Asymmetric ruthenium-catalyzed 1,4-additions of aryl thiols to enones†‡
Andrei Bădoiua, Gerald Bernardinellib, Céline Besnardb and E. Peter Kündig*a
aDepartment of Organic Chemistry, University of Geneva, 30 Quai Ernest Ansermet, CH-1211, Geneva 4, Switzerland. E-mail: peter.kundig@unige.ch; Fax: (+) 41-22-379-3215
bLaboratory of Crystallography, University of Geneva, 24 Quai Ernest Ansermet, CH-1211, Geneva 4, Switzerland
First published on 6th November 2009
Abstract
Well defined, stable, one-point binding ruthenium complexes 1 and 2 selectively bind and activate α,β-unsaturated carbonyl compounds for cycloaddition reactions. These mild Lewis acids catalyze asymmetric 1,4-addition reactions of aryl thiols to enones with product selectivities up to 87% ee. 31P NMR experiments provide an insight into the intricate equilibria governing the reaction mechanism. The absolute configuration of the major products indicates enones to react in the syn-s-trans orientation. Models based on X-ray structures of the Ru complexes can be used to rationalize selectivity.
Introduction
Life itself is the uncontested, ever-present proof of the importance of sulfur-containing and sulfur-based compounds. Many biochemical processes in nature involve sulfur, be it in organic or inorganic form.1 From a purely synthetic point of view, the words variety and richness describe best the chemistry of sulfur.2 Enzyme mimics, natural product synthesis, semi-labile ligands or building blocks for asymmetric synthesis are but a few of the applications that sulfur-containing compounds find nowadays.3Among the various methods for the generation of carbon–sulfur bonds,4 the Michael addition (or 1,4-addition) reaction is a straightforward route. This transformation takes advantage of the mild nucleophilicity of sulfur nucleophiles in general and of the thiophenolate anion in particular. Its propensity to react with activated double bonds provides thioethers as products.5 Diversity of the reaction partners, flexibility of the Michael adduct (that can be used as such or easily cleaved, reduced or oxidized) and the potential for asymmetric conjugate addition reactions are the driving forces of interest in this field.
Sulfa-Michael additions have been shown to be promoted by a number of compounds, including organic and inorganic bases, Lewis acids, water or even the absence of solvent.6 First asymmetric versions described were organocatalytic.7 Since, the field has significantly evolved and rationally designed organocatalysts now provide access to unprecedented levels of activity and selectivity for this transformation.8 By far the most efficient metal-based catalysts belong to the family of heterobimetallic complexes developed independently by Sundararajan,9 Shibasaki,10a and later by Narasimhan.11 The work of Shibasaki et al. is all the more remarkable since the sulfa-Michael addition, asymmetric protonation, and kinetic resolution10b developed were further applied in natural product synthesis.10c The innovative use of a C2-symmetrical enantiopure N-oxide/CdI2 system by Nakajima et al. is also a fine achievement.12 While mechanistic details are not yet established, this procedure allowed for the extension of the Michael acceptors to enals. Processes involving a Ca-BINOL complex,13 chiral 2-amino alkoxides14 and ethers,15 or bidentate proline-derived Michael acceptors16 were also successfully employed for the asymmetric catalytic sulfa-Michael addition.17
We have prepared iron- and ruthenium Lewis acid catalysts based on structurally well-defined monocationic half-sandwich complexes bearing chiral electron-poor diphosphinite ligands (Fig. 1).18 The ligand's perfluorinated aryl rings contribute to the Lewis acidity of these complexes and, together with the aromatic roofs and the ligand’s backbone, generate a chiral binding site that is ideal for the activation of α,β-unsaturated carbonyl compounds.
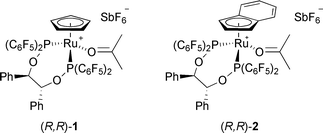 | ||
| Fig. 1 Chiral Ru Lewis acid precatalysts. | ||
These mild chiral Lewis acids proved to be excellent catalysts for the Diels–Alder reactions of dienes with enals18,19 and 1,3-dipolar cycloadditions with nitrones20 and nitrile oxides.21 In both cases enal over dipole coordination was preferred and the expected products were obtained with good yields and selectivity.
More recently, this was extended to Diels–Alder reactions of acyclic enones.22 Not only are these less reactive dienophiles, but due to the similarity of the two modes of coordination to the metal (syn or anti), stereocontrol with a single site catalyst is much more challenging.23
In the present article we probe the potential of the ruthenium catalysts in conjugate addition reactions. Specifically, we detail our study on the use of [Ru(acetone)(R,R-BIPHOP–F)Cp][SbF6] (1) as catalyst for the Michael addition of thiols to enones.
Results and discussion
Benchmark Michael additions usually involve enones since the stable products are readily analysed.17 We initially selected trans-4-(OMe)-3-buten-2-one (4), 2-cyclopenten-1-one (6) and 2-cyclohexen-1-one (8) as Michael acceptors and thiophenol (3a) as the nucleophile. Racemic reference samples were obtained from the reaction catalyzed either by diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,8-bis(dimethylamino) naphthalene (“proton sponge”).Reactions were carried out in the presence of a catalytic amount of the [Ru(acetone)(R,R-BIPHOP–F)Cp][SbF6] complex (1). Trans-4-(OMe)-3-buten-2-one (4) afforded addition product (S)-5a in moderate yields (18 h) and low enantiomeric excess (Scheme 1). Switching to more reactive 2-cyclopenten-1-one (6) only led to racemic product 7a.
