Synthesis of dihydrodehydrodiconiferyl alcohol: the revised structure of lawsonicin†
Junxiu Mengab, Tao Jiangb, Huma Aslam Bhattic, Bina S. Siddiquic, Sally Dixon*a and Jeremy D. Kilburn*a
aSchool of Chemistry, University of Southampton, Southampton, UK SO17 1BJ. E-mail: sd5@soton.ac.uk; jdk1@soton.ac.uk; Fax: +44(23)80594182; Tel: +44(23)80593596
bKey Laboratory of Marine Drugs, Ministry of Education of China, School of Pharmacy, Ocean University of China, Qingdao, 266003, China
cH.E.J Research Institute of Chemistry, International Centre for Chemical Sciences, University of Karachi, Karachi-75270Pakistan
First published on 28th October 2009
Abstract
Structural revision of lawsonicin, a natural product of Lawsonia alba, is reported based upon comparison of its spectral data with that of the naturally occurring dihydrobenzo[b]furan neolignan (rac)-trans-dihydrodehydrodiconiferyl alcohol, which is found to be identical. A concise synthesis of dihydrodehydrodiconiferyl alcohol, via Rh2[S-DOSP]4-catalysed intramolecular C–H insertion, is described.
Introduction
Isolation and structural elucidation of lawsonicin (rac-trans-1), a natural product of Lawsonia alba, was reported by one of us in 20031 (Fig. 1). Although the assignment of a core 2-aryl-(3′,4′-substituted)-2,3-trans-dihydrobenzo[b]furan is unambiguous, the 5,6-substitution pattern of the benzofuran ring was not conclusively established. Lawsonicin is of unknown biosynthesis;2 however, its formula, C20H24O6, implies that the molecule may derive from dimerisation of coniferyl alcohol (C10H12O3) and belong to a class of known 2-aryl-2,3-dihydrobenzo[b]furan lignans.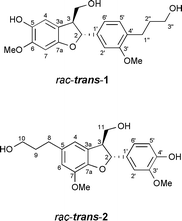 | ||
| Fig. 1 Structures of racemic 2,3-trans epimers of lawsonicin (1) and dihydrodehydrodiconiferyl alcohol (2). | ||
Lignans constitute a large group of plant secondary metabolites whose biosynthesis involves the dimerisation of phenylpropenes.3 Resonance stabilisation of a radical formed from a (phenylpropene) monolignol facilitates oxidative coupling of radical partners (or electrophilic attack of a single radical upon a second monolignol molecule),4 to give structurally diverse products, of which 8-8′-linked lignans are the most common. The established biosynthesis of dehydrodiisoeugenols, via bimolecular coupling of phenoxy radicals derived from laccase-catalysed oxidation of 2-methoxy-4-trans-propenylphenol (isoeugenol),5,6 is widely applied to 8-5′-linked lignan (neolignan) biosynthesis and can be delineated for dimerisation of coniferyl alcohol (3) (Fig. 2).6,7
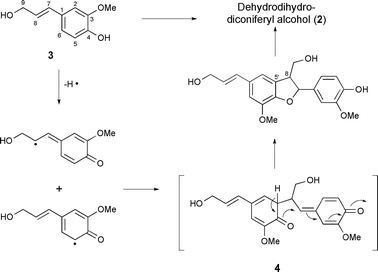 | ||
| Fig. 2 Schematised biosynthesis of dihydrodehydrodiconiferyl alcohol via 8-5′ radical coupling. | ||
8-5′-Dimerisation leads to an intermediate p-quinone methide, 4,8,9 intramolecular cyclisation of which installs the dihydrobenzo[b]furan core, prior to an enzymatic allylic alcohol reduction.10 Dihydrodehydrodiconiferyl alcohol, 2, is furnished in this overall oxidoreductive process. Added structural classes of lignan may be defined, featuring an 8-O4′, 8-3′, 8-2′ or 8-1′ linkage and potentially involving further post-dimerisation modifications. However, the ascribed connectivity of lawsonicin (rac-trans-1) cannot be rationalised within these established pathways for monolignol dimerisation, and we herein propose structural revision of lawsonicin, to the known neolignan, dihydrodehydrodiconiferyl alcohol11–14 (rac-trans-2,15Fig. 1).
