y.-W. Lam, S. Antic and D. Zecevic, Biol. Bull., 2000, 198, 1–21 CrossRef CAS; (d) A. L. Obaid, L. M. Loew, J. P. Wuskell and B. M. Salzberg, J. Neurosci. Methods, 2004, 134, 179–190 CrossRef CAS.
- Y. Tsau, P. Wenner, M. J. O'Donovan, L. B. Cohen, L. M. Loew and J. P. Wuskell, J. Neurosci. Methods, 1996, 70, 121–129 CrossRef CAS.
- (a) P. Yan, A. Xie, M. Wei and L. M. Loew, J. Org. Chem., 2008, 73, 6587–6594 CrossRef CAS; (b) J. P. Wuskell, D. Boudreau, M.-d. Wei, L. Jin, E. Reimund, R. Chebolu, A. Bullen, K. D. Hoffacker, J. Kerimo, L. B. Cohen, M. R. Zochowski and L. M. Loew, J. Neurosci. Methods, 2006, 151, 200–215 CrossRef.
- L. M. Loew, G. W. Bonneville and J. Surow, Biochemistry, 1978, 17, 4065–4071 CrossRef CAS.
- J. Y. Huang, A. Lewis and L. M. Loew, Biophys. J., 1988, 53, 665–670 CAS.
- (a) V. E. Centonze and J. G White, Biophys. J., 1998, 75, 2015–2024 CrossRef CAS; (b) P. T. C. So, C. Y. Dong, B. R. Masters and K. M. Berland, Annu. Rev. Biomed. Eng., 2000, 2, 399–429 CrossRef CAS; (c) A. Hopt and E. Neher, Biophys. J., 2001, 80, 2029–2036 CrossRef CAS; (d) L. Sacconi, D. A. Dombeck and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3124–3129 CrossRef CAS; (e) G. McConnell, J. Biomed. Opt., 2006, 11, 054020 CrossRef CAS.
- P. J. Campagnola, M. Wei, A. Lewis and L. M. Loew, Biophys. J., 1999, 77, 3341–3349 CrossRef CAS.
- L. Moreaux, O. Sandre and J. Mertz, J. Opt. Soc. Am. B, 2000, 17, 1685–1694 Search PubMed.
- L. Moreaux, O. Sandre, S. Charpak, M. Blanchard-Desce and J. Mertz, Biophys. J., 2001, 80, 1568–1574 CrossRef CAS.
- O. Bouevitch, A. Lewis, I. Pinevsky, J. P. Wuskell and L. M. Loew, Biophys. J., 1993, 65, 672–679 CrossRef CAS.
- A. C. Millard, L. Jin, M.-d. Wei, J. P. Wuskell, A. Lewis and L. M. Loew, Biophys. J., 2004, 86, 1169–1176 CrossRef CAS.
- L. Sacconi, J. Mapelli, D. Gandolfi, J. Lotti, R. P. O'Connor, E. D'Angelo and F. S. Pavone, Opt. Express, 2008, 16, 14910–14921 CrossRef CAS.
- (a) D. A. Dombeck, L. Sacconi, M. Blanchard-Desce and W. W. Webb, J. Neurophysiol., 2005, 94, 3628–3636 CrossRef; (b) T. Z. Teisseyre, A. C. Millard, P. Yan, J. P. Wuskell and M.-d. Wei, J. Biomed. Opt., 2007, 12, 044001 CrossRef.
- J. E. Reeve, H. A. Collins, K. De Mey, M. M. Kohl, K. J. Thorley, O. Paulsen, K. Clays and H. L. Anderson, J. Am. Chem. Soc., 2009, 131, 2758–2759 CrossRef CAS.
- C. Barsu, R. Fortrie, K. Nowika, P. L. Baldeck, J.-C. Vial, A. Barsella, A. Fort, M. Hissler, Y. Bretonnière, O. Maury and C. Andraud, Chem. Commun., 2006, 4744–4746 RSC.
- K. Kachel, E. Asuncion-Punzalan and E. London, Biochim. Biophys. Acta, Biomembr., 1998, 1374, 63–76 CrossRef CAS.
- For reviews concerning the function and biological role of cell surface sugars see: (a) A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS; (b) R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS. For reviews dealing with cell surface oligosaccharides and their implication in cell–cell interactions, and cell adhesion in cancer metastasis and angiogenesis see: (c) K. J. Yarema and C. R. Bertozzi, Curr. Opin. Chem. Biol., 1998, 2, 49–61 CrossRef CAS; (d) E. Gorelik, U. Galili and A. Raz, Cancer Metastasis Rev., 2001, 20, 245–277 CrossRef CAS; (e) N. Sharon and H. Lis, Glycobiology, 2004, 14, 53R–62R CrossRef CAS; (f) R. Kannagi, M. Izawa, T. Koike, K. Miyazaki and N. Kimura, Cancer Sci., 2004, 95, 377–384 CrossRef CAS.
- S. Trombotto, M. Danel, J. Fitremann, A. Bouchu and Y. Queneau, J. Org. Chem., 2003, 68, 6672–6678 CrossRef CAS.
- A. Picot, C. Feuvrie, C. Barsu, F. Malvolti, B. Le Guennic, H. Le Bozec, C. Andraud, L. Toupet and O. Maury, Tetrahedron, 2008, 64, 399–411 CrossRef CAS.
- B. E. Maryanoff, M. N. Greco, H.-C. Zhang, P. Andrade-Gordon, J. A. Kauffman, K. C. Nicolaou, A. Liu and P. H. Brungs, J. Am. Chem. Soc., 1995, 117, 1225–1239 CrossRef CAS.
- Hydrazinolysis of phthalimides 13a–b using hydrazine in ethanol without adding crotyl alcohol gave an approximately 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the desired alkynes 2a–b and of the products arising from the partial reduction of the triple bond, which could not be separated by column chromatography.
:1 mixture of the desired alkynes 2a–b and of the products arising from the partial reduction of the triple bond, which could not be separated by column chromatography. - S. Trombotto, A. Bouchu, G. Descotes and Y. Queneau, Tetrahedron Lett., 2000, 41, 8273–8277 CrossRef CAS.
- R. Cheaib, A. Listkowski, S. Chambert, A. Doutheau and Y. Queneau, Tetrahedron: Asymmetry, 2008, 19, 1919–1933 CrossRef CAS.
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lefèvre, B. Valeur, E. Gratton, M. vandeVen, N. D. J. Silva, Y. Engelborghs, K. Willaert, A. Sillen, A. J. W. G. Visser, A. van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tamai and A. Miura, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS.
- I. Liakatas, C. Cai, M. Bösch, M. Jäger, C. Bosshard, P. Günter, C. Zhang and L. R. Dalton, Appl. Phys. Lett., 2000, 76, 1368–1370 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn, Academic/Plenum, New York, 1999, pp. 205–226 Search PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- ImageJ, v.1.36 Public Domain Software, available at http://rsb.info.nih.gov/ij/.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures and characterization. Spectroscopic data for all compounds 2–5, including Lippert–Mataga plot and fit. See DOI: 10.1039/b915654b |
| This journal is © The Royal Society of Chemistry 2010 |