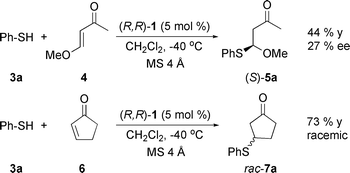 | ||
| Scheme 1 Non-optimized 1,4-addition reactions. | ||
2-Cyclohexen-1-one (8) was chosen as the Michael acceptor for the optimization of the reaction conditions. In the same conditions as above (Scheme 1), product (R)-9a was obtained in fair yield but low selectivity (Table 1, entry 1). Raising the temperature had a negative effect on the results (entry 2).
| ||||
|---|---|---|---|---|
| Entry | Solvent | T/°C | Time/h | Yieldb (eec) (%) |
| a All reactions were carried out under N2, using 3a (0.38 mmol) and 8 (0.25 mmol), with 50 mg of powdered, activated MS 4 Å, in 1 mL of dry solvent.b Isolated yield.c Determined by HPLC analysis.d Slow addition of 3a over 12 h. | ||||
| 1 | CH2Cl2 | −40 | 72 | 64 (33) |
| 2 | CH2Cl2 | −20 | 48 | 50 (11) |
| 3 | CH2Cl2–THF (1 : 1) | −20 | 48 | 40 (45) |
| 4 | THF | 0 | 24 | 54 (45) |
| 5 | THF | −20 | 48 | 60 (68) |
| 6 | THF | −40 | 64 | 48 (60) |
| 7d | THF | −20 | 48 | 82 (63) |
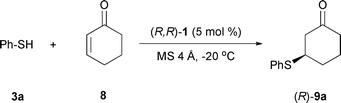
Different media were probed next and as already documented in the literature,17 THF was found to be more suitable for this reaction than CH2Cl2 (entries 2, 3, and 5).24 THF–toluene mixtures or more polar solvents (MeOH, EtOH) failed to improve the results.
With THF as the solvent of choice, temperature effects were studied next.24 Enantioselectivity slowly increased with decreasing temperature with the best result obtained at −20 °C. Further lowering the temperature led to a sluggish reaction without improvement in selectivity (entries 4 to 6).
Despite numerous attempts to optimize the conditions, the isolated product yields were limited to about 60%. Addition of a base to the reaction mixture, along with the Ru catalyst, generally led to racemic products. At the end of the reaction the ruthenium thiophenol complex was isolated; catalyst poisoning by the Michael donor is likely to occur as this complex proved to be inactive. Slow addition of excess thiophenol (over a period of 12 h) allowed us to overcome this problem and yields were now good (entry 7 vs. entry 5).
The best set of conditions (entry 7) was then applied in the screening of Michael donors (Table 2). Reactions were stopped after 48 h for ease of comparison. As before, the racemic adducts were obtained by mixing equimolar amounts of Michael donor (3a–k) and 2-cyclohexen-1-one (8) for 24 h in THF in the presence of catalytic amounts of an organic base (5 mol% DBU).
| |||
|---|---|---|---|
| Entry | RSH | Yield (%)b | ee (%)c |
| a All reactions were carried out under N2, using 3a–k (0.38 mmol) and 8 (0.25 mmol), with 50 mg of powdered, activated MS 4 Å, in 1 mL of dry THF, with slow addition of the aryl thiol over 12 h.b Isolated yield.c Determined by HPLC analysis.d Absolute configuration based on known literature data (sign of optical rotation).12,26b | |||
| 1 | PhSH (3a) | 82 | 63d |
| 2 | BnSH (3b) | 21 | 47d |
| 3 | 2-NapSH (3c) | 74 | 33d |
| 4 | 4-Cl-C6H4-SH (3d) | 86 | 52d |
| 5 | 4-Me-C6H4-SH (3e) | 57 | 51d |
| 6 | 4-t-Bu-C6H4-SH (3f) | 73 | 57d |
| 7 | 4-OMe-C6H4-SH (3g) | 58 | 53d |
| 8 | 2-OMe-C6H4-SH (3h) | 32 | 41d |
| 9 | 2-CO2Me-C6H4-SH (3i) | 39 | 53 |
| 10 | 2,6-Cl-C6H3-SH (3j) | 89 | 0 |
| 11 | 2,6-Me-C6H3-SH (3k) | 94 | 82 |

Less reactive benzyl thiol (3b) provided adduct 9b in poor yield and selectivity (Table 2, entry 2). 2-Naphthylthiol (3c) gave the expected thiol ether 9c also in poor enantioselectivity, albeit in good yield (entry 3).
Next, a series of para-substituted aryl thiols were screened. Electronic effects seem to have small, if any, influence on the outcome of the reaction; the expected Michael adducts were obtained in moderate to good yields and, with a few exceptions, with ees in the range of 50–60% (entries 4 to 7).
