Epimeric and amino disaccharide analogs as probes of an α-(1→6)-mannosyltransferase involved in mycobacterial lipoarabinomannan biosynthesis†
Pui Hang Tam and Todd L. Lowary
Alberta Ingenuity Centre for Carbohydrate Science and Department of Chemistry, The University of Alberta, Gunning-Lemieux Chemistry Centre, Edmonton, AB T6G 2G2, Canada. E-mail: tlowary@ualberta.ca
First published on 16th November 2009
Abstract
Mycobacterial lipoarabinomannan (LAM) is an important, immunologically active glycan found in the cell wall of mycobacteria, including the human pathogen Mycobacterium tuberculosis. At the core of LAM is a mannan domain comprised of α-(1→6)-linked-mannopyranose (Manp) residues. Previously, we and others have demonstrated that α-Manp-(1→6)-α-Manp disaccharides (e.g., Manp-(1→6)-α-ManpOctyl, 1) are the minimum acceptor substrates for enzymes involved in the assembly of the LAM mannan core. We report here the synthesis five epimeric and three amino analogs of 1, and their subsequent biochemical evaluation against an α-(1→6)-ManT activity present in a membrane preparation from M. smegmatis. Changing the manno- configuration of either residue of 1 to talo- or gluco- led to a reduction or loss of activity, thus confirming earlier work showing that the C-2 and C-4 hydroxyl groups of each monosaccharide were important for enzymatic recognition. Characterization of the products formed from these analogs was done using a combination of mass spectrometry and glycosidase digestion, and full substrate kinetics were also performed. The analogs in which the acceptor hydroxyl group had been replaced with an amino group were, as expected, not substrates for the enzyme, but were weak inhibitors.
Introduction
Lipoarabinomannan (LAM) is an important cell wall constituent in mycobacteria, including those that cause the human diseases tuberculosis (Mycobacterium tuberculosis) and leprosy (M. leprae).1 This glycoconjugate, which contains both arabinofuranose (Araf) and mannopyranose (Manp) residues, has been implicated in a host of biochemical and immunological processes associated with mycobacterial infection.2,3 There has consequently been significant interest not only in obtaining a molecular-level understanding of its immunomodulatory activity, but also in elucidating the biosynthetic pathway by which LAM is assembled.2–6All mycobacteria, and a number of related organisms, produce LAMs, the structure of which varies from species to species.2,3 In mycobacterial LAM (Fig. 1), the molecule is built upon an acylated phosphatidyl inositol anchor from which extends a chain of Manp residues connected viaα-(1→6) linkages. Approximately half of these core mannan residues are further elaborated by single α-(1→2)-linked Manp branching units, and this structure serves as a scaffold to which an arabinan domain containing α-(1→2)-, α-(1→3)- and α-(1→5)-linked Araf residues is bound.1–3 Depending on the species, the arabinan can be modified at its periphery by a number of capping groups such as short α-(1→2)-linked Manp oligosaccharides,2,3,7 5-deoxy-5-thiomethyl-xylofuranose residues8–11 or inositol phosphate moieties.12
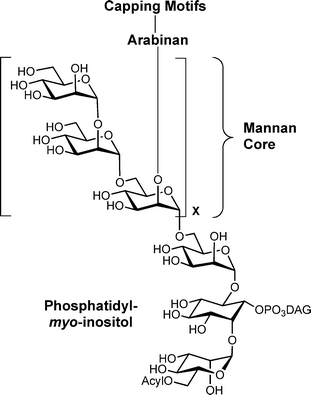 | ||
| Fig. 1 Composite structure of mycobacterial LAM; DAG = diacylglycerol. | ||
The role of each structural domain on the immunomodulatory function of LAM remains relatively poorly understood. For example, although previous studies suggested the capping motifs were important for virulence,2,3,7 more recent studies have suggested this is not the case.13 Other recent investigations have demonstrated that the ability of these glycans to bind to the toll-like receptor 2 (TLR-2) is influenced by mannan chain length14 and acylation state.15 Similarly, the recognition of LAM fragments by anti-LAM antibodies has also been studied recently.16–18
The model proposed more than a decade ago by Brennan and coworkers19 for the biosynthesis of LAM has, for the most part, been borne out through genetic and biochemical investigations carried out since that time.4,5 Of particular relevance to the work reported in this paper are a family of polyprenolphosphomannose (PPM)-dependent mannosyltransferases (ManTs) that are involved in the assembly of the mannan domain of this glycan. Earlier studies reported the use of a membrane preparation from M. smegmatis to assay for the α-(1→6)-ManT activity required for the synthesis of the LAM mannan core.20,21 This assay, in which the donor substrate for the enzyme, is generated in situ from GDP-mannose (Fig. 2), has been used to screen potential substrates and inhibitors of this enzymatic activity,22,23 and it has been demonstrated that α-Manp-(1→6)-α-Manp disaccharides (e.g., 1) are the minimum acceptor substrates.21,24 It is currently unknown how many enzymes are involved in the assembly of the mannan core. Although earlier investigations in this area assumed, without substantial experimental support, that a single enzyme was involved, recent work with mycobacterial gene knock-out strains,25 has provided evidence that more than one α-(1→6)-ManT is required.
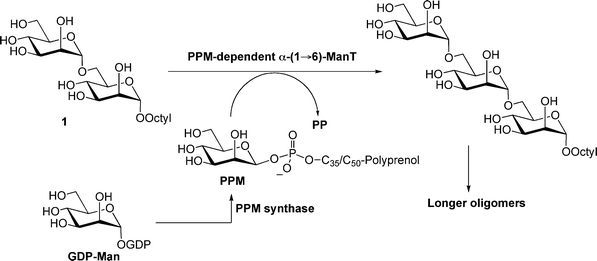 | ||
| Fig. 2 Reported assay for measuring PPM-dependent α-(1→6)-ManT activity in mycobacterial membrane preparations. PPM = polyprenolphoshomannose; GDP = guanosine disphosphate. | ||
In previous papers, we reported the preparation26 and biochemical evaluation24 of a panel of monomethoxy and monodeoxy analogs of disaccharide acceptor 1 (Fig. 3) as potential substrates and inhibitors for this α-(1→6)-ManT activity. Among the important findings from these investigations were that the enzyme appears to form critical hydrogen bonding interactions with a number of hydroxyl groups on the substrate because deoxygenation leads, in all but two cases (C-2′ and C-4), to essentially a total loss of activity. To extend our understanding of the substrate specificity of this enzymatic activity further, we describe here the synthesis five epimeric (3, 5, 7–9) and three amino (2, 4 and 6) analogs of 1 (Fig. 3), and their subsequent biochemical evaluation against the α-(1→6)-ManT activity present in the previously mentioned M. smegmatis membrane preparation.
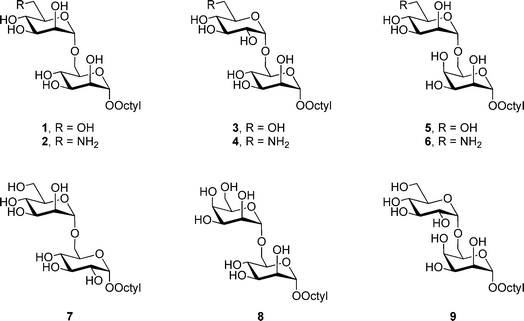 | ||
| Fig. 3 Synthetic disaccharide targets 2–9. | ||
The epimers were selected as targets to determine if the manno-configuration was absolutely required by the enzyme, or if disaccharides with rings in the gluco- (3, 7, 9) or talo- (5, 8, 9) configuration were also recognized. The results obtained from these analogs would provide additional insight into the important steric and hydrogen bonding interactions in the active site of these proteins. In addition, the amino analogs were synthesized and evaluated as potential inhibitors. We envisioned that the amino groups of 2, 4 and 6 would be protonated at physiological pH, and the resulting ammonium derivatives would serve as inhibitors via an ionic interaction with a negatively charged residue in the enzyme active site.27,28 It has been shown previously that octyl oligosaccharides are efficient substrates for glycosyltransferases from mycobacteria and other organisms.21,27–30 Moreover, the attachment of this hydrophobic group to an oligosaccharide allows for convenient product isolation using reversed-phase chromatography.30 Thus, the target compounds 2–9 were synthesized as octyl glycosides.
