Oxidative spirocyclisation routes towards the sawaranospirolides. Synthesis of ent-sawaranospirolides C and D†
Jeremy Robertson*, Praful T. Chovatia, Thomas G. Fowler, Jonathan M. Withey and Daniel J. Woollaston
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, UK OX1 3TA. E-mail: jeremy.robertson@chem.ox.ac.uk; Fax: 44 1865 285002; Tel: 44 1865 275660
First published on 4th November 2009
Abstract
Two routes are described for the synthesis of the sawaranospirolides, stereoisomeric spirolactone ascorbigenins isolated from Chamaecyparis pisifera. Trapping of the keto enal formed by oxidation of a functionalised 2-(4-hydroxybutyl)furan affords a potential butenolide spiroacetal precursor to sawaranospirolides A and C. Alternatively, epoxidation of protected 3-(dihydropyran-2-yl)-3-arylpropanoic acids results in spirolactonisation to generate ent-sawaranospirolide C; a related acid-mediated spirocyclisation gave access to ent-sawaranospirolide D.
Introduction
The sawaranospirolides are spiroacetal butyrolactones, isolated from the acetone-soluble components of the heartwood of the Japanese tree ‘Sawara’, Chamaecyparis pisifera, and shown to differ merely in the configurations at C-3 and C-5 (Fig. 1).1 In a plausible biosynthesis, outlined in Scheme 1,2 the C-6 and C-7 stereogenic centres derive from C-4 and C-5 of ascorbic acid, respectively, the C-3 centre arises on introduction of a tyrosine-derived fragment, and the C-5 configuration is established after protonation of a putative ene-diol intermediate (1). Double cyclisation initiated by the 1°-hydroxy group affords the natural products with the expected3 configuration at the spiro-centre.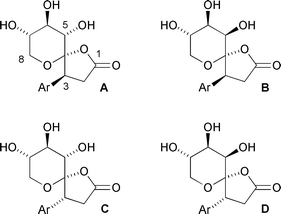 | ||
| Fig. 1 Sawaranospirolides A–D (Ar = p-HO–C6H4–). | ||
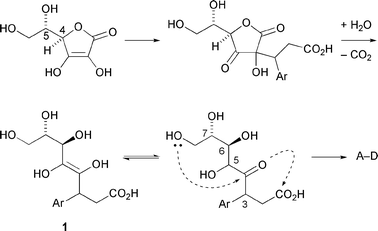 | ||
| Scheme 1 Outline biosynthesis of sawaranospirolides A–D. | ||
Furan oxidative spirocyclisation
Scheme 2 summarises, in retrosynthetic terms, an approach to the sawaranospirolides based on formation of the pyranose ring by cyclisation onto a keto enal (3) exposed by furan oxidation, with installation of the aromatic group by conjugate addition to butenolide 2.4,5 This approach is conceptually related to the outline biosynthesis (Scheme 1) in which the C-8 hydroxy group and a C-4 carbonyl participate in a tandem cyclisation event to achieve the spirocyclic ring system present in the sawaranospirolides.![Access to the sawaranospirolide system by furan oxidation (Ar = p-HO–C6H4–, [P] = protecting group).](/image/article/2010/OB/b918091e/b918091e-s2.gif) | ||
| Scheme 2 Access to the sawaranospirolide system by furan oxidation (Ar = p-HO–C6H4–, [P] = protecting group). | ||
We sought a synthesis of the tri-protected tetraol 4 that was efficient, convergent, and flexible in terms of the relative and absolute stereochemistry at the hydroxylated positions. Therefore, furfuraldehyde was olefinated with phosphonate 66 (Scheme 3), and the alkene (7) dihydroxylated with AD-mix-β to yield diol 8 with an ee > 98% (as inferred by Mosher's ester analysis of alcohol 12).7 After di-benzoylation (→9), reduction of the carbonyl group afforded alcohol 10 as a mixture of diastereomers.8 Direct benzoylation of the desired stereoisomer (10α) and benzoylation under Mitsunobu conditions of the undesired stereoisomer (10β),9 followed by deprotection of the primary hydroxy group, provided oxidative cyclisation substrate 12 (Scheme 4).
Oxidative spirocyclisation of this substrate with mCPBA alone4a could not be driven to the butenolide (13); however, direct treatment of the crude lactol mixture with TPAP/NMO completed the oxidation to give a high overall yield of the spirocycle as a single diastereomer. Fig. 2 summarises key 1H NMR resonances supporting the all-equatorial substitution pattern around the THP-ring.