In terms of steric effects, a methoxy (entry 8) or a carboxymethyl group (entry 9) ortho to the thiol only led to a decrease in both yield and selectivity. Important changes appear when bulk is added on both sides close to the thiol group (positions 2 and 6). Surprisingly, despite giving the product in good yield, 2,6-dichlorothiophenol (3j) led only to the formation of racemic adduct (entry 10). With 2,6-dimethylthiophenol (2,6-DMTP, 3k), product 9k was isolated in 94% yield and with 82% ee (entry 11). With respect to the observed selectivity, an increase in steric bulk is in agreement with the assumption of a non-orthogonal approach of the nucleophile (in contrast to the orthogonal approach in the cycloaddition reactions19–22). It should be noted that with bulky thiols 3j and 3k, a color change from yellow to orange was observed upon their addition to the reaction mixture; this was not observed with the other thiols. This suggests the appearance of charge-separated Ru-thiophenolate species that could be catalytically active.
A sterically demanding Michael donor should be beneficial not only for the selectivity, but also to the overall catalytic cycle, as the deactivation of the ruthenium catalyst (through Ru-thiol complexes) becomes less likely. Indeed, slow addition of the thiol 3k is not required, thus simplifying the procedure (Table 3, entries 1 and 2).24
| ||||
|---|---|---|---|---|
| Entry | Solvent | Time/h | Yield (%)b | ee (%)c |
| a All reactions were carried out under N2, using 3k (0.38 mmol) and 8 (0.25 mmol), with 5 mol% (R,R)-1 and 50 mg of powdered, activated MS 4 Å, in 1 mL of dry solvent, unless mentioned otherwise.b Isolated yield.c Determined by HPLC analysis.d Slow addition of the aryl thiol over 12 h.e 5 mL of dry THF.f Full conversion not reached.g Run at 0 °C.h Without activated MS 4 Å.i 5 Mol% (S,S)-1.j 5 Mol% (R,R)-2.k 1 Mol% 2,6-lutidine as additive.l 2.5 Mol% (R,R)-1.m 1 Mol% (R,R)-1.n 3k (2.2 mmol) and 8 (2 mmol), with 2 mol% (R,R)-1 and 200 mg of activated MS 4 Å in 2 mL of dry THF. | ||||
| 1 | THFd | 24 | 94 | 82 |
| 2 | THF | 24 | 95 | 85 |
| 3 | THFe | 48 | 79f | 71 |
| 4 | Neat | > 72 | — | — |
| 5g | Water | 24 | 41f | 30 |
| 6h | THF | 24 | 17f | 79 |
| 7i | THF | 24 | 95 | −87 |
| 8j | THF | 48 | 70 | 77 |
| 9k | THF | 24 | 99 | 77 |
| 10l | THF | 36 | 83 | 85 |
| 11m | THF | 72 | 75 | 85 |
| 12n | THF | 48 | 85 | 85 |

Running the reaction under more dilute conditions (x 5) had a negative effect on the results (entry 3). In the absence of solvent the reaction did not proceed at all, as indicated by TLC analysis (entry 4). Subsequent addition of 4 Å molecular sieves or solvent failed to start the reaction.
Water-catalyzed (racemic) Michael additions of thiols to enones are known.6b Interestingly, running the reaction in water led to substantial formation of the Michael adduct, albeit in low ee due to temperature limitations (entry 5). The Ru aqua complex was previously described and is known to act as an active precatalyst.19e Here, the product was formed despite the absence of “dry reaction conditions”, whereas a reaction run in THF but in the absence of molecular sieves turned out to be very sluggish (entry 6). Using the enantiomeric complex (S,S)-1 as precatalyst led, as expected, to the opposite product enantiomer (entry 7).
The indenyl analogue of the Ru precatalyst (2), designed to boost reactivity and selectivity for the Diels–Alder reactions,19b was also used for the standard reaction in the best conditions. However, it turns out that now the reaction was slower and the results were less impressive (entry 8).
2,6-Lutidine as additive in cycloaddition reactions with Fe and Ru Lewis acid catalysts is beneficial since Brønsted acid traces in the solvent are scavenged.19,20 Running the standard reaction with 5 mol% of the [Ru(acetone)(R,R-BIPHOP–F)Cp][SbF6] precatalyst (1) together with 1 mol% of 2,6-lutidine brought no increase in selectivity although the product was formed quantitatively (entry 9).
A decrease in catalyst loading was successfully accomplished without loss of selectivity, to 2.5 and then 1 mol% (entries 10 and 11), at the expense of reaction rate and a slight decrease in isolated yield. The reaction is robust and can be readily scaled-up (x 8) even with low catalyst loading (entry 12).
The optimized reaction conditions were next extended to a variety of Michael acceptors (Table 4). Thus, 2-cyclopenten-1-one (6) gives the opposite enantiomer in good yield but low ee (entry 1). 2-Cyclohepten-1-one (10) on the other hand gave the expected 7-membered adduct with results comparable to those for benchmark 2-cyclohexen-1-one (8) (entries 2 and 3). The bulkier 4,4-dimethyl-2-cyclohexen-1-one (11) not only showed poor reactivity but also afforded the product in lower selectivity, indicating possible unfavourable steric interactions between the activated enone and the Michael donor (entry 4).