Results and discussion
Preparation of monosaccharide building blocks
We envisioned that disaccharides 2–9 could be assembled from monosaccharide precursors 10–16 (Fig. 4). Previously reported methods were used to prepare octyl glycoside 1031 and 16,32 as well as thioglycosides 1133 and 12.26 The other building blocks, talopyranosides 13 and 14, and glucopyranoside 15, were synthesized as outlined below.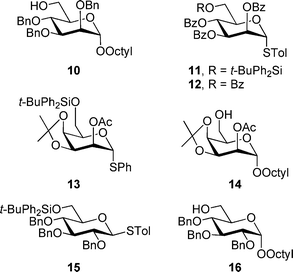 | ||
| Fig. 4 Monosaccharide building blocks used for the synthesis of 2–9. | ||
Thioglycoside 13 and octyl glycoside 14 were synthesized from the known thioglycoside 1734 as illustrated in Scheme 1. First, reaction of 17 with tert-butylchlorodiphenylsilane and imidazole provided silyl ether 18 in 94% yield. Subsequent reaction of 18 with 2,2-dimethoxypropane in the presence of a catalytic amount of p-TsOH gave a quantitative yield of alcohol 19.
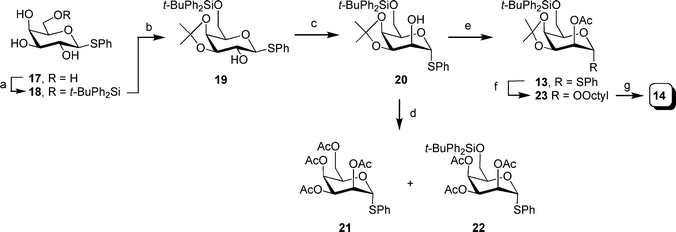 | ||
Scheme 1 Reagents and conditions: a) t-BuPh2SiCl, imidazole, DMF, 45 °C, 94%; b) 2,2-dimethoxypropane, pTsOH, acetone, quant.; c) i: oxalyl chloride, DMSO, −78 °C; then alcohol 19, warm to −60 °C; then Et3N, warm to rt.; ii: NaBH4, MeOH, 62%; d) i: 80% aq. AcOH, 50 °C; ii: Ac2O, DMAP, CH2Cl2, pyridine, 65% for 21 and 26% for 22; e) Ac2O, CH2Cl2, pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 89%; f) octanol, NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 89%; g) TBAF, THF, 74%. :1), 89%; f) octanol, NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 89%; g) TBAF, THF, 74%. | ||
Oxidation of the C-2 hydroxyl group under Swern conditions and subsequent reduction with sodium borohydride gave an anomeric mixture of thioglycoside 20 (62% yield of a 6:1 α/β mixture of isomers, which could be separated by chromatography). In the 1H NMR spectrum of the major isomer, the H-1 resonance was shifted to 5.33 ppm, which is in good agreement with the α-configuration at the anomeric center. Presumably, the basic conditions of the oxidation reaction resulted in the anomerization of the ketone product, as has been reported previously.35 However, the J1,2 (7.4 Hz) of the product obtained after borohydride reduction was larger than would be expected for thioglycosides with either the α-galacto- or α-talo-configuration.
We postulated that the larger than expected coupling constant value was due to the distortion of the chair conformation of the pyranose ring by the isopropylidene protecting group. Therefore, to confirm the stereochemistry at C-2, 20 was hydrolyzed with 80% aqueous AcOH and then reacted with acetic anhydride in the presence of pyridine and DMAP to afford a mixture of 21 and 22. The J1,2 and J2,3 of 22 (1.3 and 3.7 Hz, respectively) are consistent with the α-talo-configuration, in turn confirming the structure of 20 as that shown in Scheme 1.
Having determined that the oxidation–reduction sequence had provided the correct product, 20 was treated with acetic anhydride thus providing an 89% yield of 13. Glycosidation of 13 with n-octanol gave octyl talopyranoside 23 in 89% yield. Finally, treatment of 33 with tetra-n-butylammonium fluoride provided a 74% yield of the expected alcohol 14.
As illustrated in Scheme 2, thioglycoside 15, was obtained from the known thioglucoside 24.36 Reaction of 24 with tert-butylchlorodiphenylsilane and imidazole provided silyl ether intermediate 25. Without purification, the remaining hydroxyl groups were protected as benzyl ethers upon treatment with sodium hydride and benzyl bromide to afford 15 in 95% yield over the two steps.
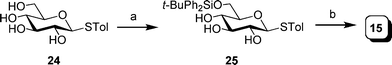 | ||
| Scheme 2 Reagents and conditions: a) t-BuPh2SiCl, imidazole, DMF, 45 °C; b) NaH, BnBr, DMF, 95% two steps. | ||
Synthesis of disaccharides
As illustrated in Scheme 3, the synthesis of amino disaccharide 2 was began with the NIS–TMSOTf activated37 glycosylation of thioglycoside 1133 and alcohol 10,31 which afforded disaccharide 26 in 85% yield. For this reaction, and all other glycosylations described in this paper using mannopyranoside and talopyranoside donors, the α-stereochemistry of the glycosidic linkages was confirmed by measurement of the one-bond heteronuclear coupling constant for the anomeric carbon atom (1JC-1,H-1). In all cases, this value was between 167 and 176 Hz, clearly indicating the α-stereochemistry.38Next, disaccharide 26 was desilyated using hydrogen fluoride in pyridine39 to give 27 in 66% yield. The resulting primary alcohol in 27 was then tosylated with tosyl chloride in pyridine (91%); treatment of the product, 28, with sodium azide gave azido-disaccharide 29 in 98% yield. That the substitution had occurred was confirmed from the 1H NMR spectrum of 29, in which the protons on C-6′ resonated between 3.38 and 3.48 ppm, as would be expected for hydrogens adjacent to an azido functionality. In addition, in the 13C NMR spectrum, a resonance at 51.2 ppm could be assigned to C-6′.
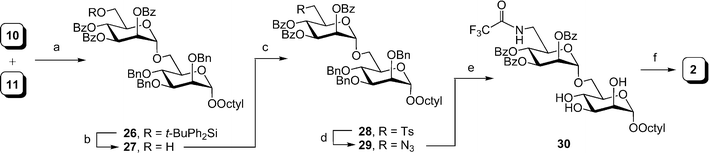 | ||
| Scheme 3 Reagents and conditions: a) NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 85%; b) HF·pyridine–pyridine–THF (1:2:20), 66%; c) TsCl, pyridine, 91%; d) NaN3, DMF, reflux, 98%; e) i: Pd(OH)2–C, pyridine, H2; ii: (CF3CO)2O, pyridine, 0 °C to rt.; iii: Pd(OH)2–C, MeOH, H2, 68% over three steps; f) NaOMe, MeOH, 61%. | ||
Similar to an earlier observation on related disaccharides,40 simultaneous reduction of the azide and deprotection of the benzyl groups by treatment of 29 with hydrogen and a palladium catalyst failed. Thin layer chromatographic analysis of the reaction mixture showed several products, presumably a series of partially-debenzylated derivatives of 29. No further reduction was observed, even after prolonged reaction times. Instead, the azido group of 29 was reduced to the amine by hydrogenation over Pd(OH)2–C in pyridine, and the product was immediately converted to its N-trifluoroacetamide using trifluoroacetic anhydride in pyridine.40 Subsequent hydrogenolysis of the benzyl groups using Pd(OH)2–C catalyst was achieved without incident and provided triol 30 in 68% yield over three steps. Final deprotection of 30 using sodium methoxide in methanol furnished a 61% yield of the desired amino disaccharide 2.
As illustrated in Scheme 4, the synthesis of disaccharides 3 and 4 were achieved by first coupling thioglycoside 15 with alcohol 10,31 under NIS–TMSOTf activation. This reaction afforded an inseparable diastereoisomeric mixture (∼5:1 α:β ratio) of products. Subsequent desilylation using hydrogen fluoride followed by chromatographic purification gave the α-linked disaccharide 31 in 50% yield over the two steps. Final deprotection of the benzyl ethers afforded 3 in 82% yield.