Results and discussion
A comparison of 1H and 13C NMR data for rac-trans-1 with that reported for rac-trans-2 was first made (Table 1). 1H NMR data is similar for the two molecules, although small differences in reported chemical shift are difficult to attribute. Integral, multiplicity of resonance and coupling constants are largely consistent, and discrepancies appear only for assignment of the 1H-11 multiplicity in a spectral region of substantial overlap, and also as disparate assignment of the 1H-3 multiplicity. Importantly, 4JH4-H6 coupling is not observed for rac-trans-2, indicating that the para-relationship, inferred on this basis between aromatic protons of the benzofuran ring for rac-trans-1, may be mistaken. 13C NMR data is an excellent match for the two molecules and aromatic δCH values support the structural assignment of rac-trans-2. Firstly, those of the benzofuran ring: δ13CH-4 (116.0 ppm) is expected to be similar for either molecule; however, δ13CH-7 (rac-trans-1) is expected at higher field than δ13CH-6 (rac-trans-2), and the observed δ13CH = 112.8 ppm is consistent with a δ13CH-6 (rac-trans-2) assignment.16 Secondly, the 2-aryl substituent: δ13CH-5′ (rac-trans-1) is expected at lower field than δ13CH-5′ (rac-trans-2), i.e.δ13CH-5′ > δ13CH-2′/δ13CH-6′ (rac-trans-1) and the 13C NMR data support the (2-aryl)-3′-methoxy-4′-hydroxy-substitution pattern of rac-trans-2.| Positionb | δ1H/ppm | δ13C/ppmc | ||
|---|---|---|---|---|
| rac-trans-1 | rac-trans-2 | rac-trans-1 | rac-trans-2 | |
| a Chemical shifts are referenced to residual solvent (δ1H) or solvent (δ13C) for rac-trans-1, and to tetramethylsilane for rac-trans-2.b Refers to the numbering of rac-trans-2 (Fig. 1) and reported assignments for rac-trans-2.2c Literature chemical shifts for both molecules were reported to 0.1 ppm. Correction of reported chemical shifts, C4 (−0.4 ppm), C10 (+0.8 ppm), C11 (+0.4 ppm) and 3′-OMe (+2.3 ppm) for rac-trans-1,1 is based upon the actual 13C NMR spectrum of lawsonicin. | ||||
| 2 | 5.41 (1H, d, J = 7.0 Hz) | 5.54 (1H, d, J = 7.6 Hz) | 88.0 | 87.9 |
| 3 | 3.45 (1H, ddd, J = 8.0, 7.0, 5.0 Hz) | 3.60 (1H, q, J = 7.6 Hz) | 53.8 | 53.8 |
| 3a | — | — | 127.9 | 127.7 |
| 4 | 6.56 (1H, s) | 6.67 (1H, s) | 116.1 | 116.0 |
| 5 | — | — | 133.1 | 133.0 |
| 6 | 6.60 (1H, s) | 6.67 (1H, s) | 112.8 | 112.4 |
| 7 | — | — | 144.2 | 144.2 |
| 7a | — | — | 146.7 | 146.6 |
| 7-OMe | 3.77 (3H, s) | 3.88 (3H, s) | 55.9 | 56.0 |
| 8 | 2.54 (2H, t, J = 7.5 Hz) | 2.67 (2H, t, J = 7.3 Hz) | 32.0 | 32.0 |
| 9 | 1.70 (2H, tt, J = 7.5, 6.5 Hz) | 1.88 (2H, tt, J = 7.3, 6.6 Hz) | 34.5 | 34.6 |
| 10 | 3.53 (2H, t, J = 6.5 Hz) | 3.69 (2H, t, J = 6.6 Hz) | 62.3 | 62.3 |
| 11 | 3.74 (2H, m) | 3.90 (2H, d, J = 7.6 Hz) | 64.0 | 63.9 |
| 1′ | — | — | 135.5 | 135.4 |
| 2′ | 6.85 (1H, d, J = 1.9 Hz) | 6.94 (1H, d, J = 1.7 Hz) | 108.9 | 108.8 |
| 3′ | — | — | 146.5 | 146.6 |
| 3′-OMe | 3.75 (3H, s) | 3.86 (3H, s) | 56.0 | 56.0 |
| 4′ | — | — | 145.7 | 145.6 |
| 5′ | 6.72 (1H, d, J = 8.1 Hz) | 6.87 (1H, d, J = 8.1 Hz) | 114.5 | 114.3 |
| 6′ | 6.77 (1H, dd, J = 8.1, 1.9 Hz) | 6.91 (1H, dd, J = 8.1, 1.7 Hz) | 119.5 | 119.4 |
In order to model δ13CH-5′, -2′ and -6′ of rac-trans-1, we prepared derivative 8, bearing the (2-aryl)-3′,4′-disubstitution pattern of lawsonicin, via a B-ring-substituted flavanone 6. 3′-Methoxy-4′-hydroxyflavanone18 (6) was prepared via Claisen–Schmidt condensation of vanillin benzyl ether (5) and 2′-hydroxy acetophenone,19 followed by cyclisation of the resulting hydroxychalcone derivative and benzyl ether hydrogenolysis. Conversion of 6 to a triflic ester was straightforward and oxidative ring contraction20 proceeded to give the expected rac-trans-dihydrobenzo[b]furan derivative 7 in moderate yield.21 Low-yielding Heck coupling with methyl acrylate, catalytic hydrogenation and methyl ester reduction steps furnished 8 (Scheme 1).
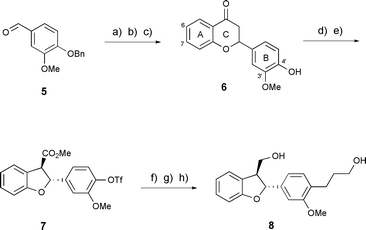 | ||
| Scheme 1 Synthesis of a (2-aryl)-3′,4′-disubstituted analogue of lawsonicin. Reagents and conditions: (a) 2′-hydroxyacetophenone, dioxane, 50% w/v KOH (aq.), EtOH, △ (75%); (b) NaOAc, MeOH, △ (70%); (c) H2 (1 atm), 10% Pd/C, CH2Cl2–MeOH, rt (100%); (d) Tf2O, Et3N, CH2Cl2, −78 °C→rt (67%); (e) HClO4, trimethyl orthoformate, Tl(NO3)2·3H2O, rt (46%); (f) PdCl2, PPh3, methyl acrylate, Et3N, DMF, 110 °C (30%); (g) H2 (1 atm), 10% Pd/C, CH2Cl2–MeOH, rt (100%); (h) LiAlH4, Et2O, rt (94%). | ||
The 13C NMR spectrum of model compound 8 (CDCl3, 400 MHz) correlates poorly with that of lawsonicin; 13C-1′, 13C-3′ and 13CH-5′ appear substantially downfield of the corresponding nuclei in lawsonicin, whilst 13C-4′ is upfield (Table 2).