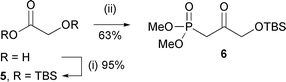 | ||
| Scheme 3 Reagents and conditions: (i) TBSCl, imidazole, DMF, 20 °C, 16 h; (ii) MePO(OMe)2, BuLi, THF, −78 °C, 2 h. | ||
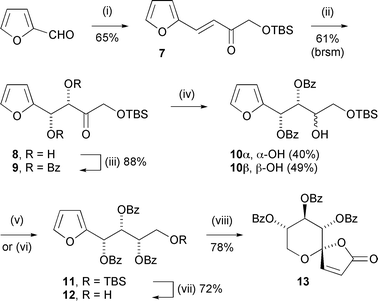 | ||
| Scheme 4 Reagents and conditions: (i) add to 6/NaH, THF, 0 °C, 2 h; (ii) AD-mix-β, MsNH2, aq. t-BuOH, 20 °C, 16 h; (iii) BzCl, DMAP, pyridine, CH2Cl2, 20 °C, 16 h; (iv) Zn(BH4)2, Et2O, −25 °C, 2 h; (v) BzCl, DMAP, pyridine, CH2Cl2, 20 °C, 16 h (88% from 10α); (vi) BzOH, DEAD, PPh3, C6H6, 20 °C, 48 h (57% from 10β); (vii) H2SiF6, aq. CH3CN, 20 °C, 5 min; (viii) mCPBA, CH2Cl2, 0→20 °C, 18 h then TPAP, NMO, CH2Cl2, 20 °C, 18 h. | ||
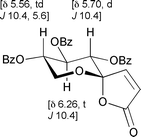 | ||
| Fig. 2 Diagnostic 1H NMR coupling constants for spirolactone 13. | ||
In order to complete syntheses of sawaranospirolides A and C, all that remained at this point was to effect conjugate addition of a p-hydroxyphenyl equivalent for which various approaches were screened. We had been successful in effecting such transformations in the context of our work on the lituarines4b but substrate 13 proved to be remarkably unreactive towards conjugate addition under the mild conditions necessary to prevent degradation of the starting material, and variants of the Heck reaction and radical additions were also unproductive.10 Ultimately, this had to be abandoned and we anticipate an eventual solution to this problem to be based on oxidative spirocyclisation of a furan substrate already bearing the phenol substituent.11
Dihydropyran oxidative spirocyclisation
Our second approach was based on the construction of the C-3–C-4 bond by conjugate addition of a metallated glycal (14, Scheme 5) to a chiral 4-coumaric acid equivalent12 (15) in order to control the C-3 configuration (pathway a). Subsequent epoxidation of the enol ether anti to the allylic alkoxy substituent (→17), and cyclisation13 to the spirolactone (18) in situ, would lead directly to sawaranospirolides A and C following deprotection. Alternatively, acidic treatment of enol ether 16 with loss of the C-6 hydroxy group, then lactonisation14 (→20) and dihydroxylation (→21) would lead to sawaranospirolides B and D. Attractions in this approach included the ready availability of suitably protected glycals, the possibility of total control of the C-3 centre (by the chiral auxiliary in A*) and the divergent entry to the four natural product configurations towards the end of the synthesis.![Synthetic strategies based on dihydropyran oxidative or acid-mediated spirolactonisation ([M] = metal, [P] = protecting group, Ar = p-hydroxyphenyl equivalent, A* = chiral carboxylic acid equivalent).](/image/article/2010/OB/b918091e/b918091e-s5.gif) | ||
| Scheme 5 Synthetic strategies based on dihydropyran oxidative or acid-mediated spirolactonisation ([M] = metal, [P] = protecting group, Ar = p-hydroxyphenyl equivalent, A* = chiral carboxylic acid equivalent). | ||
Expecting that dihydropyran-2-yl cuprate reagents would be insufficiently reactive to form conjugate adducts with coumarate-type electrophiles, we assessed Meyers' 2-alkenyl-1,3-oxazolines as the chiral conjugate acceptor because these react with organolithium reagents with a predictable sense of stereoselectivity (anti addition with respect to the oxazoline substituent in an s-cis diene conformation).15 Preliminary experiments16 with model oxazoline 22 (Scheme 6) showed that 2-lithiodihydropyran was unable to generate 1,4-adducts under a variety of conditions. This led to an early revision of the strategy; viz., introduction of the p-hydroxyphenyl substituent to a pre-formed (C-1)–(C-8) system (19, pathway b, Scheme 5) to link up with the first strategy at key intermediate 16.
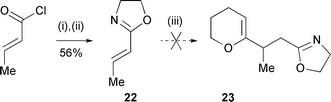 | ||
| Scheme 6 Reagents and conditions: (i) HOCH2CH2NH2, CHCl3, 0 °C, 30 min; (ii) PPh3, DIAD, THF, 0 → 20 °C, 4.5 h; (iii) DHP-Li. | ||
Following a preliminary feasibility study,17 the sawaranospirolide synthesis was initiated by formylation and Horner–Wadsworth–Emmons reaction of dihydropyran derivative 2418 with phosphonate 3119 to give dienyloxazoline 26 in good overall yield (Scheme 7). Treatment of this acceptor with p-(benzyloxy)phenyllithium at −48 °C resulted in the formation of a single diastereomer (27)20 along with a second component, tentatively assigned as an oxidation product of the intermediate α-lithio-oxazoline. Other p-hydroxyphenyllithium equivalents (32–34, Fig. 3) also gave acceptable results in this reaction but the phenolic protecting groups were either incompatible with subsequent steps (32, 33) or did not allow release of the free phenol at the end of the synthesis (34).