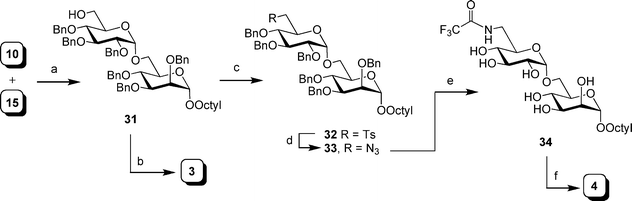 | ||
| Scheme 4 Reagents and conditions: a) i: NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 5:1 of α:β isomers; ii: HF·pyridine–pyridine–THF (1:2:20), 50%; b) Pd(OH)2–C, MeOH, H2, 82%; c) TsCl, pyridine, 89%; d) NaN3, DMF, reflux, 85%; e) i: Pd(OH)2–C, pyridine, H2; ii: (CF3CO)2O, pyridine, 0 °C to rt.; iii: Pd(OH)2–C, MeOH, H2, 72% over three steps; f) NaOMe, MeOH, 79%. | ||
The amino disaccharide 4 was synthesized via a route analogous to 2, starting with 31. Thus, the primary hydroxyl group in 31 was first tosylated to provide 32, which upon heating in reflux in DMF with sodium azide gave azidosugar 33 in 76% yield over the two steps. As was seen for compound 29, the primary azido functionality in 33 was evident in the 13C NMR spectrum, which showed a resonance for C-6′ at 51.2 ppm. The azido group of 33 was then converted to the corresponding N-trifluoroacetamide derivative in two steps (azide reduction and trifluoroacetylation of the product amine). Without purification, the crude intermediate was hydrogenated to provide 34 in 72% yield over three steps. Final deprotection by treatment with sodium methoxide furnished a 79% yield of the target analog 4.
The synthesis of 4 and 5, starting from octyl talopyranoside 14, is shown in Scheme 5. Coupling of this acceptor with thioglycoside 1133 under NIS–TMSOTf activation afforded the corresponding protected disaccharide 35 in 90% yield. Deprotection of the silyl ether proceeded under standard conditions to give a 77% yield of 36. Cleavage of the isopropylidene acetal and treatment with sodium methoxide in methanol gave 5 in 91% overall yield. To prepare 6, alcohol 36 was converted to azido disaccharide 37via the two step sequence (91% and 84% yields, respectively) described for the preparation of 29. Reduction of azide 37, following removal of the isopropylidene acetal and acyl protecting groups, provided aminosugar 6 in 86% yield.
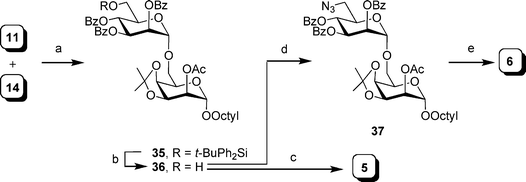 | ||
| Scheme 5 Reagents and conditions: a) NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 90%; b) HF·pyridine–pyridine–THF (1:2:20), 77%; c) i: 80% aq. AcOH, 50 °C; ii: NaOMe, MeOH, 91%; d) i: TsCl, pyridine, 91%; ii: NaN3, DMF, reflux, 84%; e) i: 80% aq. AcOH, 50 °C; ii: NaOMe, MeOH; iii: Pd(OH)2–C, MeOH, H2, 86% over three steps. | ||
The preparation of disaccharides 7–9 is illustrated in Scheme 6. Glycosylation of alcohols 10,3114, 1632 with thioglycosides 12,2613, 15 using NIS–TMSOTf activation, gave disaccharides 38, 39, and 40 in excellent yields (92%, 81% and 88%, respectively). Debenzoylation and hydrogenolysis of 38 provided the desired compound 7 (84% yield). To obtain 8 and 9, disaccharides 39 and 40 were subjected to a four-step sequence (desilylation, deacetylation, acetal cleavage and hydrogenolysis) to give the expected products in overall yields of 45% and 62%, respectively. NMR analysis of the final compounds suggested that the modified oligosaccharides have a conformation similar to the parent compounds. For example, the chemical shifts and coupling constants were consistent with all of the pyranose rings adopting a 4C1 conformation.
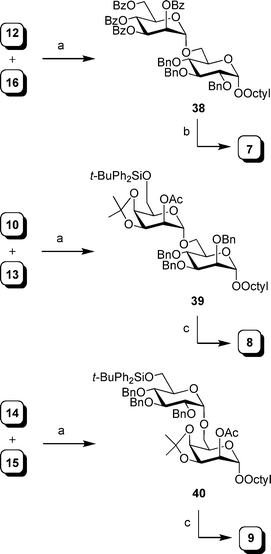 | ||
| Scheme 6 Reagents and conditions: a) NIS, TMSOTf, 4 Å MS, CH2Cl2, 0 °C, 92% for 38, 81% for 39, 88% for 40 (5:1 of α:β isomers); b) i: NaOMe, MeOH; ii: Pd(OH)2–C, MeOH, H2, 84%; c) i: TBAF, THF; ii: NaOMe, MeOH; iii: 80% aq. AcOH, 50 °C; iv: Pd(OH)2–C, MeOH, H2, 45% for 8; 62% for 9. | ||
Screening analogs as substrates and inhibitors
Once synthesized, 2–9 were screened as potential substrates and inhibitors of the PPM-dependent α-(1→6)-ManT activity, present in the M. smegmatis membrane preparation.20,21 Each analog was incubated with [3H]-labeled GDP-mannose (Fig. 2) and the membrane extracts, the radiolabeled products were recovered by solvent extraction and reversed-phase chromatography, and the radioactivity was measured by scintillation counting.The ability of 2–9 to act as acceptor substrates for ManT were compared with the parent compound 1 and the results are summarized in Table 1. The initial screening revealed that analogs 3 (α-Glcp-(1→6)-α-Manp) and 5 (α-Manp-(1→6)-α-Talp) are moderate substrates of ManT with 41% and 57% activity, relative to 1 (α-Manp-(1→6)-α-Manp). Our previous data suggested that the hydroxyl groups at C-2′ and C-4 of 1 were not crucial for substrate–enzyme interaction, as the replacement with hydrogen had no dramatic effect on ManT catalysis.24 However, it appears that epimerization at either position, leading to analog 3 or 5, significantly affects the efficiency to serve as a substrate. This finding is further indicated by the apparent kinetic parameters shown in Table 1. These acceptor analogs, have KM values of 1.8 mM (3) and 1.2 mM (5), which are 13- and 20-fold larger, respectively, than that of 1 (0.09 mM). The Vmax values of 3 or 5 are comparable to that of the parent compound.
| Analog | Relative activitya (%) | Apparent KMb/mM | Apparent Vmaxb/pmol mg min−1 | Remaining ManT activityc (%) | Mass of oligosaccharide productd |
|---|---|---|---|---|---|
| a Relative activities were measured at 2.0 mM acceptor concentration with 0.2 μCi of [3H]GDP-Man and are expressed with respect to disaccharide 1. 100% activity corresponds to 29.8 pmol mg−1 h−1.b Kinetic parameters were determined using a range of acceptor concentrations by nonlinear regression analysis of the Michaelis–Menten equation with the GraphPad Prism 4.0 program.c Compounds were screened at a concentration of 2.0 mM with 1 as the substrate at 0.2 mM.d The enzymatic products were isolated from larger-scale incubations and their masses were determined by MALDI mass spectrometry. The found values correspond to the sodium adducts, which were in good agreement with the calculated values.e Not determined. | |||||
| 1 | 100 | 0.09 ± 0.01 | 0.37 ± 0.005 | — | 639.2, 801.3 |
| 2 | 4 | –e | — | 21 | — |
| 3 | 41 | 1.80 ± 0.71 | 0.34 ± 0.071 | — | 639.1, 801.1 |
| 4 | 0 | — | — | 9 | — |
| 5 | 57 | 1.20 ± 0.24 | 0.40 ± 0.031 | — | 639.2, 801.2 |
| 6 | <1 | — | — | 30 | — |
| 7 | 10 | 3.04 ± 0.69 | 0.10 ± 0.014 | — | 639.3, 801.3, 963.3 |
| 8 | <1 | — | — | — | — |
| 9 | 10 | 2.27 ± 0.12 | 0.11 ± 0.003 | — | 639.3, 801.3 |
The lower relative activity of 3 and 5 compared to 1 appears to be due to the steric requirements of the ManT. For both compounds, the hydroxyl group is placed in the opposite orientation in the parent compound (changed from axial to equatorial in 3, or vice versa in 5). This change appears to lead to a potential negative steric interaction with amino acid residues in the active site thus resulting in an unfavorable enzyme–substrate interaction and a higher KM. These observations suggest that while the enzyme preferentially recognizes disaccharides in which both residues are in the manno-configuration, it will also accept analogs of different stereochemical configurations, but with lower affinity.