| |||||
|---|---|---|---|---|---|
| Entry | Enoneb | Product | Time/h | Yield (%)c | ee (%)d |
a All reactions were carried out under N2, using 3k (0.38 mmol) and enone (0.25 mmol), with 50 mg of powdered, activated MS 4 Å, in 1 mL of dry THF.b Enones: 2-cyclopenten-1-one (6), 2-cyclohexen-1-one (8), 2-cyclohepten-1-one (10), 4,4-dimethyl-2-cyclohexen-1-one (11), trans-1,3-diphenyl-2-propen-1-one (12), trans-4-phenyl-3-buten-2-one (13).c Isolated yield.d Determined by HPLC analysis.e Full conversion not reached. 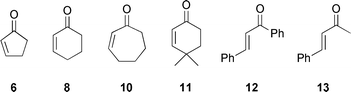 | |||||
| 1 | 6 | 7k | 48 | 86 | −16 |
| 2 | 8 | 9k | 24 | 95 | 85 |
| 3 | 10 | 14 | 36 | 95 | 83 |
| 4 | 11 | 15 | 48 | 33e | 52 |
| 5 | 12 | 16 | 16 | 35 | −8 |
| 6 | 13 | 17 | 16 | 31 | −4 |
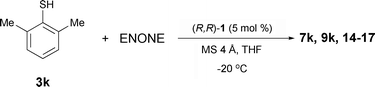
Acyclic enones proved to be extremely poor Michael acceptors. Chalcone (12) and trans-4-phenyl-3-buten-2-one (13) afforded addition products in low yield and enantiomeric excess (entries 5 and 6). Results with 3-alkyl enones were equally poor.
Another limitation of this transformation is encountered with cyclic 2- and/or 3-substituted enones. For the 3-substituted enones, no products could be isolated even upon addition of a base or temperature increase; these substrates are known for their low reactivity in 1,4-addition reactions. On the other hand, for 2-substituted enones we believe steric factors impede efficient coordination, and thus activation, at the metal center.
Extension to 2,6-dimethylphenol (14) and 2,6-dimethylaniline (19) as Michael donors was not met by success (Scheme 2). The deprotonation is expected to be more difficult (pKa values in DMSO : thiophenol = 10.3, phenol = 18, aniline = 30.6), and, once formed, nucleophiles derived from phenols and anilines would have a greater propensity to irreversibly bind the Lewis acidic ruthenium center. For the reasons stated above, the asymmetric catalytic Michael additions with amine- and/or alcohol-based donors are scarce.9d,25 Lowering the pKa by using electron-poor aryl substituents or changing the reaction medium to water could make this transformation possible and it is worth further investigation.
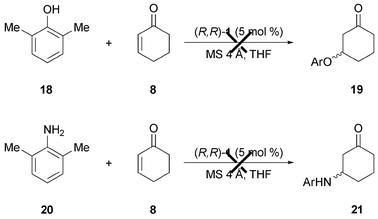 | ||
| Scheme 2 Attempts to use phenols and anilines as Michael donors for the Ru-catalyzed 1,4-addition. | ||
The general picture of the mechanism of the Michael addition is well known and supported by a wealth of theoretical and experimental data.5 More specific, for the tertiary amine-catalyzed 1,4-addition of thiols to enones, early mechanistic studies suggested a rate-limiting transfer of the thiolate from of a 1 : 1 complex of thiol and base, to form an enolate intermediate which is rapidly protonated to give the product (Scheme 3). The reaction thus follows first order kinetics in each of the donor, acceptor, and base.26
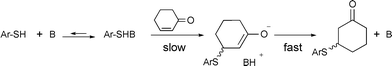 | ||
| Scheme 3 Mechanism of the amine catalyzed 1,4-addition of thiols to enones. | ||
The absence of a base in our case suggests a more complex mechanism, in which several interconnected equilibria are playing a role on the reaction outcome and mechanism (Scheme 4).27 We encountered a similar case when investigating asymmetric Lewis acid-catalyzed 1,3-dipolar cycloadditions with nitrones.20a31P NMR data showed competitive nitrone-enal coordination to the Ru-Lewis acid 1 with preferential binding of the enal and readily reversible binding of both enal and nitrone.
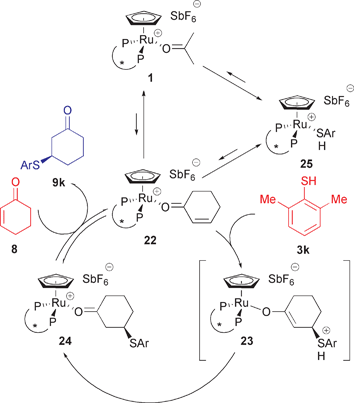 | ||
| Scheme 4 Proposed catalytic cycle for the Ru-catalyzed 1,4-addition of thiols to enones. Complexes 1, 22, 24 and 25 were observed by 31P NMR spectroscopy. | ||
The 31P NMR spectrum of the [Ru(acetone)(R,R-BIPHOP–F)Cp][SbF6] complex (1) (18 mg, 0.025 mmol, 10 mol% based on enone) in THF-d8 shows the expected peaks corresponding to the BIPHOP–F ligand bound to the monocationic CpRu fragment (doublets at 125 and 128 ppm, Fig. 2).28
![31P NMR experiments showing the formation of [Ru(R,R-BIPHOP–F)Cp(2-cyclohexen-1-one)][SbF6] (22) from the acetone complex 1.28](/image/article/2010/OB/b918877k/b918877k-f2.gif) | ||
| Fig. 2 31P NMR experiments showing the formation of [Ru(R,R-BIPHOP–F)Cp(2-cyclohexen-1-one)][SbF6] (22) from the acetone complex 1.28 | ||
Addition of 2-cyclohexen-1-one (8) (10 eq., 0.25 mmol) leads to an important decrease of the intensity of signals for the acetone complex 1, along with the appearance of two new doublets (124 and 130 ppm, Fig. 2). These are assigned to the 2-cyclohexen-1-one complex 22. The ratio of 4 : 1 in favour of 22 shows that in THF-d8, at −20 °C, the ruthenium Lewis acid prefers coordinating acetone over 2-cyclohexen-1-one (8) by a factor of 2.5 (10 : 1 reagent ratio acetone : 2-cyclohexen-1-one).