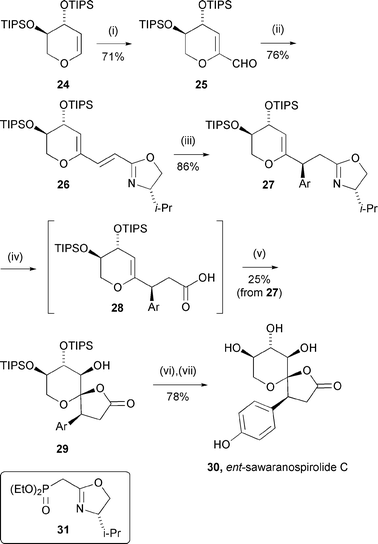 | ||
| Scheme 7 (i) t-BuLi, THF, −78 → 0 °C, 40 min then DMF, −78 °C, 10 h; (ii) add to 31, DBU, LiCl, CH3CN, 20 °C, 38 h; (iii) add to p-BnO–C6H4Li, THF, −48 °C, 30 min; (iv) a. MeOTf, CH2Cl2, 20 °C, 5 min; b. LiOH, aq. THF, 45 °C, 6 h; c. KOH, aq. MeOH/t-BuOH, reflux, 30 h; d. buffer (pH = 5); (v) mCPBA, CH2Cl2, 0 °C, 30 min; (vi) H2SiF6, aq. CH3CN, 20 °C, 20 h; (vii) H2, Pd/C, EtOH, 20 °C, 18 h. (Ar = p-BnO–C6H4–). | ||
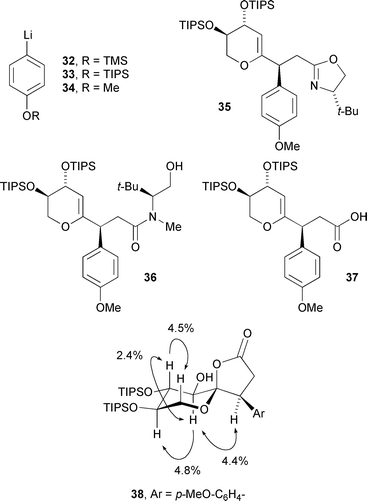 | ||
| Fig. 3 Reference structures and NOE data from model studies. | ||
Conditions for the overall hydrolysis of the oxazoline and subsequent oxidative spirocyclisation were optimised on a model substrate (35, Fig. 3) and, after extensive experimentation, a sequence of methyl triflate activation of the oxazoline, followed by partial hydrolysis with LiOH in warm aq. THF, gave amide 36.21 More forcing alkaline conditions completed amide hydrolysis, and acid 37 was obtained without apparent loss of stereochemical integrity, on protonation at pH 5. Pleasingly, the crucial oxidative spirocyclisation step progressed smoothly on treatment with mCPBA at 0 °C, the spirolactone (38) being isolated in 72% yield. The relative stereochemistry in this molecule was supported by 1H–1H coupling constants—all CHOH and CHOTIPS methine protons are axial, showing 3J 9.1 Hz—and the NOE interactions shown.
Application of these conditions to adduct 27 provided spirolactone 29 in 25% overall yield, somewhat down on the optimised yield obtained for the equivalent transformation in 35→38 (63%). Although we could not improve upon this yield, it was gratifying that the subsequent TIPS-deprotection and hydrogenolysis of the phenolic O-benzyl substituent proceeded efficiently to produce ent-sawaranospirolide C (30). Both 1H and 13C NMR data were in excellent agreement with those reported1 for the natural product, and the specific rotation for the synthetic material was, as expected, opposite in sign and of comparable magnitude to that reported for the natural enantiomer.
We also examined the possibility of extending this general methodology to encompass the synthesis of ent-sawaranospirolide D, the C-5 epimer of sawaranospirolide C (Scheme 8). An attempted Mitsunobu inversion at the C-5 centre (29→40) was unsuccessful; therefore, spirolactone 29 was oxidised efficiently to ketone 39 with Dess–Martin periodinane, with the intention of reducing it to the epimer (40). However, reduction of this ketone led either to return of the original alcohol (29) (with NaBH4) or non-productive reactions (with L-Selectride, Luche reduction, ‘kinetic’ Meerwein–Pondorff–Verley conditions22) and this idea was abandoned.
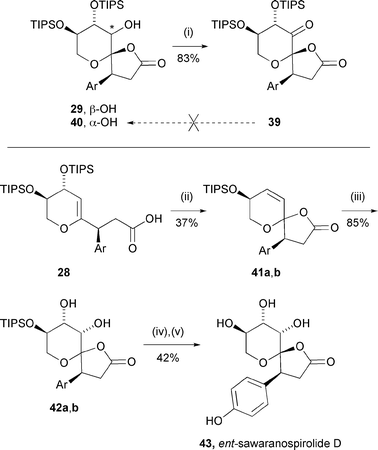 | ||
| Scheme 8 Reagents and conditions: (i) Dess–Martin periodinane, CH2Cl2, 20 °C, 18 h; (ii) PPTS, CH2Cl2, 20 °C, 6 h; (iii) a. OsO4, TMEDA, CH2Cl2, −78 °C, 3 h then 20 °C, 12 h; b. H2S (g), THF, 0 °C, 30 min; (iv) H2SiF6, aq. CH3CN, 20 °C, 24 h; (v) H2, Pd/C, EtOH, 20 °C, 18 h. | ||
Alternatively, acid-mediated spirocyclisation of acid 28 could be achieved, albeit in only moderate yield.23 Problems in this approach included simple enol ether protonation and cyclisation, giving 5-deoxyspirolactones, or slow reactions which offered opportunities for loss of stereochemical integrity at the benzylic (C-3) position. Fortunately, both ‘anomers’ (41a,b) of the spirocyclic product could be dihydroxylated to give diols 42a,b in high yield, with the stereochemistry apparently being dictated in both cases by the bulky C-7 silyloxy substituent. The stereochemistry in these two series converged during the silyl ether deprotection and the enantiomer of the second natural product isomer, ent-sawaranospirolide D (43), was obtained after hydrogenolysis. There was again complete correspondence between the NMR data for synthetic and natural material and the respective specific rotations were of opposite signs as expected, although their magnitude differed somewhat.