This finding is further supported by the results obtained from analogs 7–9, in which α-Talp-(1→6)-α-Manp analog 8 is not a substrate, and both the α-Manp-(1→6)-α-Glcp7 and α-Glcp-(1→6)-α-Talp analogs 9 are poor substrates with only 10% relative activity compared to 1. Consistent with the results for 3 and 5, subsequent kinetic analysis provided larger apparent KM values for 7 and 9, 3.04 mM and 2.27 mM, respectively. However, the Vmax values for these substrates was only marginally less than the native substrate, 1. The poor substrate activity of 7, and the lack of activity of 8, is consistent with our previous work,24 which indicated that the C-4′ and C-2 hydroxyl groups are essential for ManT catalysis. In 3 and 5, the orientations of the C-4′ and C-2 hydroxyl groups were the same as in the parent compound, while the stereochemistry at one other centre was inverted (C-2′ in 3 and C-4 in 5), and these compounds were weak substrates for the enzyme. However, evaluation of an analog in which the stereochemistry at both C-2′ and C-4 were inverted, the α-Glcp-(1→6)-α-Talp isomer 9, showed even weaker ManT substrate activity.
In summary, the change of the configuration of the disaccharide motif, regardless of whether the change was at the reducing or non-reducing end, did not enhance but rather reduced the ManT activity. This finding indicates that the enzyme requires a manno- configuration of both rings for optimal activity.
Characterization of enzymatic products by mass spectrometry
In addition to the radiochemical assays, milligram-scale enzymatic incubations of 1, 3, 5, 7 and 9 with unlabelled GDP-Man and the membrane fraction were carried out to determine the structure of the oligosaccharide products that were produced from these analogs. After the incubations, the enzymatic products were purified and analyzed by MALDI mass spectrometry. As shown in Fig. S1, these reactions all resulted in the formation of the corresponding trisaccharides and tetrasaccharides as the major and minor products, respectively. Contrary to the proposed processive nature of this enzyme,20 a homologous series of products was not observed. The absence of these larger oligosaccharides is probably due to much lower acceptor concentrations of the resulting tri- and tetrasaccharide products, resulting in slower rates of the subsequent elongation reactions. It is also possible that the larger enzymatic products (e.g., tetra- and pentasaccharide) are degraded by an α-(1→6)-endo-mannosidase present in the membrane preparation.24Characterization of enzymatic products by glycosidase digestion
In addition to the PPM-dependent α-(1→6)-mannosyltransferases, the crude membrane extract of M. smegmatis used in these assays also contains α-(1→2)-ManT's involved in mannan core branching and the capping of the arabinan domain.25,41–43 To confirm that the observed addition of radiolabeled mannose to 3, 5, 7 and 9 arose from α-(1→6)-ManT activity, the radiochemical assays were repeated on a larger scale with [3H]-labelled GDP-Man. After the reaction, the [3H]Man-labeled enzymatic products were purified, divided evenly and digested with glycosidases.As illustrated in Fig. 5, there is no significant difference in the radioactivities between the control experiments and the samples that have undergone treatment with the α-Man-(1→2)-α-Man-specific Aspergillus saitoiα-(1→2)-mannosidase (AS). These results demonstrate that none of the mannosylated products contained an α-Man-(1→2)-α-Man linkage. On the other hand, digestions of the radiolabeled products using α-mannosidases from jack bean (JB, α-(1→2,3,6)-specific) and Xanthomonas manihotis (XM, unbranched α-(1→6)-specific) removed essentially all the [3H]-labeled mannose units. The combination of these mannosidases enabled the relative proportions of the α-(1→6)- and α-(1→3/4)-linkages to be determined. These results further suggested that the observed mannosylations, in all cases except 3, resulted exclusively from the action of an α-(1→6)-specific ManT in the membrane preparation.
![Mannosidase digestion of products formed from 3, 5, 7 and 9. Each acceptor at 2 mM was incubated with [3H] GDP-Man under the assay conditions as described in the experimental section. The radiolabeled enzymatic products were divided evenly after purification and treated with exo-mannosidases including Aspergillus saitoiα-(1→2)-mannosidase (AS), jack bean α-(1→2,3,6)-mannosidase (JB) and Xanthomonas manihotisα-(1→6)-mannosidase (XM). The mannosidase-digested samples were purified using C18 reversed-phase column and the radioactivities were compared with the controls (without any mannosidase treatment).](/image/article/2010/OB/b916580k/b916580k-f5.gif) | ||
| Fig. 5 Mannosidase digestion of products formed from 3, 5, 7 and 9. Each acceptor at 2 mM was incubated with [3H] GDP-Man under the assay conditions as described in the experimental section. The radiolabeled enzymatic products were divided evenly after purification and treated with exo-mannosidases including Aspergillus saitoiα-(1→2)-mannosidase (AS), jack bean α-(1→2,3,6)-mannosidase (JB) and Xanthomonas manihotisα-(1→6)-mannosidase (XM). The mannosidase-digested samples were purified using C18 reversed-phase column and the radioactivities were compared with the controls (without any mannosidase treatment). | ||
In the case of 3, the small difference of the residual radioactivity between the initial product and that treated with mannosidases from jack bean and X. manihotis suggests that a small amount of the enzymatic products from 3 might contain α-(1→3/4)-linkages. Besra and co-workers have recently demonstrated that in M. tuberculosis an enzyme, MgtA is able to utilize GlcpAGroAc2 (1,2-di-O-C16/C18:1-α-D-glucopyranosyluronic acid-(1→3)-glycerol as a substrate to form α-Manp-(1→4)-GlcpAGroAc2 (Fig. 6).44
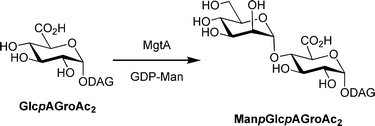 | ||
| Fig. 6 Reaction catalyzed by MgtA. DAG = diacylglycerol. | ||
It is therefore plausible to speculate that MgtA has a relaxed substrate specificity, which allows it to recognize both disaccharides such as 3 as well as GlcAGroAc2. With regard to this proposal, it is important to note that the non-reducing residue in 3, is a Glcp residue and this may be a reasonable substrate for this newly discovered mannosyltransferase activity. It is also important to consider that an MgtA-like enzymatic activity has not, to date, been identified in M. smegmatis, the source of the membrane fraction used in these investigations. Although disaccharide 9 also carries a Glcp residue at its non-reducing terminus, no α-(1→3/4)-linkage was detected from the exo-mannosidase treatments. Unfortunately, we were unable to carry out any further investigation due to the small turnover of 3, and the negligible amount of the observed α-(1→3/4)-linked products obtained in the assay.