Addition of 2,6-DMTP (3k, 15 eq., 0.38 mmol) to the mixture above led to the appearance of two new sets of doublets (130 and 165 ppm, Fig. 3). The reaction was monitored by repeating a sequence of 1H and 31P analyses for 20 h at −20 °C. No change was observed in the spectra other than formation of the Michael adduct.
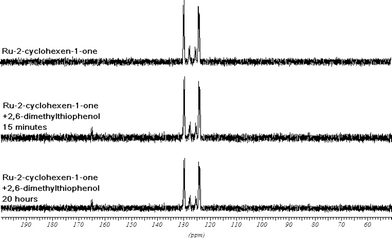 | ||
| Fig. 3 31P NMR experiments showing the complexes 1, 22, 24 and 25 present during the Ru-catalyzed 1,4-addition of 2,6-DMTP (3k) to 2-cyclohexen-1-one (8).28 | ||
In order to determine the nature of the complexes formed during the reaction, 2,6-DMTP (3k) was added to a solution of the ruthenium acetone complex 1 in THF-d8. This produced a color change from yellow to orange, suggesting appearance of charge-separated Ru-thiophenolate complex 26, as in the case of usual catalytic reactions with thiol 3k. However, the disappearance of peaks corresponding to the acetone complex 1 is accompanied by the formation of two pairs of doublets, the major complex at 130 and 165 ppm and the minor one at 126 and 163 ppm respectively (Fig. 4). The two complexes are in a ratio of approximately 5 to 1. While the minor complex cannot be assigned yet, the chemical shifts of the major complex confirm the formation of the Ru-2,6-DMTP complex (25) during the Michael addition.
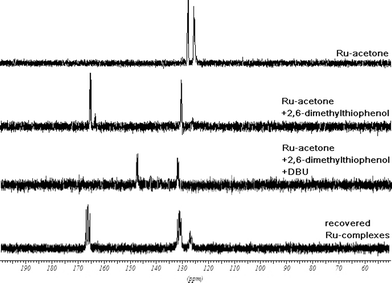 | ||
| Fig. 4 31P NMR experiments showing the existence of Ru-2,6-dimethylthiophenol (25) and Ru-2,6-dimethylthiophenolate (26) complexes, possible intermediates in the catalytic cycle.28 | ||
Several attempts to isolate the Ru-thiophenolate complex 26 were carried out. Using NaH or NaOH in the presence of the ruthenium complex 1 led to decomposition. Deprotonation of 2,6-DMTP (3k) with DBU prior to adding the ruthenium acetone complex 1 gave quantitative formation of a single species, with clear doublets at 131 and 149 ppm, which were tentatively assigned to the [Ru(R,R-BIPHOP–F)Cp][2,6-dimethylthiophenolate] complex (26) (Fig. 4). To confirm this, isolated 2,6-DMTPNa salt was mixed with the Ru-acetone complex in THF-d8. The 31P NMR spectrum showed quantitative formation of 26.
The standard workup after a catalytic run involves addition of hexanes. The resulting suspension is filtered through a pad of Celite. The yellow–orange solution is then concentrated and purified to yield pure Michael adducts. The precipitate on the Celite contains a mixture of ruthenium complexes and can be eluted with acetone. The 31P NMR spectrum shows the mixture to contain the Ru-2,6-DMTP complex (25, major, quartets at 131 and 166 ppm) along with an unidentified complex (minor, doublets at 127 and 131 ppm, Fig. 4).
Next, the possibility of the Michael adduct complexing the monocationic complex was considered. To a mixture of the ruthenium complexes 1 and 22 (ratio 4 : 1) in THF-d8, excess Michael adduct (9k) was added (10 eq.). Only two doublets could be observed (124 and 127 ppm) that were attributed to the overlapping signals of the Ru-acetone 1 and Ru-adduct 24 complexes.
A Ru-enolate complex (23) was never observed; since protonation in the classical mechanism is the fastest step, such a complex is expected to rapidly lead to the Ru-adduct complex (25) (Scheme 4). While identification of all species involved in the catalytic cycle has not been realized, these NMR experiments provide insight to the complex nature of the equilibria present in the reaction mixture.
X-ray structures of Lewis acid substrate complexes help in for the rationalization of the observed selectivity in cycloaddition reactions.19–23 Crystals suitable for X-ray analysis of the Ru-2-cyclohexen-1-one complex 22 were obtained from a dichloromethane–pentane solvent mixture (Fig. 5).20 Complex 22 closely resembles the structure containing methylvinyl ketone described previously.22 In both cases, the enone is coordinated in an anti-s-trans conformation and the Ru–O bond length (2.613 Å) and tilt angle of the bound enone are similar.24
![X-Ray structure representation of [Ru(R,R-BIPHOP–F)Cp(2-cyclohexen-1-one)][SbF6] (22) showing the Ru-enone coordination to be anti-s-trans.24](/image/article/2010/OB/b918877k/b918877k-f5.gif) | ||
| Fig. 5 X-Ray structure representation of [Ru(R,R-BIPHOP–F)Cp(2-cyclohexen-1-one)][SbF6] (22) showing the Ru-enone coordination to be anti-s-trans.24 | ||
Based on the X-ray structure of 22 the addition of the Michael donor would be expected to occur to the exposed alkene Si face of the enone, leading to the S-(–) adduct.