Observed differences, Δδ (ppm), are as expected for the comparison of a 3′-methoxy-4′-(3-hydroxypropyl)-aryl substituent with the 3′-methoxy-4′-hydroxy aryl group of dihydrodehydrodiconiferyl alcohol. However, due to small inconsistencies in the 13C NMR data reported elsewhere for rac-trans-2,12,22 we wished to finally verify the structural revision of lawsonicin by comparison of authentic samples.
Although the oxidative rearrangement of a simple flavanone had proved successful (Scheme 1), attempted rearrangement of various 6,7-disubstituted flavanones failed to yield a functionalised 2,3-dihydrobenzo[b]furan core precursor to the reported structure of lawsonicin, and so we wished to avoid the potentially difficult cyclisation of an A-ring-substituted flavanone for preparation of rac-trans-dihydrodehydrodiconiferyl alcohol (rac-trans-2). Therefore, a synthetic approach to rac-trans-2 involving intramolecular C–H insertion of an α-diazoester, as the key step for formation of the dihydrobenzo[b]furan ring, was adopted. The presence of a (pivaloate) protected 5-(3-hydroxypropyl) side chain was detrimental to the yield in preparation of a closely related α-diazo methyl ester,23 and we chose, therefore, to prepare a 5-bromo-2,3-dihydrobenzo[b]furan, trans-14, in order to later introduce the 3-hydroxypropyl side chain under Pd0 catalysis.
Benzyl iodide 10 was prepared in 77% yield from 4-hydroxy-3-methoxybenzyl alcohol. Coupling of 10 with 5-bromo-2-hydroxy-3-methoxybenzaldehyde (9) was carried out, followed by Wittig olefination, enol ether hydrolysis, oxidation and methylation with diazomethane, yielding methyl ester 12. Diazo transfer, using p-acetamidobenzenesulfonyl azide (p-ABSA) and DBU,24 enabled conversion of 12 to 13 in good yield. Catalysis of the intramolecular C–H insertion of 13 was effected upon treatment with Rh2[S-DOSP]4,25,26 and a 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (trans:cis) ratio of separable 2,3-dihydroxybenzo[b]furan isomers, 14, resulted, also in good yield. Pleasingly, epimerisation of cis-14 to its thermodynamically more stable trans-14 isomer proceeded smoothly upon treatment with sodium methoxide, following Hashimoto's conditions27 (Scheme 2). Sonogashira coupling between trans-14 and prop-2-ynyloxymethyl benzene28 (15) took place to give 16 in good yield.29 Corresponding alkyne reduction and benzyl ether hydrogenolysis was then carried out, and required careful control of conditions in order to avoid dihydrobenzofuran ring-opening. Finally, methyl ester reduction using LiAlH4 resulted in partial C3-epimerisation30 and dihydrodehydrodiconiferyl alcohol 2 was obtained as a 10:3 (2,3-trans:2,3-cis) diastereomeric product mixture, which we were unable to separate using HPLC (Scheme 3). Nonetheless, direct comparison of 1H and 13C NMR, and EIMS data for the product mixture, 2, with that of lawsonicin, indicated the major isomer rac-trans-2 to be identical to lawsonicin.
:1 (trans:cis) ratio of separable 2,3-dihydroxybenzo[b]furan isomers, 14, resulted, also in good yield. Pleasingly, epimerisation of cis-14 to its thermodynamically more stable trans-14 isomer proceeded smoothly upon treatment with sodium methoxide, following Hashimoto's conditions27 (Scheme 2). Sonogashira coupling between trans-14 and prop-2-ynyloxymethyl benzene28 (15) took place to give 16 in good yield.29 Corresponding alkyne reduction and benzyl ether hydrogenolysis was then carried out, and required careful control of conditions in order to avoid dihydrobenzofuran ring-opening. Finally, methyl ester reduction using LiAlH4 resulted in partial C3-epimerisation30 and dihydrodehydrodiconiferyl alcohol 2 was obtained as a 10:3 (2,3-trans:2,3-cis) diastereomeric product mixture, which we were unable to separate using HPLC (Scheme 3). Nonetheless, direct comparison of 1H and 13C NMR, and EIMS data for the product mixture, 2, with that of lawsonicin, indicated the major isomer rac-trans-2 to be identical to lawsonicin.