In summary, we have described the first total syntheses of sawaranospirolides C and D, as their enantiomers, in 7 and 9 steps, respectively, from known glycal derivative 24.
Experimental
(3R,4R)-3,4-Bis(triisopropylsilyloxy)-3,4-dihydro-2H-pyran-6-carbaldehyde 25
To a stirred solution of dihydropyran derivative 24 (911 mg, 2.13 mmol) in THF (1 mL) at −78 °C was added dropwise tert-butyllithium (1.25 mL of a 1.8 M solution in pentane, 2.25 mmol). After 10 min the reaction was warmed to 0 °C for 30 min then re-cooled to −78 °C and a solution of DMF (0.17 mL, 2.19 mmol) in THF (5 mL) added dropwise. After a further 10 h at −78 °C the reaction mixture was then poured into water (30 mL) and extracted with ether (3 × 30 mL). The combined organic extracts were washed successively with water and brine before being dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (petrol/ether, 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded starting material (24, 102 mg, 11%) and aldehyde 25 (693 mg, 71%) as a colourless oil. Rf 0.64 (petrol/ether, 4:1); [α]23D−116.5 (c 1.3, CHCl3); νmax (thin film)/cm−1 1718 s, 1638 s, 1464 s, 1344 m, 1245 s, 1176 s, 1141 s, 1070 s, 923 s, 882 s, 738 s, 681 s; δH (400 MHz, CDCl3) 0.95–1.15 (42 H, m, 2 × TIPS), 3.96–3.97 (1 H, m, H-3), 4.07 (1 H, d, J 11.2, H-2), 4.16–4.19 (1 H, m, H-4), 4.21–4.25 (1 H, m, H-2′), 5.84 (1 H, dd, J 5.3, 1.5, H-5), 9.19 (1 H, s, CHO); δC (100 MHz, CDCl3) 12.3 (complex), 18.0 (complex), 64.3, 66.4, 68.8, 118.7, 151.7, 187.6; m/z (CI) 474 (MNH4+, 23%), 300 (38), 283 (100), 241 (22), 213 (17); HRMS (CI) found 474.3439, C24H52NO4Si2 (MNH4+) requires 474.3429.
:1) afforded starting material (24, 102 mg, 11%) and aldehyde 25 (693 mg, 71%) as a colourless oil. Rf 0.64 (petrol/ether, 4:1); [α]23D−116.5 (c 1.3, CHCl3); νmax (thin film)/cm−1 1718 s, 1638 s, 1464 s, 1344 m, 1245 s, 1176 s, 1141 s, 1070 s, 923 s, 882 s, 738 s, 681 s; δH (400 MHz, CDCl3) 0.95–1.15 (42 H, m, 2 × TIPS), 3.96–3.97 (1 H, m, H-3), 4.07 (1 H, d, J 11.2, H-2), 4.16–4.19 (1 H, m, H-4), 4.21–4.25 (1 H, m, H-2′), 5.84 (1 H, dd, J 5.3, 1.5, H-5), 9.19 (1 H, s, CHO); δC (100 MHz, CDCl3) 12.3 (complex), 18.0 (complex), 64.3, 66.4, 68.8, 118.7, 151.7, 187.6; m/z (CI) 474 (MNH4+, 23%), 300 (38), 283 (100), 241 (22), 213 (17); HRMS (CI) found 474.3439, C24H52NO4Si2 (MNH4+) requires 474.3429.(S)-2-{(E)-2-[(3R,4R)-3,4-Bis(triisopropylsilyloxy)-3,4-dihydro-2H-pyran-6-yl]vinyl}-4-isopropyl-4,5-dihydrooxazole 26
To stirred a solution of diisopropylamine (6.20 mL, 43.9 mmol) in THF (25 mL) at 0 °C was added dropwise butyllithium (25.5 mL of a 1.7 M solution in hexanes, 43.4 mmol). After 15 min the solution was cooled to −78 °C and a solution of (S)-4-isopropyl-2-methyl-4,5-dihydrooxazole24 (1.06 g, 8.35 mmol) in THF (8 mL) was added. After 1 h diethylphosphochloridate (1.51 mL, 10.5 mmol) was added; stirring was continued for 1 h at −78 °C then warmed to 0 °C and quenched with saturated NH4Cl solution (50 mL). The mixture was poured onto water (15 mL) and extracted with ether (3 × 25 mL), dried over MgSO4, and concentrated in vacuo to give the crude phosphonate (31),19 as a yellow oil, that was used directly in the next step. The phosphonate (31, 8.35 mmol assumed) was dissolved in acetonitrile (30 mL), and LiCl (416 mg, 9.82 mmol) and DBU (1.43 mL, 9.58 mmol) were then added to this stirred solution. A solution of aldehyde 25 (3.65 g, 8.00 mmol) in acetonitrile (20 mL) was added dropwise and, after 38 h, the mixture was poured onto water (100 mL) and extracted with ether (3 × 50 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue that was purified by column chromatography (petrol/ether, 9:1) to give the oxazoline 26 (3.43 g, 76%) as a viscous clear light yellow oil. Rf 0.39 (petrol/ether, 9:1); νmax (thin film)/cm−1 2867 s, 1640 s, 1464 s, 1385 s, 1248 s, 1143 s, 1060 s, 995 s, 921 s, 682 s; δH (400 MHz, CDCl3) 0.89 and 0.97 (2 × 3 H, 2 × d, J 6.8, oxazoline-CH(CH3)2), 0.99–1.10 (42 H, m, 2 × TIPS), 1.72–1.85 (1 H, m, oxazoline-CH(CH3)2), 3.82–3.91 (1 H, m, CH(OTIPS)CH2), 3.95–4.01 (2 H, m, OCHH′CHN), 4.02–4.06 (2 H, m, CHH′CHOTIPS and CH(OTIPS)CH=), 4.10 (1 H, br d, J 11.3, CHH′CHOTIPS), 4.22–4.32 (1 H, m, OCHH′CHN), 5.09 (1 H, d, J 4.3, =CH), 6.46 and 6.64 (2 × 1 H, 2 × d, J 15.7, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH); δC (100 MHz, CDCl3) 12.4–12.7 and 17.9–18.8 (complex), 32.8, 65.2, 66.0, 69.1, 69.7, 72.6, 106.8, 115.6, 135.0, 150.3, 163.0; HRMS (ESI+) found 566.4041, C31H60NO4Si2 (MH+) requires 566.4055.