The assignment of absolute configuration for the Michael adducts 9a–g was made by comparison of [α]D values with literature data.12 For the other products, the absolute configuration was assigned by analogy. To our surprise, measurements indicated Michael adduct 9k to be R-(+). We came upon the same issue when assigning the absolute configuration for the Diels–Alder adducts of some enones and dienes.22 We note here that enals react exclusively in the anti-s-trans orientation. In this case the ground state structure and the reacting conformation are identical. With enones, the two coordination modes anti-s-trans and syn-s-trans are sterically and electronically very similar. The results presented here for the 1,4-additions show that, as in some of the Diels–Alder reactions, the reactive coordination conformation of the Ru-enone is not the one adopted in the crystal structure but it is the syn-s-trans conformation. This being the case, the Michael donor approaches the more available alkene Re face of the enone, leading to the observed R-(+) product (see Fig. 6).
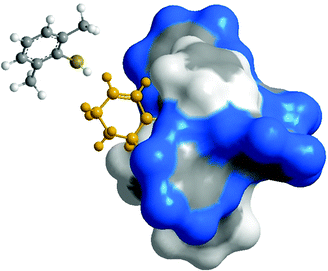 | ||
| Fig. 6 Modelled approach of the thiol 3k to the 2-cyclohexen-1-one (8) coordinated at the Ru center in a syn-s-trans conformation.24 | ||
Conclusions
The ruthenium half-sandwich Lewis-acidic complexes 1 and 2 can efficiently catalyze 1,4-additions of aryl thiols to enones. Whilst still somewhat limited in substrate scope, the ruthenium catalyzed asymmetric catalytic 1,4-addition of aryl thiols to enones shows the potential of such monocationic complexes for future applications. Remarkable levels of activity and selectivity are observed in spite of complex stereocontrol and potential catalyst inhibition.Experimental Section24
General experimental procedure
In a 50 mL oven-dried Schlenk tube equipped with a magnetic stirring bar, the catalyst (18 mg, 0.025 mmol, 5 mol%) was loaded along with powdered, activated MS 4 Å (50 mg) and the desired solvent (1 mL) was added. The mixture was stirred at the appropriate temperature and the enone (0.25 or 0.5 mmol, 1 eq.) was added. To the stirred solution, thiophenol (0.38 or 0.75 mmol, 1.5 eq.) was added dropwise by a syringe (or by means of a syringe pump, as a solution in THF, for the period indicated). The advancement of the reaction was followed by TLC (SiO2, CH2Cl2). The reaction was stopped by precipitation of the [Ru(R,R-BIPHOP–F)Cp(PhSH)][SbF6] complex with excess hexanes (8–10 mL), the mixture was filtered on a plug of Celite 545 (on frit) and then and volatiles were removed in vacuo to give the product as an oil. The crude product was further purified by FCC (SiO2, pentanes/CH2Cl2 1/1, 0/1). Enantiomeric excess was determined by means of HPLC analysis (CHIRALPAK AD or CHIRACEL OD-H, ISO hexane–isopropanol 99.5 : 0.5). Units: [α]D values are given in 10−1 deg cm2 g−1, c values are given in g 100 mL−1, δ values are given in ppm, J values are given in Hz, tR (retention times) are given in minutes.Acknowledgements
We thank the Swiss National Science Foundation for financial support.References
- (a) F. da Silva, Jr, R. J. Williams, The Biological Chemistry of the Elements, Oxford University Press, New York, 2001 Search PubMed; (b) C. Chatgilialoglu, K.-D. Asmus, Sulfur-Centered Reactive Intermediates in Chemistry and Biology, Springer, New York, 1991 Search PubMed.
- (a) A. L. Fluharty, in The Chemistry of the Thiol Group, part 2, ed. S. Patai, Wiley, New York, 1974, pp. 589-668 Search PubMed; (b) M. Mikolajczyk, J. Drabowicz, A. Kielbasinski, in Methods of Organic Chemistry, 1995, vol. E21e, ch. 5, Georg Thime Verlag, Stuttgart Search PubMed.
- (a) R. J. Parry, in Comprehensive Natural Products Chemistry, eds. D. Barton, K. Nakanishi, 1999, vol. 1, p. 825, Pergamon Press, Oxford Search PubMed; (b) R. E. Gawley, J. Aube, in Principles of Asymmetric synthesis, Tetrahedron Organic Chemistry Series, 1996, vol. 14, p. 161, Oxford, Pergamon Press Search PubMed; (c) R. Okazaki, in Organosulfur Chemistry, ed. P. Page, 1995, vol. 1, p. 225, Academic Press, London Search PubMed; (d) Q. L. Zhou and A. Pfaltz, Tetrahedron, 1994, 50, 4467 CrossRef CAS; (e) J. C. Anderson and M. Harding, Chem. Commun., 1998, 393 RSC , and references therein; (f) C. E. Aroyan and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 256 CrossRef; (g) S. Fanjul, A. N. Hulme and J. W. White, Org. Lett., 2006, 8, 4219 CrossRef CAS.