![Synthesis of an aryl bromide functionalised precursor to dihydrodehydrodiconiferyl alcohol. Reagents and conditions: (a) KH, 18-crown-6, THF, 60 °C, 1.5 h (98%); (b) nBuLi, (methoxymethyl) triphenylphosphonium chloride, THF, −78 °C→rt, 5 h (70%); (c) Hg(OAc)2, MeCN–H2O, rt, 1 h (67%); (d) NaClO2, 2-methyl-2-butene, NaH2PO4·2H2O, tBuOH–H2O, 0 °C→rt then rt, 14 h (86%); (e) CH2N2, Et2O–THF, −78 °C→rt (95%); (f) p-ABSA, DBU, MeCN, 0 °C→rt then rt, 24 h (90%); (g) Rh2[S-DOSP]4 (1.3 mol%), toluene, 0 °C, 2 h (95%); (h) NaOMe, MeOH, −60 °C, 26 h (96%).](/image/article/2010/OB/b918179b/b918179b-s2.gif) | ||
| Scheme 2 Synthesis of an aryl bromide functionalised precursor to dihydrodehydrodiconiferyl alcohol. Reagents and conditions: (a) KH, 18-crown-6, THF, 60 °C, 1.5 h (98%); (b) nBuLi, (methoxymethyl) triphenylphosphonium chloride, THF, −78 °C→rt, 5 h (70%); (c) Hg(OAc)2, MeCN–H2O, rt, 1 h (67%); (d) NaClO2, 2-methyl-2-butene, NaH2PO4·2H2O, tBuOH–H2O, 0 °C→rt then rt, 14 h (86%); (e) CH2N2, Et2O–THF, −78 °C→rt (95%); (f) p-ABSA, DBU, MeCN, 0 °C→rt then rt, 24 h (90%); (g) Rh2[S-DOSP]4 (1.3 mol%), toluene, 0 °C, 2 h (95%); (h) NaOMe, MeOH, −60 °C, 26 h (96%). | ||
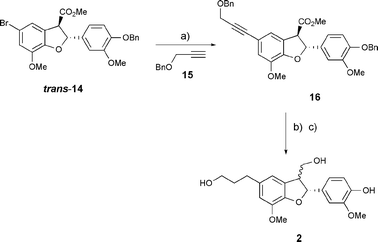 | ||
| Scheme 3 Synthesis of dihydrodehydrodiconiferyl alcohol. Reagents and conditions: (a) Pd(PPh3)4 (13 mol%), CuI (0.53 equiv.), Et3N, 90 °C, 6 h (73%); (b) H2 (1 atm), 10% Pd/C, MeOH, rt, 3 h; then formic acid, 20 min (97%); (c) LiAlH4, THF, −15 °C→0 °C, 3 h (100%). | ||
Conclusions
An efficient synthesis of dihydrodehydrodiconiferyl alcohol 2, in twelve linear steps and 16% yield, has been completed, although attenuated by partial C3-epimerisation in the final step. Valuable advantages of the described route are high-yielding diazo transfer and C–H insertion, for formation of the 2,3-dihydrobenzo[b]furan core, and a late-stage Pd0-catalysed functionalisation, which permits the synthesis of analogues with varying C5-substitution. Of most importance, the major 2,3-trans-epimer of 2 has identical spectral data to the 2,3-dihydrobenzo[b]furan natural product, lawsonicin, whose earlier reported structure is revised to 2,3-trans-dihydrodehydrodiconiferyl alcohol (rac-trans-2). The identity of (rac-trans-2) as a constituent of Lawsonia alba is confirmed.Experimental
General techniques
Reagents and solvents were obtained from commercial suppliers and, if necessary, dried and distilled before use. THF was freshly distilled from sodium benzophenone ketal under argon. Toluene was distilled from sodium under argon. Dichloromethane, acetonitrile and triethylamine were freshly distilled from CaH2. N,N-Dimethylformamide and hexamethyldisilazane were distilled from CaH2 and stored over 4 Å molecular sieves. Reactions requiring a dry atmosphere were conducted in oven dried glassware under argon. Petrol refers to the fraction boiling between 40 and 60 °C. 1H and 13C NMR spectra were recorded on Bruker 300 or 400 MHz spectrometers. 1H chemical shifts are reported as values in ppm referenced to residual solvent. The following abbreviations are used to denote multiplicity and may be compounded: s = singlet, d = doublet, t = triplet, q = quartet, qn = quintuplet, sext = sextet. Coupling constants, J, are measured in Hertz (Hz). 13C spectra were proton decoupled and referenced to solvent. Signals are reported as s, d, t, q, depending on the number of directly attached protons (0, 1, 2 and 3, respectively), this being determined by DEPT experiments. Infra-red spectra were recorded either as neat solids or as oils on a Bio-Rad Golden Gate ATR FT–IR spectrometer fitted with an ATR accessory. Absorptions are given in wavenumbers (cm−1) and the following abbreviations used to denote peak intensities: s = strong, m = medium, w = weak and/or br (broad). Low resolution mass spectra were recorded on a Micromass platform single quadrupole mass spectrometer in methanol or acetonitrile. Accurate mass spectra were recorded on a double focusing mass spectrometer.Synthetic procedures