CH); δC (100 MHz, CDCl3) 12.4–12.7 and 17.9–18.8 (complex), 32.8, 65.2, 66.0, 69.1, 69.7, 72.6, 106.8, 115.6, 135.0, 150.3, 163.0; HRMS (ESI+) found 566.4041, C31H60NO4Si2 (MH+) requires 566.4055.(S)-2-{(S)-2-[4-(Benzyloxy)phenyl]-2-[(3R,4R)-3,4-bis(triisopropylsilyloxy)-3,4-dihydro-2H-pyran-6-yl]ethyl}-4-isopropyl-4,5-dihydrooxazole 27
To a stirred solution of 4-(benzyloxy)bromobenzene (827 mg, 3.14 mmol) in dry THF (10 mL) at −78 °C was added dropwise tert-butyllithium (1.89 mL of a 1.7 M solution in pentane, 3.21 mmol). After 30 min the solution was warmed up to −48 °C and a solution of oxazoline 26 (890 mg, 1.58 mmol) in THF (10 mL) was added over 30 min. After a further 10 min, the reaction was quenched with methanol (2 mL) and allowed to warm to RT. The mixture was poured onto water (50 mL) and extracted with ether (3 × 50 mL). The combined organic extracts were dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (petrol/ether, 9:1) gave the title compound (27) as a colourless oil (980 mg, 86%). Rf 0.4 (petrol/ether, 4:1); νmax (thin film)/cm−1 2943 s, 2866 s, 1668 s, 1611 m, 1511 s, 1464 s, 1384 m, 1244 s, 1058 s, 883 s, 681 s; δH (400 MHz, CDCl3) 0.65 and 0.73 (2 × 3 H, 2 × d, J 6.7, oxazoline-CH(CH3)2), 0.95–1.10 (42 H, m, 2 × TIPS), 1.45–1.55 (1 H, m, oxazoline-CH(CH3)2), 2.76 (1 H, dd, J 14.7, 6.0) and 2.84 (1 H, dd, J 14.7, 10.2, CH2C(=N)O), 3.71–3.80 (4 H, m), 3.90–4.01 (3 H, m) and 4.05–4.12 (1 H, m, CH2CH(OTIPS)CH(OTIPS), CH2CHN and CHAr), 4.77 (1 H, d, J 5.2, =CH), 5.03 (2 H, s, CH2Ph), 6.85 and 7.20 (2 × 2 H, 2 × d, J 8.6, Ar), 7.29–7.46 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.0–12.5 and 17.9–18.3 (complex), 32.2 (2 peaks), 46.1, 65.3, 66.3, 69.0, 69.7, 70.0, 71.8, 96.7, 114.4, 127.4, 127.8, 128.5, 129.2, 132.9, 137.3, 156.7, 157.5, 165.4; HRMS (ESI+) found 750.4944, C44H72NO5Si2 (MH+) requires 750.4944.(S)-3-[4-(Benzyloxy)phenyl]-3-[(3R,4R)-3,4-bis(triisopropylsilyloxy)-3,4-dihydro-2H-pyran-6-yl]propanoic acid 28
To stirred a solution of oxazoline 27 (558 mg, 0.774 mmol) in dichloromethane (5 mL) at RT was added methyl triflate (93 μL, 0.82 mmol). After 5 min the mixture was poured onto water (50 mL) and extracted with dichloromethane (3 × 50 mL). The combined organic layers were then dried over MgSO4 and concentrated in vacuo. This material was then dissolved in THF (5 mL), treated with LiOH solution (0.97 mL of a 1 M solution, 0.97 mmol), and the mixture warmed to 45 °C for 6 h. The mixture was cooled to RT, most of the THF evaporated in vacuo, the residue diluted with B(OH)3 solution (0.033 M, 25 mL), and extracted with dichloromethane (3 × 50 mL). The combined organic portions were dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (dichloromethane/methanol, 50:1) afforded a mixture of amide and ester products of oxazoline hydrolysis (595 mg, 98%) as a viscous, colourless oil. A portion of this amide/ester mixture (356 mg, 0.456 mmol) was dissolved in methanol (2.5 mL) and tert-butanol (2.5 mL) then KOH solution (4.60 mL, 2 M, 9.20 mmol) was added. The mixture was heated at reflux for 30 h then, after cooling to RT, was concentrated in vacuo, diluted with B(OH)3 solution (0.033 M, 20 mL) and extracted with dichloromethane (3 × 100 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. This crude material was