- (a) T. Kondo and T.-A. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS; (b) M. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS.
- P. Perlmutter, Conjugate Addition Reactions in Organic Syntheses, 1992, Pergamon Press Search PubMed.
- (a) M.-C. Chu, S. Gao, S. Sastry, C.-W. Kuo, C. Lu, J.-T. Liu and C.-F. Yao, Tetrahedron, 2007, 63, 1863 CrossRef CAS , and references therein; (b) G. L. Khatik, R. Kumar and A. K. Chakraborti, Org. Lett., 2006, 8, 2433 CrossRef CAS; (c) B. Movassagh and P. Shaygan, ARKIVOC, 2006, XII, 130; (d) T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem.–Eur. J., 2004, 10, 484 CrossRef CAS; (e) M. Meciarova and S. Toma, Lett. Org. Chem., 2006, 3, 794 Search PubMed.
- (a) H. Pracejus, F.-W. Wilke and K. Hanemann, J. Prakt. Chem., 1977, 319, 219 CAS; (b) H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 16, 4057 CrossRef; (c) N. Kobayashi and K. Iwai, J. Org. Chem., 1981, 46, 1823 CrossRef CAS; (d) T. Mukaiyama, A. Ikegawa and K. Suzuki, Chem. Lett., 1981, 165 CAS.
- (a) P. McDaid, Y. Chen and L. Deng, Angew. Chem., Int. Ed., 2002, 41, 338 CrossRef CAS; (b) D. Leow, S. Lin, S. K. Chittimalla, X. Fu and C.-H. Tan, Angew. Chem., Int. Ed., 2008, 47, 5641 CrossRef CAS; (c) Y. Liu, B. Sun, B. Wang, M. Wakem and L. Deng, J. Am. Chem. Soc., 2009, 131, 418 CrossRef CAS , and references therein.
- (a) G. Manickam and G. Sundararajan, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1997, 36, 516; (b) G. Manickam and G. Sundararajan, Tetrahedron: Asymmetry, 1997, 8, 2271 CrossRef CAS; (c) G. Manickam and G. Sundararajan, Tetrahedron, 1999, 55, 2721 CrossRef CAS; (d) G. Sundararajan and N. Prabagaran, Org. Lett., 2001, 3, 389 CrossRef CAS; (e) N. Prabagaran and G. Sundararajan, Tetrahedron: Asymmetry, 2002, 13, 1053 CrossRef CAS; (f) N. Prabagaran, S. Abraham and G. Sundararajan, ARKIVOC, 2002, VII, 212.
- (a) E. Emori, T. Arai, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 4043 CrossRef CAS; (b) E. Emori, T. Iida and M. Shibasaki, J. Org. Chem., 1999, 64, 5318 CrossRef CAS; (c) D. Sawada, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 10521 CrossRef CAS.
- (a) S. Narasimhan and S. Velmathi, Synth. Commun., 2002, 32, 3791 CrossRef CAS; (b) S. Narasimhan and S. Velmathi, Molecules, 2003, 8, 256 Search PubMed; (c) S. Narasimhan, S. Swarnalakshmi and S. Velmathi, Tetrahedron: Asymmetry, 2003, 14, 113 CrossRef CAS.
- (a) M. Saito, M. Nakajima and S. Hashimoto, Chem. Commun., 2000, 1851 RSC; (b) M. Saito, M. Nakajima and S. Hashimoto, Tetrahedron, 2000, 56, 9589 CrossRef CAS.
- G. Kumaraswamy, M. N. V. Sastry and N. Jena, Tetrahedron Lett., 2001, 42, 8515 CrossRef CAS.
- T. Kumamoto, S. Aoki, M. Nakajima and K. Koga, Tetrahedron: Asymmetry, 1994, 5, 1431 CrossRef CAS.
- (a) K. Nishimura, M. Ono, Y. Nagaoka and K. Tomioka, J. Am. Chem. Soc., 1997, 119, 12974 CrossRef CAS; (b) K. Tomioka, M. Okuda, K. Nishimura, S. Manabe, M. Kanai, Y. Nagaoka and K. Koga, Tetrahedron Lett., 1998, 39, 2141 CrossRef CAS; (c) K. Nishimura, M. Ono, Y. Nagaoka and K. Tomioka, Angew. Chem., Int. Ed., 2001, 40, 440 CrossRef CAS; (d) K. Nishimura and K. Tomioka, J. Org. Chem., 2002, 67, 431 CrossRef CAS; (e) K. Nishimura and K. Tomioka, Yakugaku Zasshi, 2003, 123, 9 CrossRef CAS.
- (a) S. Kobayashi, C. Ogawa, M. Kawamura and M. Sugiura, Synlett, 2001, 983 CrossRef CAS; (b) K. Matsumoto, A. Watanabe, T. Uchida, K. Ogi and T. Katsuki, Tetrahedron Lett., 2004, 45, 2385 CrossRef CAS; (c) S. J. Sauerland, E. Kiljunen and A. M. Koskinen, Tetrahedron Lett., 2006, 47, 1291 CrossRef CAS.
- (a) N. N. Joshi and S. C. Jha, ARKIVOC, 2002, VII, 167; (b) D. Enders, K. Luttgen and A. Narine, Synthesis, 2007, 959 CrossRef CAS.
- M. E. Bruin and E. P. Kündig, Chem. Commun., 1998, 2635 RSC.