:1) gave the title compound 10 as white needles (2.70 g, 80%). m.p. 82–83 °C. IR (solid): 2877 (w), 1587 (m), 1513 (m, br), 1257 (s) cm−1. 1H NMR (300 MHz, CDCl3): δ = 7.43-7.29 (5H, m), 6.90 (1H, d, J = 2.0 Hz), 6.89 (1H, dd, J = 2.0, 8.1 Hz), 6.76 (1H, d, J = 8.1 Hz), 5.13 (2H, s), 4.45 (2H, s), 3.88 (3H, s) ppm. 13C NMR (75 MHz, CDCl3): δ = 149.83 (s), 148.17 (s), 137.09 (s), 132.31 (s), 128.76 (d), 128.08 (d), 127.40 (d), 121.19 (d), 113.93 (d), 112.60 (d), 71.15 (t), 56.19 (q), 7.12 (t) ppm. MS (ES+): m/z (%) = 377 (100%) [M+Na]+.:1) gave the title compound 11 as a white solid (3.17 g, 98%). m.p. 126 °C. IR (solid): 2938 (w), 1684 (m), 1475 (m, br), 736 (m, br) cm−1. 1H NMR (400 MHz, CDCl3): δ = 10.09 (1H, s), 7.48 (1H, d, J = 2.1 Hz), 7.42-7.25 (5H, m), 7.23 (1H, d, J = 2.1 Hz), 6.89 (1H, d, J = 1.3 Hz), 6.81 (1H, t, J = 8.0 Hz), 6.77 (1H, dd, J = 8.0, 1.3 Hz), 5.13 (2H, s), 5.07 (2H, s), 3.92 (3H, s), 3.85 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 188.89 (d), 154.09 (s), 150.28 (s), 149.94 (s), 148.81 (s), 137.07 (s), 131.40 (s), 129.10 (s), 128.73 (d), 128.06 (d), 127.44 (d), 121.89 (d), 121.72 (d), 120.95 (d), 117.23 (s), 113.98 (d), 112.75 (d), 76.63 (t), 71.18 (t), 56.57 (q), 56.22 (q) ppm. MS (ES+): m/z (%) = 479 (98%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C23H21BrNaO5: 479.0470; found 479.0469.:1) gave the title compound 17 as a white solid (1.11 g, 70%). m.p. 87 °C. IR (solid): 2935 (w), 2832 (w), 1638 (m), 1585 (w), 1513 (m), 1463 (s), 1265 (s, br), 736 (m) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.41 (2H, d, J = 7.4 Hz), 7.35 (2H, t, J = 7.4 Hz), 7.29 (1H, t, J = 7.4 Hz), 7.28 (1H, m), 7.03 (1H, d, J = 13.0 Hz), 7.01 (1H, s), 6.86-6.82 (3H, m), 5.87 (1H, d, J = 13.0 Hz), 5.16 (2H, s), 4.84 (2H, s), 3.89 (3H, s), 3.83 (3H, s), 3.55 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 153.90 (s), 150.74 (d), 149.81 (s), 148.28 (s), 143.60 (s), 137.29 (s), 132.81 (s), 130.84 (s), 128.71 (d), 128.00 (d), 127.41 (d), 121.21 (d), 120.11 (d), 116.87 (s), 113.99 (d), 113.03 (d), 112.66 (d), 99.40 (d), 75.02 (t), 71.23 (t), 56.62 (q), 56.21 (q), 56.16 (q) ppm. MS (ES+): m/z (%) = 507 (93%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C25H25BrNaO5: 507.0783; found 507.0778.:1) gave the title compound 18 as a white solid (133 mg, 67%). m.p. 125 °C. IR (solid): 2938 (w), 2727 (w), 1721 (m), 1591 (m), 1514 (m), 1265 (s), 1206 (m), 849(w, br) cm−1. 1H NMR (400 MHz, CDCl3): δ = 9.53 (1H, t, J = 1.8 Hz), 7.43-7.33 (2H, m), 7.28 (1H, t, J = 7.5 Hz), 7.27–7.20 (2H, m) 6.95 (1H, d, J = 2.5 Hz), 6.85 (1H, d, J = 1.5 Hz), 6.80 (1H, d, J = 2.1 Hz), 6.76 (1H, d, J = 8.1 Hz), 6.73 (1H, dd, J = 8.1, 1.5 Hz), 5.15 (2H, s), 4.90 (2H, s), 3.89 (2 × 3H, s), 3.46 (2H, d, J = 1.8 Hz) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 198.97 (d), 153.68 (s), 149.87 (s), 148.44 (s), 145.53 (s), 137.21 (s), 130.29 (s), 128.81 (s), 128.72 (d), 128.03 (d), 127.44 (d), 125.69 (d), 121.29 (d), 116.66 (s), 115.61 (d), 114.00 (d), 112.64 (d), 74.89 (t), 71.22 (t), 56.28 (q), 56.21 (q), 45.18 (t) ppm. MS (ES+): m/z (%) = 493 (87%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C24H23BrNaO5: 493.0627; found 493.0621.:1 mixture) dropwise. Concentration in vacuo gave a yellow residue. Purification by column chromatography (SiO2 eluted with EtOAc/petrol, 1:8→1:4) gave the title compound 12 as a white solid (184 mg, 95%). m.p. 98 °C. IR (solid): 2948 (w), 1734 (m), 1591 (m), 697 (w) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.42-7.40 (2H, m), 7.35 (2H, t, J = 7.5 Hz), 7.30 (1H, d, J = 7.0 Hz), 6.96–6.95 (2H, m), 7.00-6.94 (3H, m), 5.15 (2H, s), 4.90 (2H, s), 3.90 (3H, s), 3.86 (3H, s), 3.60 (3H, s), 3.50 (2H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 172.08 (s), 153.97 (s), 150.24 (s), 148.66 (s), 145.91 (s), 137.71 (s), 131.13 (s), 130.84 (s), 129.12 (d), 128.42 (d), 127.85 (d), 126.00 (d), 121.46 (d) 116.77 (s), 115.74 (d), 114.39 (d), 112.87 (d), 75.21 (t), 71.64 (t), 56.65 (q), 56.58 (q), 52.61 (q), 36.00 (t) ppm. MS (ES+): m/z (%) = 523 (73%) [M+Na]+. HRMS (ES+): m/z [M+Na] + calcd for C25H25BrNaO6: 523.0732; found 523.0729.:8) gave the title compound 13 as a yellow oil (52 mg, 90%). IR (film): 2951 (w), 1701 (m), 1268 (m, br), 740 (w) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.43-7.27 (6H, m), 6.94-6.93 (2H, m), 6.81 (1H, d, J = 8.0 Hz), 6.74 (1H, d (fine splitting), J = 8.0 Hz), 5.13 (2H, s), 4.87 (2H, s), 3.87 (3H, s), 3.86 (3H, s), 3.75 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 165.78 (s), 153.40 (s), 149.75 (s), 148.58 (s), 142.88 (s), 137.24 (s), 129.52 (s), 128.73 (d), 128.02 (d), 127.43 (d), 123.91 (d), 122.47 (s), 121.57 (d), 117.15 (s), 114.67 (d), 113.72 (d), 112.47 (d), 75.69 (t), 71.16 (t), 56.35 (q), 56.04 (q), 52.21 (q) ppm. MS (ES+): m/z (%) = 549 (93%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C25H23BrN2NaO6: 549.0637; found 549.0632.:8) gave the title compound, a colourless oil, as a 1.3:1 (2,3-trans:2,3-cis) ratio of isomers, which were separated by column chromatography (SiO2 eluted