then re-dissolved in ether (50 mL) and washed with B(OH)3 solution (0.033 M, 3 × 20 mL) and brine (10 mL). The organic solution was dried over MgSO4 and concentrated in vacuo to give acid 28 as a gluey oil (256 mg, 82%) that was used in this crude form. νmax (thin film)/cm−1 3500–2500 br m, 2866 s, 1713 s, 1663 s, 1612 m, 1511 s, 1463 s, 1245 m, 883 m, 681 m; δH (400 MHz, CDCl3) 0.95–1.10 (42 H, m, 2 × TIPS), 2.79 (1 H, dd, J 16.3, 8.5) and 2.92 (1 H, dd, J 16.3, 6.7, CHCO2H), 3.75–3.88 (2 H, m) and 3.91–4.07 (3 H, m, CH2CH(OTIPS)CH(OTIPS) and CHAr), 4.76 (1 H, d, J 5.1, CH), 5.04 (2H, s, CH2Ph), 6.90 and 7.21 (2 × 1 H, 2 × d, J 8.6, Ar), 7.31–7.48 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.2–12.5 and 17.7–18.1 (complex), 29.7, 44.9, 65.0, 66.3, 68.9, 70.0, 96.9, 114.5, 127.5, 127.9, 128.6, 128.9, 133.2, 137.2, 156.3, 157.6, 177.7; HRMS (ESI+) found 683.4149, C39H63O6Si2 (MH+) requires 683.4158.(4S,5R,8R,9R,10R)-4-[4-(Benzyloxy)phenyl]-10-hydroxy-8,9-bis(triisopropylsilyloxy)-1,6-dioxaspiro[4.5]decan-2-one 29
To a stirred solution of crude acid 28 (256 mg, 0.375 mmol) in dichloromethane (5 mL) at 0 °C was added dropwise a solution of mCPBA (101 mg, ca. 75% by weight, 0.439 mmol) in dichloromethane (4 mL). After 30 min the reaction mixture was mixed with saturated NaHCO3 solution (25 mL) and extracted with dichloromethane (3 × 25 mL). The combined organic extracts were dried over MgSO4, concentrated in vacuo, and purified by column chromatography (petrol/ether, 7:3) to afford spirocycle 29 (82 mg, 31%) as an oil. Rf 0.5 (petrol/ether, 6:4); νmax (thin film)/cm−1 3480 br m, 2945 s, 2867 s, 1785 s, 1613 m, 1514 s, 1465 m, 1383 m, 1241 m, 1090 m, 1016 m, 916 s, 884 s, 735 s, 682 s; δH (400 MHz, CDCl3) 1.02–1.20 (42 H, m, 2 × TIPS), 1.88 (1 H, d, J 9.1, OH), 2.79 (1 H, dd, J 17.1, 8.3) and 3.01 (1 H, dd, J 17.1, 12.5, CH2CO), 3.31 (1 H, t, J 9.1, CHOH), 3.57–3.66 (2 H, m, CHH′CHOTIPS), 3.69–3.76 (1 H, m, CHH′CHOTIPS), 3.85 (1H, app. t, J 8.1, CH(OTIPS)CHOH), 4.00 (1 H, dd, J 12.5, 8.3, CHAr), 5.07 (2 H, s, CH2Ph), 6.97 and 7.24 (2 × 2 H, 2 × d, J 8.8, Ar), 7.32–7.48 (5 H, m, Ph); δC (100 MHz, CDCl3) 13.4–13.6 and 18.2–18.4 (complex), 34.1, 44.6, 64.8, 70.0, 71.7, 71.8, 78.2, 108.6, 114.6, 126.1, 127.5, 128.1, 128.6, 130.5, 136.8, 158.5, 174.9; HRMS (ESI+) found 721.3929, C39H62NaO7Si2 (MNa+) requires 721.3926.(4S,5R,8R,9S,10R)-4-[4-(Benzyloxy)phenyl]-8,9,10-trihydroxy-1,6-dioxaspiro[4.5]decan-2-one (30-OBn)
Aqueous H2SiF6 solution (7.0 μL, 25% by weight, 0.011 mmol) was added to a stirred solution of spirocycle 29 (39 mg, 0.056 mmol) in acetonitrile (2 mL) at RT. After 2 h a further portion of H2SiF6 solution (7 μL, 25% by weight, 0.011 mmol) was added and the mixture stirred for 18 h. The solution was then concentrated in vacuo and purified by column chromatography (dichloromethane/methanol, 49:1) to provide the title compound (18 mg, 83%). Rf 0.2 (dichloromethane/methanol, 95:5); νmax (thin film)/cm−1 3600–3400 br m, 2943 s, 2866 s, 1784 s, 1514 s, 1240 m, 1109 s, 915 s; δH (400 MHz, CDCl3) 2.74 (1 H, dd, J 17.3, 8.6) and 2.97 (1 H, dd, J 17.3, 12.2, CH2CO), 3.35–3.42 (1 H, m, CH(OH)-spiro), 3.43–3.51 (1 H, m, CH2CHOH), 3.56 (1 H, app. t, J 10.8, CHH′CHOH), 3.71–3.83 (2 H, m, CHH′CH(OH)CHOH), 4.00 (1 H, dd, J 12.2, 8.6, CHAr), 4.59 (1 H, bs, OH), 4.96 (2 H, app. s, CH2Ph), 5.04 (1 H, bs, OH), 5.40 (1 H, bs, OH), 6.90 and 7.23 (2 × 2 H, J 8.5, Ar), 7.31–7.42 (5 H, m, Ph); δC (100 MHz, CDCl3) 33.9, 44.2, 64.1, 69.3, 69.9, 70.3, 75.2, 108.9, 114.6, 125.9, 127.5, 128.0, 128.6, 130.5, 136.8, 158.5, 176.1; HRMS (ESI+) found 409.1257, C21H22NaO7 (MNa+) requires 409.1258.(4S,5R,8R,9S,10R)-8,9,10-Trihydroxy-4-(4-hydroxyphenyl)-1,6-dioxaspiro[4.5]decan-2-one (ent-sawaranospirolide C 30)