- (a) E. P. Kündig, C. M. Saudan and G. Bernardinelli, Angew. Chem., 1999, 111, 1297 CrossRef; E. P. Kündig, C. M. Saudan and G. Bernardinelli, Angew. Chem., Int. Ed., 1999, 38, 1219 CrossRef; (b) E. P. Kündig, M. C. Saudan, V. Alezra, F. Viton and G. Bernardinelli, Angew. Chem., 2001, 113, 4613 CrossRef; E. P. Kündig, M. C. Saudan, V. Alezra, F. Viton and G. Bernardinelli, Angew. Chem., Int. Ed., 2001, 40, 4481 CrossRef; (c) E. P. Kündig, C. M. Saudan and F. Viton, Adv. Synth. Catal., 2001, 343, 51 CrossRef CAS; (d) P. G. A. Kumar, P. S. Pregosin, M. Vallet, G. Bernardinelli, R. F. Jazzar, F. Viton and E. P. Kündig, Organometallics, 2004, 23, 5410 CrossRef CAS; (e) V. Alezra, G. Bernardinelli, C. Corminboeuf, U. Frey, E. P. Kündig, A. E. Merbach, C. M. Saudan, F. Viton and J. Weber, J. Am. Chem. Soc., 2004, 126, 4843 CrossRef CAS.
- (a) F. Viton, G. Bernardinelli and E. P. Kündig, J. Am. Chem. Soc., 2002, 124, 4968 CrossRef CAS; (b) A. Bădoiu, Y. Brinkmann, F. Viton and E. P. Kündig, Pure Appl. Chem., 2008, 80, 1013 CrossRef CAS; (c) A. Bădoiu, G. Bernardinelli, J. Mareda, E. P. Kündig and F. Viton, Chem.–Asian J., 2008, 3, 1298 CrossRef CAS; (d) A. Bădoiu, G. Bernardinelli, J. Mareda, E. P. Kündig and F. Viton, Chem.–Asian J., 2009, 4, 1021 CrossRef CAS (this article corrects Chem.–Asian J., 2008, 3, 1298).
- Y. Brinkmann, J. M. Reniguntala, R. Jazzar, G. Bernardinelli and E. P. Kündig, Tetrahedron, 2007, 63, 8413 CrossRef CAS.
- J. Rickerby, M. Vallet, G. Bernardinelli, F. Viton and E. P. Kündig, Chem.–Eur. J., 2007, 13, 3354 CrossRef CAS.
- (a) K. J. Corvy and C. J. Helal, Tetrahedron Lett., 1995, 36, 9153 CrossRef CAS; (b) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458 CrossRef CAS; (c) J. M. Hawkins, M. Nambu and S. Loren, Org. Lett., 2003, 5, 4293 CrossRef CAS; (d) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, L. A. Oro, F. J. Lahoz, A. I. Balana, T. Tejero and P. Merino, J. Am. Chem. Soc., 2005, 127, 13386 CrossRef CAS; (e) D. Carmona, M. P. Lamata, F. Viguri, J. Ferrer, N. Garcia, F. J. Lahoz, M. L. Martin and L. A. Oro, Eur. J. Inorg. Chem., 2006, 3155 CrossRef CAS; (f) R. Gordillo and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 3543 CrossRef CAS.
- See the electronic supplementary information for general experimental information, a complete table of solvent and temperature optimization, full experimental procedures and characterization data for all compounds, and a discussion on the X-ray structure of 22. See DOI: 10.1039/b918877k.
- (a) X. Lu and L. Deng, Angew. Chem., Int. Ed., 2008, 47, 7710 CrossRef CAS; (b) F. Pesciaioli, F. De Vincentiis, P. Galzerano, G. Bencivenni, G. Bartoli, A. Mazzanti and P. Melchiorre, Angew. Chem., Int. Ed., 2008, 47, 8703 CrossRef CAS; (c) D. R. Li, A. Murugan and J. R. Falck, J. Am. Chem. Soc., 2008, 130, 46 CrossRef CAS; (d) J. Alemán, A. Milelli, S. Cabrera, E. Reyes and K. A. Jørgensen, Chem.–Eur. J., 2008, 14, 10958 CrossRef CAS.
- (a) B. Dmuchovsky, B. D. Vineyard and F. B. Zienty, J. Am. Chem. Soc., 1964, 86, 2874 CrossRef CAS; (b) H. Hiemstra and H. Wynberg, J. Am. Chem. Soc., 1981, 103, 417 CrossRef CAS.
- V. van Axel Castelli, A. Dalla Cort and L. Mandolini, J. Am. Chem. Soc., 1998, 120, 12688 CrossRef CAS.
- NMR experiments were carried out in degassed, dry [D8]THF, at −20 °C and under N2, keeping the ratios between components as in the catalytic reactions. For practical reasons, no molecular sieves were used. Peaks of residual THF were used as internal standard in the 1H NMR analyses that preceded all 31P NMR spectra. Analyses were carried out on a Bruker Avance 500 spectrometer.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental information, a complete table of solvent and temperature optimization, full experimental procedures and characterization data for all compounds, and a discussion on the X-ray structure of 22. CCDC reference numbers 747701 and 750039. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b918877k |
| ‡ Dedicated to the memory of Christophe M. Saudan |
| This journal is © The Royal Society of Chemistry 2010 |