with hexane/EtOAc, 10:1), (47 mg, 95%). To effect epimerisation of rac-cis-14; to a solution of rac-cis-14 (23 mg, 0.046 mmol) in THF (1 mL) was added NaOMe (0.2 mL of a 1.11 M solution in MeOH, 0.22 mmol) at −60 °C. The mixture was stirred at −60 °C for 26 h before dropwise addition of 0.2 mL of sodium phosphate buffer (1 M, pH 7). The mixture was then warmed to room temperature and EtOAc (10 mL) was added. The organic phase was separated and the aqueous phase extracted with EtOAc (2 × 10 mL). The combined organic extracts were dried over MgSO4 and concentrated in vacuo. Purification by flash column chromatography (SiO2 eluted with EtOAc/petrol, 1:4) gave rac-trans-14 as a white solid (22 mg, 96%). rac-cis-14: m.p. 102–104 °C. IR (solid): 2947 (w), 1736 (m), 1616 (w), 1261 (m, br), 1202 (m, br), 733 (m, br) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.40 (2H, d J = 7.5 Hz), 7.33 (2H, t, J = 7.5 Hz), 7.28 (1H, m), 6.96 (2H, m), 6.89 (1H, m), 6.82 (2H, m), 5.93 (1H, d, J = 9.8 Hz), 5.13 (2H, s), 4.50 (1H, d, J = 9.8 Hz), 3.89 (3H, s), 3.84 (3H, s), 3.22 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 170.05 (s), 149.66 (s), 148.44 (s), 148.32 (s), 145.34 (s), 137.12 (s), 129.43 (s), 128.69 (d), 128.04 (d), 127.48 (d), 120.85 (d), 119.20 (d), 116.18 (d), 113.87 (d), 113.20 (s), 110.29 (d), 87.01 (d), 71.13 (t), 56.47 (q), 56.27 (q), 54.29 (d), 52.13 (q) ppm. MS (ES+): m/z (%) = 521 (99%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C25H23BrNaO6: 521.0576; found 521.0573. rac-trans-14: m.p. 70 °C (EtOH). IR (solid): 2953 (w), 1738 (m), 1261 (m, br), 733 (w) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.41 (2H, d, J = 7.5 Hz), 7.36 (2H, t, J = 7.5 Hz), 7.29 (1H, m), 7.09 (1H s), 6.95 (1H, s), 6.92 (1H, s), 6.87 (1H, broad d, J = 8.5 Hz), 6.83 (1H, d, J = 8.5 Hz), 6.05 (1H, d, J = 8.3 Hz), 5.14 (2H, s), 4.30 (1H, d, J = 8.3 Hz), 3.86 (2 × 3H, s), 3.81 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 170.75 (s), 150.09 (s), 148.65 (s), 147.32 (s), 145.27 (s), 137.13 (s), 132.73 (s), 128.72 (d), 128.03 (d), 127.38 (d), 126.54 (s), 120.12 (d), 118.83 (d), 116.13 (d), 114.16 (d), 112.83 (s), 110.00 (d), 87.02 (d), 71.18 (t), 56.49 (q), 56.29 (q), 55.78 (d), 53.04 (q) ppm. MS (ES+): m/z (%) = 521 (99%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C25H23BrNaO6: 521.0576; found 521.0557.:9) to give the title compound 16 as a light yellow oil (33 mg, 73%). IR (film): 2951 (w), 1739 (m), 1597 (m), 1225 (s), 741 (m, br) cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.42-7.27 (10H, m), 7.11 (1H, s), 6.95 (1H, s), 6.93 (1H, d, J = 1.6 Hz), 6.88 (1H, dd, J = 8.0, 1.6 Hz) 6.84 (1H, d, J = 8.3 Hz), 6.08 (1H, d, J = 8.4 Hz), 5.14 (2H, s), 4.67 (2H, s), 4.39 (2H, s), 4.31 (1H, d, J = 8.4 Hz), 3.87 (3H, s), 3.86 (3H, s), 3.81 (3H, s) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 170.92 (s), 150.09 (s), 148.65 (s), 144.36 (s), 137.71 (s), 137.15 (s), 132.81 (s), 128.73 (d), 128.64 (d), 128.28 (d), 128.06 (d), 128.05 (d), 127.39 (d), 125.31 (s), 121.33 (d), 118.88 (d), 116.35 (d), 115.78 (s), 114.17 (d), 110.04 (d), 87.17 (d), 86.66 (s), 83.62 (s), 71.94 (t), 71.20 (t), 58.20 (t), 56.30 (2 × q), 55.70 (d), 53.01 (q) ppm. MS (ES+): m/z (%) = 587 (100%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C35H32NaO7: 587.2046; found 587.2050.:2) gave the title compound 20 as a colourless oil (6 mg, 97%). IR (film): 2926 (w), 1734 (m, br), 755 (w, br) cm−1. 1H NMR (400 MHz, CDCl3): δ = 6.88–6.84 (2H, m), 6.81 (1H, d, J = 8.0 Hz), 6.78 (1H, s), 6.68 (1H, s), 6.02 (1H, d, J = 8.5 Hz), 5.60 (1H, s)), 4.30 (1H, d, J = 8.5 Hz), 3.88 (3H, s), 3.87 (3H, s), 3.80 (3H, s), 3.71 (2H, t, J = 6.5 Hz), 2.69 (2H, m), 1.91 (2H, tt, J = 7.5, 6.5 Hz) ppm. 13C NMR (100.5 MHz, CDCl3): δ = 171.49 (s), 146.84 (s), 146.28 (s), 146.06 (s), 144.46 (s), 135.75 (s), 132.21 (s), 125.32 (s), 119.68 (d), 116.72 (d), 114.60 (d), 113.22 (d), 109.01 (d), 86.98 (d), 62.50 (t), 56.35 (2 × q), 56.23 (d), 52.83 (q), 34.83 (t), 32.21 (t) ppm. MS (ES+): m/z (%) = 411 (100%) [M+Na]+. HRMS (ES+): m/z [M+Na]+ calcd for C21H24NaO7: 411.1420; found 411.1425.:2) gave the title compound 2, a colourless oil, as a 10:3 (2,3-trans:17 2,3-cis32) diastereomeric product mixture (3 mg, ca. 100%).Acknowledgements
This work was supported by the Education Commission Pakistan under the Split Ph.D. program (HAB) and the Chinese Government Scholarship Programme 2007-2009 (JM).Notes and references
- B. S. Siddiqui, M. N. Kardar, S. T. Ali and S. Khan, Helv. Chim. Acta, 2003, 86, 2164 CrossRef CAS.