Inhibition effects of amino analogs
As expected, analogs 2, 4 and 6, which lack a hydroxyl group at C-6′, are not substrates of ManT; all compounds show relative activities below 5% (Table 1). These amino derivatives were next tested as inhibitors against ManT using 1 as the substrate. In these studies, analogs 2, 4 and 6 were screened at a concentration of 2.0 mM with the parent compound 1 at 0.2 mM. As shown in Table 1, disaccharides 2, 4 and 6 inhibit the mannosylation of 1 by 79, 91 and 70%, respectively. These compounds are thus very weak inhibitors (at 10-fold excess of 1) and not comparable to other aminosugar-containing acceptor analogs, which have been demonstrated to be very potent glycosyltransferase inhibitors.28 To examine if the inhibition was proportional to an increase in inhibitor concentration, we studied the mannosylation of 1 (fixed concentration of 0.2 mM) by the α-(1→6)-ManT in the presence of these amino analogs at concentrations up to 4 mM. In all cases, 50% inhibition was observed at about 1 mM under these conditions, as shown in Fig. 7.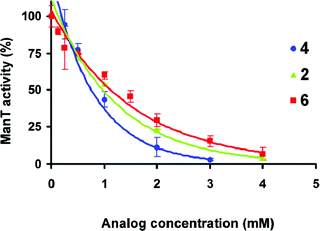 | ||
| Fig. 7 Inhibition effects of amino analogs 2, 4 and 6 on ManT activity. The activities were determined using aminosugar analog concentrations up to 4.0 mM with 1 as the acceptor substrate at 0.2 mM. All other reaction conditions were identical to the cell-free assay as described in the experimental section. | ||
Based on the data shown in Table 1, the substrate preference for ManT, based on the relative activity of the epimeric substrate analogs, appears to follow the order of α-Manp-(1→6)-α-Manp1 (100%) > α-Manp-(1→6)-α-Talp5 (57%) > α-Glcp-(1→6)-α-Manp3 (41%). We were therefore surprised to see that the 6-amino-α-Glcp-(1→6)-α-Manp analog 4, rather than 6-amino-α-Manp-(1→6)-α-Manp analog 2 is a comparably better inhibitor. One explanation for this result is that these amino analogs may not be competing with 1 in the active site, but rather act in a noncompetitive or uncompetitive fashion. However, given that these analogs are at best modest inhibitors of α-(1→6)-mannosylation, we chose not to further probe the mechanism by which they inhibit the enzyme.
Conclusions
In this paper we have described the synthesis of a panel of epimeric and amino analogs of disaccharide 1 and their evaluation as potential substrates and inhibitors of a PPM-dependent α-(1→6)-ManT involved in LAM/LM biosynthesis. The results presented here are in agreement with our previous findings that the hydroxyl groups at C-4′ and C-2 positions in 1 are important for both the polar interactions and steric requirements. In particular, a change of the manno- configuration of the parent substrate 1, at either the reducing or non-reducing end, to the gluco- or talo-configuration, results in poor turnover by the ManT, with the most pronounced loss of activity being observed for a compound in which the stereochemistry at C-4′ and C-2 are both inverted. Analogs 2, 4 and 6, in which the OH group at C-6′ has been substituted with an amino group, are weak inhibitors against ManT. Although the observed inhibition may result from an ionic interaction of the protonated amino groups of 2, 4 and 6 with a negatively charged residue in the enzyme active site, the substrate specificity trends observed in the epimeric substrates are different from those seen with the aminosugar analogs. This suggests that the mode of inhibition may not be competitive. Regardless of the mode of action, the α-Manp-(1→6)-α-Manp (2), α-Glcp-(1→6)-α-Manp (4), and α-Manp-(1→6)-α-Talp (6) analogs all show 50% inhibition at a concentration of ∼1 mM. A summary of these results in presented graphically in Fig. 8.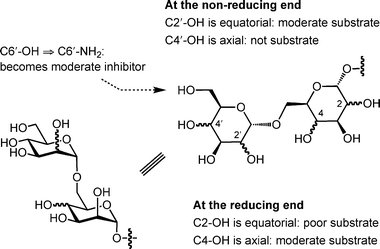 | ||
| Fig. 8 Graphical summary of activity of analogs 1–9 with α-(1→6)-ManT. | ||
Experimental section
General methods for chemical synthesis
All reagents used were purchased from commercial sources and were used without further purification unless noted. Solvents used in reactions were purified by successive passage through columns of alumina and copper under nitrogen. Unless indicated otherwise, all reactions were performed at room temperature (rt) and under a positive pressure of argon. The reactions were monitored by analytical TLC on silica gel 60-F254 (0.25 mm, Silicycle) and spots were detected under UV light or by charring with acidified anisaldehyde solution in ethanol. Organic solvents were evaporated under reduced pressure at <40 °C. Products were purified by chromatography using silica gel (40–60 μM), Iatrobeads (Iatron Laboratories, Tokyo) or SepPak C18 reversed-phase cartridges (Waters). Before use, the SepPak cartridges were prewashed with 10 mL of MeOH followed by 10 mL of H2O. The crude residue in water was loaded onto the prewashed column. After washing the column with water (10 mL), the desired product was eluted with MeOH (4 mL). Optical rotations were measured at 22 ± 2 °C and are in units of degrees mL/(g dm). 1H NMR spectra were recorded at 400, 500 or 600 MHz, and chemical shifts are referenced to either TMS (0.0, CDCl3), or HOD (4.78, D2O and CD3OD). 13C NMR spectra were recorded at 100 or 125 MHz and chemical shifts are referenced to internal CDCl3 (77.23, CDCl3), or CD3OD (48.9, CD3OD). Assignments of NMR spectra were made based on two-dimensional (1H–1H COSY and HMQC) experiments. The stereochemistry at the anomeric centres of the mannopyranose and talopyranose rings were proven by measuring the 1JC1-H1.719F spectra were recorded at 376 MHz, and chemical shifts were referenced to external CFCl3. Samples were prepared by the cast film method and infrared spectra were measured on a FT-IR spectrometer. Electrospray mass spectra were recorded on samples suspended in mixtures of THF with MeOH and added NaCl.:1 CH2Cl2–CH3OH) to give 3 (14 mg, 82%) as clear glass. Rf 0.54 (4:1 CH2Cl2–MeOH); [α]D = +99.1 (c 0.3, CH3OH); 1H NMR (500 MHz, CD3OD) δH 4.82 (d, 1H, J = 3.7 Hz, H-1′), 4.70 (d, 1H, J = 1.7 Hz, H-1), 4.00 (dd, 1H, J = 10.6, 4.0 Hz, H-6a), 3.61–3.82 (m, 9H, H-3′, H-5′, H-6a′, H-6b′, H-2, H-3, H-4, H-5, octyl OCH2), 3.60 (dd, 1H, J = 10.6, 2.3 Hz, H-6b), 3.39 (dt, 1H, J = 9.6, 6.3 Hz, octyl OCH2), 3.35 (dd, 1H, J = 9.6, 3.7 Hz, H-2′), 3.30 (overlap with residual CD2HOD peak, 1H, H-4′), 1.52–1.62 (m, 2H, octyl OCH2CH2), 1.23–1.42 (m, 10H, octyl CH2), 0.89 (t, 3H, J = 7.0 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 101.8 (C-1), 100.0 (C-1′), 74.3 (C-5′/C-5), 73.9 (C-2′), 73.5 (C-5′/C-5), 72.8 (C-3′, C-3), 72.3 (C-2), 71.8 (C-4′), 68.8 (octyl OCH2), 68.4 (C-4), 67.3 (C-6), 62.6 (C-6′), 33.0 (octyl CH2), 30.6(3) (octyl CH2), 30.5(6) (octyl CH2), 30.4 (octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.4 (octyl CH3). HRMS (ESI) calcd. for (M + Na) C20H38O11: 477.2306. Found: 477.2305.:1 AcOH–H2O (5 mL) and heated at 50 °C for 3 h. The mixture was diluted with EtOAc (20 mL), washed with satd aq NaHCO3 (5 mL), dried (Na2SO4), and concentrated. The resulting product was subsequently dissolved in MeOH (5 mL) and NaOMe (54 mg) was added. After 2 h, the solution was neutralized with AcOH and, following concentration, the crude product was purified by chromatography on Iatrobeads (4:1 CH2Cl2–CH3OH) to give 5 (21 mg, 91% over three steps) as clear glass. Rf 0.54 (4:1 CH2Cl2–MeOH); [α]D = +78.2 (c 0.4, CH3OH); 1H NMR (600 MHz, CD3OD) δH 4.80 (br s, 1H, H-1), 4.79 (d, 1H, J = 1.8 Hz, H-1′), 3.60–3.74 (m, 2H, H-5, H-6a), 3.82 (dd, 1H, J = 11.8, 2.3 Hz, H-6a′), 3.80 (dd, 1H, J = 3.3, 1.8 Hz, H-2′), 3.58–3.74 (m, 9H, H-3′, H-4′, H-5′, H-6b′, H-2, H-3, H-4, H-6b, octyl OCH2), 3.43 (dt, 1H, J = 9.7, 6.3 Hz, octyl OCH2), 1.52–1.62 (m, 2H, octyl OCH2CH2), 1.24–1.41 (m, 10H, octyl CH2), 0.89 (t, 3H, J = 7.0 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 102.2 (C-1), 101.6 (C-1′), 74.6 (C-5′), 72.7, 72.6 (C-3′, C-3), 72.2 (C-2), 71.7 (C-2′), 71.0 (C-5), 68.9 (octyl OCH2), 68.5 (C-4′), 67.9 (C-6), 67.2 (C-4), 62.9 (C-6′), 33.0 (octyl CH2), 30.6 (octyl CH2), 30.5 (octyl CH2), 30.4 (octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.4 (octyl CH3). HRMS (ESI) calcd. for (M + Na) C20H38O11: 477.2306. Found: 477.2307.:1 AcOH–H2O (5 mL) and heated at 50 °C for 6 h. The mixture was cooled, diluted with EtOAc (20 mL), washed with satd aq NaHCO3 (5 mL), dried (Na2SO4), and concentrated. The crude product was subsequently dissolved in MeOH (4 mL) and 1 M NaOMe (1 mL) was added. After stirring overnight, the solution was neutralized with Amberlite 120 resin (H+ form), filtered and concentrated. The azidosugar intermediate was dissolved in CH3OH (8 mL) and 20% Pd(OH)2–C (50 mg) was added. The mixture was stirred overnight under a H2 atmosphere and the catalyst was separated by filtration through a short pad of Celite. The eluant was concentrated, redissolved in satd aq NaHCO3 (5 mL) and extracted with EtOAc (5 mL). The aqueous layer was purified by on a SepPak C18 reversed-phase cartridge to give 6 (24 mg, 86%), after lyophilization, as white solid; [α]D = +100.0 (c 0.2, CH3OH); 1H NMR (500 MHz, CD3OD) δH 4.81 (br s, 1H, H-1), 4.79 (d, 1H, J = 1.6 Hz, H-1′), 3.86–3.94 (m, 2H, H-5, H-6a), 3.80 (dd, 1H, J = 3.3, 1.8 Hz, H-2′), 3.77–3.81 (m, 1H, H-2), 3.58–3.75 (m, 6H, H-3′, H-5′, H-3, H-4, H-6b, octyl OCH2), 3.52 (dd, 1H, J = 9.5, 9.5 Hz, H-4′), 3.42 (dt, 1H, J = 9.6, 6.3 Hz, octyl OCH2), 3.05 (dd, 1H, J = 13.5, 2.9 Hz, H-6a′), 2.83 (dd, 1H, J = 13.5, 7.0 Hz, H-6b′), 1.52–1.64 (m, 2H, octyl OCH2CH2), 1.23–1.42 (m, 10H, octyl CH2), 0.90 (t, 3H, J = 6.9 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 102.2 (C-1), 101.6 (C-1′), 73.7 (C-5′), 72.6, 72.5 (C-3′, C-3), 72.1 (C-2′), 71.8 (C-2), 71.1 (C-5), 69.8 (C-4′), 68.9 (octyl OCH2), 68.2 (C-6), 67.2 (C-4), 43.4 (C-6′), 33.0 (octyl CH2), 30.6 (octyl CH2), 30.5 (octyl CH2), 30.4 (octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.4 (octyl CH3). HRMS (ESI) calcd. for (M + H) C20H40NO10: 454.2647. Found: 454.2649.:1 CH2Cl2–CH3OH). The pure fractions were collected, concentrated, and redissolved in CH3OH (8 mL) and 20% Pd(OH)2–C (30 mg) was added. The mixture was stirred overnight under a H2 atmosphere and the catalyst was separated by filtration through a short pad of Celite. The filtrate was concentrated to give 7 (16 mg, 84%) as clear glass. Rf 0.36 (4:1 CH2Cl2–MeOH); [α]D = +95.7 (c 0.5, CH3OH); 1H NMR (600 MHz, CD3OD) δH 4.80 (d, 1H, J = 1.7 Hz, H-1′), 4.74 (d, 1H, J = 3.9 Hz, H-1), 3.89 (dd, 1H, J = 11.1, 5.3 Hz, H-6a), 3.78–3.84 (m, 2H, H-2′, H-6a′), 3.65–3.74 (m, 5H, H-3′, H-6b′, H-5, H-6b, octyl OCH2), 3.58–3.65 (m, 3H, H-4′, H-5′, H-3), 3.43 (dt, 1H, J = 9.6, 6.3 Hz, octyl OCH2), 3.37 (dd, 1H, J = 9.7, 3.9 Hz, H-2), 3.33 (dd, 1H, J = 10.0, 9.0 Hz, H-4), 1.56–1.69 (m, 2H, octyl OCH2CH2), 1.24–1.46 (m, 10H, octyl CH2), 0.89 (t, 3H, J = 6.6 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 101.5 (C-1′), 100.2 (C-1), 75.3 (C-3), 74.4 (C-2), 73.6, 72.7, 72.2, 72.1 (4C, C-2′, C-3′, C-5′, C-5), 71.7 (C-4), 69.3 (octyl OCH2), 68.6 (C-4′), 67.1 (C-6), 62.9 (C-6′), 33.0 (octyl CH2), 30.7 (octyl CH2), 30.6 (octyl CH2), 30.4 (octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.4 (octyl CH3). HRMS (ESI) calcd. for (M + Na) C20H38O11: 477.2306. Found: 477.2305.:1 HOAc–H2O (5 mL) at 50 °C overnight. The mixture was diluted with CH2Cl2 (15 mL), washed with satd aq NaHCO3 (5 mL × 2), and the organic phase was dried (Na2SO4) and concentrated. The resulting crude product was redissolved in CH3OH (8 mL) and 20% Pd(OH)2–C (15 mg) was added. The mixture was stirred overnight under a H2 atmosphere and the catalyst was separated by filtration through a short pad of Celite. The filtrate was concentrated and the residue purified by chromatography on Iatrobeads (4:1 CH2Cl2–MeOH) to give 8 (8 mg, 45%) as clear glass. Rf 0.38 (4:1 CH2Cl2–MeOH); [α]D = +83.5 (c 0.3, CH3OH); 1H NMR (600 MHz, CD3OD) δH 4.90 (d, 1H, J = 1.5 Hz, H-1′), 4.70 (d, 1H, J = 1.7 Hz, H-1), 3.80–3.93 (m, 3H, H-3′, H-4′, H-6a), 3.77 (dd, 1H, J = 3.2, 1.7 Hz, H-2), 3.71–3.77 (m, 5H, H-2′, H-5′, H-6a′, H-6b′, H-6b), 3.60–3.69 (m, 4H, H-3, H-4, H-5, octyl OCH2), 3.40 (dt, 1H, J = 9.6, 6.4 Hz, octyl OCH2), 1.52–1.62 (m, 2H, octyl OCH2CH2), 1.24–1.42 (m, 10H, octyl CH2), 0.90 (t, 3H, J = 7.2 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 102.0 (C-1′), 101.7 (C-1), 73.1, 72.8, 72.5, 72.2 (5C, C-2′, C-3′, C-2, C-3, C-4), 71.8 (C-4′), 68.6 (octyl OCH2), 68.6 (C-5), 67.5 (C-6), 67.3 (C-5′), 62.9 (C-6′), 33.0 (octyl CH2), 30.6 (octyl CH2), 30.5 (octyl CH2), 30.4 (octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.4 (octyl CH3). HRMS (ESI) calcd. for (M + Na) C20H38O11: 477.2306. Found: 477.2303.:1 HOAc–H2O (5 mL) at 50 °C overnight. The mixture was diluted with CH2Cl2 (15 mL), washed with sat. aq. NaHCO3 (5 mL x 2), and the organic phase was dried (Na2SO4) and concentrated. The resulting crude product was redissolved in CH3OH (8 mL) and 20% Pd(OH)2–C (20 mg) was added. The mixture was stirred overnight under a H2 atmosphere and the catalyst was separated by filtration through a short pad of Celite. The filtrate was concentrated and the residue was purified by chromatography on Iatrobeads (4:1 CH2Cl2–MeOH) to give 9 (23 mg, 62%) as clear glass. Rf 0.33 (4:1 CH2Cl2–MeOH); [α]D = +102.9 (c 1.3, CH3OH); 1H NMR (500 MHz, CD3OD) δH 4.82 (d, 1H, J = 3.7 Hz, H-1′), 4.80 (d, 1H, J = 1.5 Hz, H-1), 3.93 (dd, 1H, J = 6.3, 6.3 Hz, H-5), 3.84–3.90 (m, 2H, H-4, H-6a), 3.79 (dd, 1H, J = 11.9, 2.4 Hz, H-6a′), 3.66–3.76 (m, 5H, H-6b′, H-2, H-3, H-6b, octyl OCH2), 3.59–3.66 (m, 2H, H-3′, H-5′), 3.41 (dt, 1H, J = 9.7, 6.4 Hz, octyl OCH2), 3.40 (dd, 1H, J = 9.8, 3.7 Hz, H-2′), 3.31 (dd, 1H, J = 9.8, 8.9 Hz, H-4′), 1.52–1.62 (m, 2H, octyl OCH2CH2), 1.24–1.42 (m, 10H, octyl CH2), 0.89 (t, 3H, J = 7.2 Hz, octyl CH3); 13C NMR (125 MHz, CD3OD) δC 102.3 (C-1), 100.2 (C-1′), 75.2 (C-3′), 73.8 (C-5′), 73.6 (C-2′), 72.7 (C-2), 71.7 (C-4′), 71.4 (C-3), 70.8 (C-5), 69.0 (octyl OCH2), 68.0 (C-6), 67.1 (C-4), 62.6 (C-6′), 33.0 (octyl CH2), 30.6 (octyl CH2), 30.4 (2C, octyl CH2), 27.4 (octyl CH2), 23.7 (octyl CH2), 14.5 (octyl CH3). HRMS (ESI) calcd. for (M + Na) C20H38O11: 477.2306. Found: 477.2299.Bacterial strains and growth conditions