A solution of the spirocycle obtained by TIPS-deprotection of precursor 29 (18 mg, 46.6 μmol) and 5% Pd/C (20 mg) in ethanol (2 mL) was purged with argon and then stirred under hydrogen at RT for 18 h. After flushing with argon, the reaction mixture was filtered though Celite and concentrated in vacuo to give ent-sawaranospirolide C (13 mg, 94%) as a viscous oil. Rf 0.28 (dichloromethane/methanol, 9:1); [α]23D +40.5 (c 1.4, MeOH), lit.1 (for enantiomer) −36 (c 2.25, MeOH); νmax (thin film)/cm−1 3600–3000 br s, 2926 m, 1769 s, 1700 m, 1651 m, 1519 s, 1367 m, 1260 m, 1105 m, 1047 m, 917 m; δH (500 MHz, d6-DMSO) 2.73 (1 H, dd, J 17.1, 8.4) and 3.01 (1 H, dd, J 17.1, 12.9, CH2CO), 3.07–3.15 (2 H, m, CH(OH)CH(OH)-spiro), 3.30 (1 H, t, J 11.3, CHH′O), overlaying 3.28–3.35 (1 H, m, CH(OH)CH2O), 3.53 (1 H, dd, J 11.3, 6.4, CHH′O), 3.89 (1 H, dd, J 12.9, 8.4, CHAr), 5.01 (1 H, d, J 6.0, one of CH(OH)CH(OH)-spiro), 5.13 (1 H, d, J 6.0, CH(OH)CH2O), 5.74 (1 H, d, J 6.0, one of CH(OH)CH(OH)-spiro), 6.70 and 7.12 (2 × 2H, 2 × d, J 8.5, Ar), 9.40 (1 H, br s, ArOH); δC (125 MHz, d6-DMSO) 32.6, 43.3, 64.2, 69.0, 69.7, 74.3, 108.7, 114.8, 124.8, 130.2, 156.6, 175.1; HRMS (FI+) found 296.0901, C14H16O7 (M+) 296.0891.(4S,5R,8R,9S)-4-[4-(Benzyloxy)phenyl]-8,9-bis(triisopropylsilyloxy)-1,6-dioxaspiro[4.5]decane-2,10-dione 39
Dess–Martin periodinane (44 mg, 0.10 mmol) was added to a solution of spirocycle 29 (36 mg, 0.052 mmol) in dichloromethane (5 mL) at RT and the mixture was stirred for 18 h. The mixture was diluted with ether (20 mL) then filtered through Celite; the filtrate was concentrated in vacuo and the residue purified by column chromatography (petrol/ether, 9:1) to give ketone 39 (29 mg, 83%) as a colourless oil. Rf 0.82 (petrol/ether, 4:1); νmax (thin film)/cm−1 2947 s, 1814 s, 1752 s, 1614 m, 1514 s, 1464 s, 1383 m, 1124 s, 899 m, 793 m; δH (400 MHz, CDCl3) 1.03–1.12 (42 H, m, 2 × TIPS), 2.88 (1 H, dd, J 17.5, 8.7) and 3.00 (1 H, dd, J 17.5, 11.8, CH2CO), 3.87–4.02 (3 H, m, CH2CHO), 4.31 (1 H, dd, J 11.8, 8.7, CHAr), 4.82 (1 H, dd, J 4.8, 3.7, CHCO),25 5.06 (2 H, s, CH2Ph), 6.94 and 7.26 (2 × 2 H, 2 × d, J 8.8, Ar), 7.31–7.47 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.9–13.1 and 18.0–18.2 (complex), 33.9, 42.7, 65.4, 70.0, 75.1, 81.4, 105.4, 114.5, 125.9, 127.5, 128.0, 128.6, 130.6, 136.8, 158.3, 173.4, 196.5; HRMS (ESI+) found 787.4747, C42H71N2O8Si2 (M·CH3CN·MeOH·NH4+) requires 787.4743.(4S,5S,8S)- and (4S,5R,8S)-4-[4-(Benzyloxy)phenyl]-8-(triisopropylsilyloxy)-1,6-dioxaspiro[4.5]dec-9-en-2-one 41a,b