- Biosynthesis of a simple analogue, 6-methoxy-3-methyl-2-phenyl-2,3-dihydrobenzofuran-5-ol (obtusafuran), via coupling of phenolic (C6) and cinnamyl (C9) precursors has been advanced; however, to our knowledge, the requisite 4-(3-hydroxypropyl)-substituted cinnamyl alcohol precursor, for lawsonicin biosynthesis according to this pathway, has not been identified. See: (a) L. Jurd, G. Manners and K. Stevens, J. Chem. Soc., Chem. Commun., 1972, 992 RSC; (b) W. D. Ollis and O. R. Gottlieb, Chem. Commun. (London), 1968, 1396 RSC.
- L. B. Davin, M. Jourdes, A. M. Patten, K-W. Kim, D. G. Vassao and N. G. Lewis, Nat. Prod. Rep., 2008, 25, 1015 RSC.
- M. Sefkow, Synthesis, 2003, 2595 CrossRef CAS.
- (a) H. Erdtman, Justus Liebigs Ann. Chem., 1933, 503, 283 CrossRef CAS; (b) N. G. Lewis, L. B. Davin, Lignin and Lignan Biosynthesis, ACS Symposium Series, ed. N. G. Lewis and S. Sarkanen, American Chemical Society, Washington DC, USA, 1998, 697, 334 Search PubMed.
- T. Shiba, L. Xiao, T. Miyakoshi and C. L. Chen, J. Mol. Catal. B: Enzym., 2000, 10, 605 CrossRef CAS.
- (a) S. Y. Guan, J. Mlynar and S. Sarkanen, Phytochemistry, 1997, 45, 911 CrossRef CAS; (b) K. Syrjänen and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1998, 3425–3429 RSC.
- (a) S. R. Angle and J. D. Rainier, J. Org. Chem., 1992, 57, 6883 CrossRef; (b) S. R. Angle and K. D. Turnbull, J. Am. Chem. Soc., 1990, 112, 3698 CrossRef CAS; (c) S. R. Angle and K. D. Turnbull, J. Org. Chem., 1993, 58, 5360 CrossRef CAS.
- For examples of 1,6-nucleophilic addition to p-quinone methide derivatives in biomimetic neolignan syntheses, see: (a) S. Antus, R. Bauer, A. Gottsegen, O. Seligmann and H. Wagner, Liebigs Ann. Chem., 1987, 357 CrossRef CAS; (b) L. Juhász, L. Kurti and S. Antus, J. Nat. Prod., 2000, 63, 866 CrossRef CAS.
- To our knowledge, presumed reductase catalysis of this transformation has not yet been specified. For a discussion of the overall oxidoreductive biosynthesis, see: M. Nose, M. A. Bernards, M. Furlan, J. Zajicek, T. L. Eberhardt and N. G. Lewis, Phytochemistry, 1995, 39, 71 Search PubMed.
- (a) P. K. Agrawal, S. K. Agarwal and R. P. Rastogi, Phytochemistry, 1980, 19, 1260 CrossRef CAS; (b) J. M. Fang, C. K. Lee and Y. S. Cheng, Phytochemistry, 1992, 31, 3659 CrossRef CAS; (c) A. Jutiviboonsuk, H. Zhang, G. T. Tan, C. Ma, N. Van Hung, N. M. Cuong, N. Bunyapraphatsara, D. D. Soejarto and H. H. S. Fong, Phytochemistry, 2005, 66, 2745 CrossRef CAS; (d) S. Kawai, K. Sugishita and H. Ohashi, Phytochemistry, 1999, 51, 243 CrossRef CAS; (e) M. Ono, Y. Nishida, C. Masuoka, J.-C. Li, M. Okawa, T. Ikeda and T. Nohara, J. Nat. Prod., 2004, 67, 2073 CrossRef CAS.
- P. M. Pauletti, A. R. Araujo, M. C. M. Young, A. M. Giesbrecht and V. D. Bolzani, Phytochemistry, 2000, 55, 597 CrossRef CAS.