M. smegmatis mc2155 was a generous gift from Professor William R. Jacobs, Jr. at the Albert Einstein College of Medicine. The bacteria were grown at 37 °C in 100 mL of Luria Bertoni (LB) broth medium containing 0.05% Tween 80 to an A600nm of < 1.0 (∼two days from a frozen bacterial stock). 50 mL liquid cultures were then transferred to 2 × 1 L of fresh media and cultured further for 24 h at 37 °C. Cells were harvested by centrifugation, washed with phosphate buffered saline (PBS) and stored at −20 °C until use.Preparation of membrane fractions from M. smegmatis
The M. smegmatis cell pellet (∼10 g wet weight) was washed and resuspended in 100 mL of 50 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (adjusted to pH 7.9 with KOH) containing 5 mM β-mercaptoethanol and 10 mM MgCl2 supplemented with Complete Protease Inhibitor Cocktail Tablets (Roche) at 4 °C. The cells were subjected to two passes through a Thermo Spectronic French Pressure Cell Press at 20000 psi. The cell lysate was centrifuged at 600 ×g for 15 min and then at 27000 ×g for 20 min. The resulting supernatant was centrifuged at 100000 ×g for 60 min. The supernatant was carefully removed and the membrane pellets were gently resuspended in 1 mL of 50 mM MOPS buffer, pH 7.9, containing 5 mM β-mercaptoethanol and 10 mM MgCl2. Protein concentration (31.6 mg mL−1) was determined by the BCA(tm) Protein Assay (Pierce) using bovine serum albumin as the standard.Radiochemical activity assays
The ManT enzyme activity was determined using the previously established cell-free system.21 Unless indicated otherwise, the synthetic acceptor analogs at a concentration of 2.0 mM were incubated with 0.20 μCi of guanosine diphosphate mannopyranose (GDP-mannose), [mannose-2-3H] (American Radiolabeled Chemicals, Inc., 20 Ci mmol−1) in 50 mM MOPS buffer, pH 7.9, containing 1 mM ATP, 10 mM MgCl2, 5 mM β-mercaptoethanol, and membrane fraction (94.8 μg of protein) in a total volume of 80 μL. All assays were performed in duplicate and control assays without acceptor were also performed in parallel to correct for the presence of endogenous acceptor. The enzymatic activities were determined using radiochemical SepPak C18 assays.30 Briefly, after incubation at 37 °C for 1 h, the reactions were stopped by adding 100 μL of CHCl3–MeOH (2:1 v/v) and the mixtures were centrifuged. The supernatants were recovered and further diluted with H2O before loading onto SepPak C18 cartridges (Waters). The unreacted donor was removed by washing the cartridges with H2O (50 mL) and the radiolabeled products were eluted with MeOH (4.0 mL). The isolated products in the eluants were quantified by liquid scintillation counting on a Beckman LS6500 Scintillation Counter using 10 mL of Ecolite cocktail. For kinetic analysis, the ManT activities were determined using a range of acceptor concentrations. All other reaction conditions were identical to the cell-free assay as described above. Assays were performed under the conditions in which the formations of radiolabeled products were linear for both time and protein concentration. The kinetic parameters KM and Vmax were obtained by nonlinear regression analysis using the Michaelis–Menten equation with the GraphPad Prism 4.0 program (GraphPad Software, San Diego, CA).Product characterizations from milligram-scale incubations
Larger-scale ManT reactions were performed for the structural characterization using acceptor substrates 3, 5, 7 and 9. A typical reaction containing 50 mM MOPS buffer, pH 7.9, 1 mM ATP, 10 mM MgCl2, 5 mM β-mercaptoethanol, 2 mM acceptor, 2 mM GDP-mannose, and the M. smegmatis membrane preparation (3.2 mg) in a total volume of 0.5 mL, was incubated at 37 °C with gentle rotation for 2 days. The reaction was stopped by adding equal volume of CHCl3–MeOH (2:1, v/v) and the mixtures were centrifuged. The supernatant was recovered and further diluted with H2O before loading onto the C18 reversed-phase cartridge and the unreacted donor was washed away with H2O (50 mL) and the product was eluted subsequently with MeOH (4 mL). The solvent was evaporated and the residue was redissolved in H2O (50 μL). The conversion of the acceptor substrate to the enzymatic product was analyzed by MALDI mass spectrometry on a Voyager Elite time-of-flight spectrometer on sample suspended in 2,5-dihydroxy benzoic acid, using the delayed-extraction mode and positive-ion detection.Glycosidic linkage analysis by exo-mannosidases
Larger-scale ManT reactions (160 μL) were performed using acceptor substrates 3, 5, 7 and 9. A typical reaction contained 50 mM MOPS buffer, pH 7.9, 1 mM ATP, 10 mM MgCl2, 5 mM β-mercaptoethanol, 2 mM acceptor, 2 mM GDP-mannose, and the M. smegmatis membrane preparation (189.6 μg) in a total volume of 160 μL. The radioactive enzymatic products were purified using SepPak C18 cartridges. After removal of methanol, the residues were redissolved in 200 μL H2O. Ten microliters of the suspension were digested with exo-mannosidases, Aspergillus saitoiα-(1→2)-mannosidase (AS) (Glyko), jack bean α-(1→2,3,6)-mannosidase (JB) (Glyko) and Xanthomonas manihotisα-(1→6)-mannosidase (XM) (New England Biolabs), according to the manufacturers procedures. After incubation for one day, the reaction mixtures were purified using a C18 reversed-phase cartridge as described above.30Acknowledgements
This work was supported by the Alberta Ingenuity Centre for Carbohydrate Science, the University of Alberta and the Natural Sciences and Engineering Research Council of Canada. PHT thanks Alberta Ingenuity for an Ingenuity PhD Student Scholarship.References
- P. J. Brennan, Tuberculosis, 2003, 83, 91–97 CrossRef CAS.
- V. Briken, S. A. Porcelli, G. S. Besra and L. Kremer, Mol. Microbiol., 2004, 53, 391–403 CrossRef CAS.
- M. Nigou, M. Gilleron and G. Puzo, Biochimie, 2003, 85, 153–166 CrossRef CAS.
- S. Berg, D. Kaur, M. Jackson and P. J. Brennan, Glycobiology, 2007, 17, 35R–56R CrossRef CAS.
- P. H. Tam and T. L. Lowary, Curr. Opin. Chem. Biol., 2009 DOI:10.1016/j.cbpa.2009.09.012.
- L. J. Alderwick, H. L. Birch, A. Mishra, L. Eggeling and G. S. Besra, Biochem. Soc. Trans., 2007, 35, 1325–1328 CrossRef CAS.
- D. Chatterjee, K. Lowell, B. Rivoire, M. R. McNeil and P. J. Brennan, J. Biol. Chem., 1992, 267, 6234–6239 CAS.
- W. B. Turnbull, K. H. Shimizu, D. Chatterjee, S. W. Homans and A. Treumann, Angew. Chem., Int. Ed., 2004, 43, 3918–3922 CrossRef CAS.
- A. Treumann, X. D. Feng, L. McDonnell, P. J. Derrick, A. E. Ashcroft, D. Chatterjee and S. W. Homans, J. Mol. Biol., 2002, 316, 89–100 CrossRef CAS.
- M. Joe, D. Sun, H. Taha, G. C. Completo, J. E. Croudace, D. A. Lammas, G. S. Besra and T. L. Lowary, J. Am. Chem. Soc., 2006, 128, 5059–5072 CrossRef CAS.
- P. Ludwiczak, M. Gilleron, Y. Bordat, C. Martin, B. Gicquel and G. Puzo, Microbiology, 2002, 148, 3029–3037 CAS.
- K. H. Khoo, A. Dell, H. R. Morris, P. J. Brennan and D. Chatterjee, J. Biol. Chem., 1995, 270, 12380–12389 CrossRef CAS.
- B. J. Appelmelk, J. den Dunnen, N. N. Driessen, R. Ummels, M. Pak, J. Nigou, G. Larrouy-Maumus, S. S. Gurcha, F. Movahedzadeh, J. Geurtsen, E. J. Brown, M. M. Eysink Smeets, G. S. Besra, P. T. J. Willemsen, T. L. Lowary, Y. van Kooyk, J. J. Maaskant, N. G. Stoker, P. von der Ley, G. Puzo, C. M. J. E. Vandenbroucke-grauls, C. W. Wieland, T. von der Poll, T. B. H. Geijtenbeek, A. M. van der Sar and W. Bitter, Cell. Microbiol., 2008, 10, 930–944 CrossRef CAS.
- J. Nigou, T. Vasselon, A. Ray, P. Constant, M. Gilleron, G. Besra, I. C. Sutcliffe, G. Tiraby and G. Puzo, J. Immunol., 2008, 180, 6696–6702 CAS.
- E. Doz, S. Rose, J. Nigou, M. Gilleron, G. Puzo, F. Erard, B. Ryffel and V. Quesniaux, J. Biol. Chem., 2007, 282, 26014–26025 CrossRef CAS.
- T. Murase, R. B. Zheng, M. Joe, Y. Bai, S. L. Marcus, T. L. Lowary and K. K. S. Ng, J. Mol. Biol., 2009, 392, 381–392 CrossRef CAS.
- C. Rademacher, G. K. Shoemaker, H. S. Kim, R. B. Zheng, H. Taha, C. Liu, R. C. Nacario, D. C. Schriemer, J. S. Klassen, T. Peters and T. L. Lowary, J. Am. Chem. Soc., 2007, 129, 10489–10502 CrossRef CAS.
- D. Kaur, T. L. Lowary, V. D. Vissa, D. C. Crick and P. J. Brennan, Microbiology, 2002, 148, 3049–3057 CAS.
- G. S. Besra, C. B. Morehouse, C. M. Rittner, C. J. Waechter and P. J. Brennan, J. Biol. Chem., 1997, 272, 18460–18466 CrossRef CAS.
- K. Yokoyama and C. E. Ballou, J. Biol. Chem., 1989, 264, 21621–21628 CAS.
- J. R. Brown, R. A. Field, A. Barker, M. Guy, R. Grewal, K. H. Khoo, P. J. Brennan, G. S. Besra and D. Chatterjee, Bioorg. Med. Chem., 2001, 9, 815–824 CrossRef CAS.
- A. K. Pathak, V. Pathak, J. M. Riordan, S. S. Gurcha, G. S. Besra and R. C. Reynolds, Carbohydr. Res., 2004, 339, 683–691 CrossRef CAS.
- M. R. Guy, P. A. Illarionov, S. S. Gurcha, L. G. Dover, K. J. C. Gibson, P. W. Smith, D. E. Minnikin and G. S. Besra, Biochem. J., 2004, 382, 905–912 CrossRef CAS.
- P. H. Tam, G. S. Besra and T. L. Lowary, ChemBioChem, 2008, 9, 267–278 CrossRef CAS.
- D. Kaur, M. R. McNeil, K. H. Khoo, D. Chatterjee, D. C. Crick, M. Jackson and P. J. Brennan, J. Biol. Chem., 2007, 282, 27133–27140 CrossRef CAS.
- P. H. Tam and T. L. Lowary, Carbohydr. Res., 2007, 342, 1741–1772 CrossRef CAS.
- R. U. Lemieux, A. P. Venot, U. Spohr, P. Bird, G. Mandal, N. Morishima, O. Hindsgaul and D. R. Bundle, Can. J. Chem., 1985, 63, 2664–2668 CrossRef CAS.
- T. L. Lowary and O. Hindsgaul, Carbohydr. Res., 1994, 251, 33–67 CrossRef CAS.
- (a) R. E. Lee, P. J. Brennan and G. S. Besra, Glycobiology, 1997, 7, 1121–1128 CrossRef CAS; (b) N. L. Rose, G. C. Completo, S.-J. Lin, M. McNeil, M. M. Palcic and T. L. Lowary, J. Am. Chem. Soc., 2006, 128, 6721–6729 CrossRef CAS.
- M. M. Palcic, L. D. Heerze, M. Pierce and O. Hindsgaul, Glycoconjugate J., 1988, 5, 49–63 CAS.
- V. Subramaniam, S. S. Gurcha, G. S. Besra and T. L. Lowary, Bioorg. Med. Chem., 2005, 13, 1083–1094 CrossRef CAS.
- C. Wing, J. C. Errey, B. Mukhopadhyay, J. S. Blanchard and R. A. Field, Org. Biomol. Chem., 2006, 4, 3945–3950 RSC.
- A. Watts and S. J. Williams, Org. Biomol. Chem., 2005, 3, 1982–1992 RSC.
- J. Ohlsson and G. Magnusson, Carbohydr. Res., 2000, 329, 49–55 CrossRef CAS.
- T. L. Lowary, E. Eichler and D. R. Bundle, Can. J. Chem., 2002, 80, 1112–1130 CrossRef CAS.
- J. Broddefalk, K.-E. Bergquist and J. Kihlberg, Tetrahedron, 1998, 54, 12047–12070 CrossRef CAS.
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- M. Nazare and H. Waldmann, Angew. Chem., Int. Ed., 2000, 39, 1125–1128 CrossRef CAS.
- V. Subramaniam, S. S. Gurcha, G. S. Besra and T. L. Lowary, Tetrahedron: Asymmetry, 2005, 16, 553–567 CrossRef CAS.
- P. Dinadayala, D. Kaur, S. Berg, A. G. Amin, V. D. Vissa, D. Chatterjee, P. J. Brennan and D. C. Crick, J. Biol. Chem., 2006, 281, 20027–20035 CrossRef CAS.
- D. Kaur, S. Berg, P. Dinadayala, B. Gicquel, D. Chatterjee, M. R. McNeil, V. D. Vissa, D. C. Crick, M. Jackson and P. J. Brennan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13664–13669 CrossRef CAS.
- D. Kaur, A. Obregon-Henao, H. Pham, D. Chatterjee, P. J. Brennan and M. Jackson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17973–17977 CrossRef CAS.
- R. V. V. Tatituri, P. A. Illarionov, L. G. Dover, J. Nigou, M. Gilleron, P. Hitchen, K. Krumbach, H. R. Morris, N. Spencer, A. Dell, L. Eggeling and G. S. Besra, J. Biol. Chem., 2007, 282, 4561–4572 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and data for new compounds and 1H and 13C NMR spectra. See DOI: 10.1039/b916580k |
| This journal is © The Royal Society of Chemistry 2010 |