- (a) T. Deyama, T. Ikawa, S. Kitagawa and S. Nishibe, Chem. Pharm. Bull., 1987, 35, 1785 CAS; (b) W.-L. Lo, Y.-C. Wu, T.-J. Hsieh, S.-H. Kuo, H.-C. Lin and C.-Y. Chen, Chin. Pharm. J., 2004, 56, 69 Search PubMed; (c) V. Seidel, F. Bailleul and P. G. Waterman, J. Nat. Prod., 2000, 63, 6 CrossRef CAS.
- Y. Fukuyama, M. Nakahara, H. Minami and M. Kodama, Chem. Pharm. Bull., 1996, 44, 1418.
- For elucidation of the 2,3-trans-configuration of dihydrodehydrodiconiferyl alcohol, see: ref. 13 and 14.
- Compare the 7-methoxy-5-methyl-2,3-dihydrobenzofuran analogue of rac-trans-2 (δ13CH-6 = 111.9 ppm: D. L. J. Clive, S. P. Fletcher and M. Z. Zhu, Chem. Commun., 2003, 526 Search PubMed and the 5-hydroxy-6-methoxy-2,3-dihydrobenzofuran analogue of rac-trans-1 (δ13CH-7 = 94.1 ppm: J. W. Benbow and R. Katoch-Rouse, J. Org. Chem., 2001, 66, 4965 RSC.
- Data reproduced in Table 1 is from the isolated natural product, see: ref. 14. Corresponding data and structural verification has been reported for synthetic rac-trans-2, see: L. Pieters, S. Van Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemiere, J. Med. Chem., 1999, 42, 5475 Search PubMed.
- B. Sen, J. Am. Chem. Soc., 1952, 74, 3445 CrossRef CAS.
- (a) M. Cabrera, M. Simoens, G. Falchi, M. L. Lavaggi, O. E. Piro, E. E. Castellano, A. Vidal, A. Azqueta, A. Monge, A. L. de Cerain, G. Sagrera, G. Scoane, H. Cerecetto and M. Gonzalez, Bioorg. Med. Chem., 2007, 15, 3356 CrossRef CAS; (b) C. X. Qin, X. Q. Chen, R. A. Hughes, S. J. Williams and O. L. Woodman, J. Med. Chem., 2008, 51, 1874 CrossRef CAS.
- M. S. Khanna, O. V. Singh, C. P. Garg and R. P. Kapoor, Synth. Commun., 1993, 23, 585 CrossRef CAS.
- 3JH2,H3 Coupling constants are diagnostic in the assignment of relative trans-configuration from the 1H NMR spectrum of 7 (CDCl3, 300 MHz); δ = 6.27 (1H, d, 3JH2,H3 = 7.7 Hz, H2), 4.42 (1H, d, 3JH3,H2 = 7.7 Hz, H3). Comparative 3JH2,H3 = 7.7 Hz coupling has also been reported for (rac)trans-3-carboxymethyl-2-phenyl-2,3-dihydrobenzofuran; 3JH2,H3 (rac)cis-isomer = 10.0 Hz: L. Juhasz, L. Szilagyi, S. Antus, J. Visy, F. Zsila and M. Simonyi, Tetrahedron, 2002, 58, 4261 Search PubMed.
- L. Pieters, T. Debruyne, A. Degroot, G. Mei, R. Dommisse, G. Lemiere and A. Vlietinck, Magn. Reson. Chem., 1993, 31, 692 CAS.
- S. García-Muñoz, M. Alvarez-Corral, L. Jimenez-Gonzalez, C. Lopez-Sanchez, A. Rosales, M. Munoz-Dorado and I. Rodriguez-Garcia, Tetrahedron, 2006, 62, 12182 CrossRef CAS.
- H. M. L. Davies, W. R. Cantrell, K. R. Romines and J. S. Baum, Org. Synth., 1991, 70, 93.
- H. M. L. Davies, M. V. A. Grazini and E. Aouad, Org. Lett., 2001, 3, 1475 CrossRef CAS.
- For applications of Rh2[S-DOSP]4-mediated intramolecular C–H insertion in 2,3-dihydroxybenzofuran natural product synthesis, see: (a) W. Kurosawa, T. Kan and T. Fukuyama, Synlett, 2003, 7, 1028; (b) W. Kurosawa, H. Kobayashi, T. Kan and T. Fukuyama, Tetrahedron, 2004, 60, 9615 CrossRef CAS.
- H. Saito, H. Oishi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Org. Lett., 2002, 4, 3887 CrossRef CAS.
- B. M. Trost and J. Xie, J. Am. Chem. Soc., 2006, 128, 6044 CrossRef CAS.
- S. Y. Lin, C. L. Chen and Y. J. Lee, J. Org. Chem., 2003, 68, 2968 CrossRef CAS.
- C3-Epimerisation of a similar 2,3-cis-precursor to dihydrodehydrodiconiferyl alcohol during reduction with NaBH4/MeOH, and stability to epimerisation under LiAlH4/THF reducing conditions at −60 °C, has been reported, see: ref. 32.
- J. W. Bode, M. P. Doyle, M. N. Protopova and Q.-L. Zhou, J. Org. Chem., 1996, 61, 9146 CrossRef CAS.
- S. García-Muñoz, L. Jimenez-Gonzalez, M. Alvarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Synlett, 2005, 3011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR spectra for compounds 2, 8, 10–14, 16–20 and the isolated natural product. See DOI: 10.1039/b918179b |
| This journal is © The Royal Society of Chemistry 2010 |