To a stirred solution of acid 28 (121 mg, 0.177 mmol) in dichloromethane (4 mL) at RT was added a solution of PPTS (30 mg, 0.12 mmol) in dichloromethane (2 mL). Stirring was continued for 6 h, then the mixture was mixed with water (20 mL) and the separated aqueous layer was extracted with dichloromethane (3 × 20 mL). The combined organic portions were dried over MgSO4, filtered and concentrated in vacuo. Purification by flash chromatography (petrol/ether, 4:1) afforded spirocycles 41a (22 mg, 24%) and 41b (12 mg, 13%) as oils. Data for 41a: Rf 0.53 (petrol/ether, 7:3); νmax (thin film)/cm−1 2942 s, 2890 s, 1784 s, 1659 m, 1612 m, 1583 m, 1514 s, 1463 m, 1427 m, 1400 m, 1383 m, 1245 s, 781 s; δH (400 MHz, CDCl3) 1.04–1.12 (21 H, m, TIPS), 2.77 (1 H, dd, J 17.5, 5.1) and 3.25 (1 H, dd, J 17.5, 8.6, CH2CO), 3.73 (1 H, dd, J 8.6, 5.1, CHAr), 3.98 (1 H, d, J 12.1, CHH′O), 4.04–4.07 (1 H, m, CHOTIPS), 4.15 (1 H, dd, J 12.1, 2.6, CHH′O), 5.05 (2 H, s, CH2Ph), 5.30 (1 H, d, J 10.2) and 6.02 (1 H, ddd, J 10.2, 5.2, 1.0, CHCH), 6.94 and 7.13 (2 × 2 H, 2 × d, J 8.7, Ar), 7.32–7.47 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.3, 18.0 (2 peaks), 35.2, 49.7, 61.0, 68.4, 70.0, 106.0, 115.0, 125.3, 127.5, 128.1, 128.6, 128.9, 129.4, 131.0, 136.8, 158.3, 175.6; HRMS (ESI+) found 509.2717, C30H41O5Si (MH+) requires 509.2718. Data for 41b: Rf 0.45 (petrol/ether, 7:3); νmax (thin film)/cm−1 2943 s, 2867 s, 1799 s, 1514 s, 1463 m, 1395 m, 1221 s, 1181 s, 1125 m, 899 m, 688 m; δH (400 MHz, CDCl3) 0.98–1.10 (21 H, m, TIPS), 2.78 (1 H, dd, J 16.9, 7.9) and 3.13 (1 H, dd, J 16.9, 12.9, CH2CO), 3.57 (1 H, dd, J 12.9, 7.9, CHAr), 3.69 (1 H, t, J 10.3) and 3.80 (1 H, dd, J 10.3, 5.7, CH2O), 3.99–4.07 (1 H, m, CHOTIPS), 5.06 (2 H, s, CH2Ph), 5.78 (1 H, dd, J 10.2, 1.9) and 6.14 (1 H, d, J 10.2, CHCH), 6.94 and 7.18 (2 × 2 H, 2 × d, J 8.7, Ar), 7.32–7.46 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.1–12.3 and 17.9–18.0, 33.5, 50.1, 62.9, 65.5, 70.0, 104.5, 114.5, 124.7, 126.7, 127.5, 128.1, 128.6, 129.6, 136.8, 139.1, 158.3, 175.1; HRMS (ESI) found 509.2723, C30H41O5Si (MH+) requires 509.2718.(4S,5R,8R,9R,10S)- and (4S,5S,8R,9R,10S)-4-[4-(Benzyloxy)phenyl]-9,10-dihydroxy-8-(triisopropylsilyloxy)-1,6-dioxaspiro[4.5]decan-2-one 42a,b
A solution of spirocycle 41a (39 mg, 76.7 μmol) and TMEDA (13 μL, 0.084 mmol) in dichloromethane (3 mL) was cooled to −78 °C under argon. A solution of OsO4 (21 mg, 0.084 mmol) in dichloromethane (2 mL) was added and the solution was stirred at −78 °C for 3 h then allowed to warm up to RT. The reaction mixture was stirred for a further 12 h and then concentrated in vacuo to give a residue that was subsequently dissolved in THF (5 mL). H2S (g) was bubbled through the solution at 0 °C for 30 min then the reaction mixture was concentrated in vacuo and the residue purified by chromatography (dichloromethane/methanol, 98:2) to give diol 42a (35 mg, 85%) as an oil. Rf 0.67 (dichloromethane/methanol, 95:5); νmax (thin film)/cm−1 3600–3300 br m, 2941 s, 2865 s, 1774 s, 1513 s, 1456 s, 1238 s, 1124 s, 1054 s, 845 s; δH (500 MHz, CDCl3) 0.95–1.05 (21 H, m, TIPS), 2.31 (1 H, br s, OH), 2.55 (1 H, br s, OH), 2.85 (1 H, dd, J 17.1, 9.2) and 3.32 (1 H, dd, J 17.1, 12.8, CH2CO), 3.71 (1 H, dd, J 12.8, 9.2, CHAr), 3.73 (1 H, br s, CH(OH)-spiro) overlaying 3.78 (1 H, d, J 12.6, CHH′O), 3.80 (1 H, br s, CH(OH)CHOTIPS), 3.99 (1 H, d, J 3.6, CHOTIPS), 4.21 (1 H, d, J 12.6, CHH′O), 5.06 (2 H, AB q, J 11.8, CH2Ph), 6.96 and 7.33 (2 × 2 H, 2 × d, J 8.7, Ar), 7.30–7.46 (5 H, m, Ph); δC (125 MHz, CDCl3) 12.1–12.2 and 17.9 (2 peaks), 34.3, 50.8, 64.0, 64.6, 70.0, 70.1, 71.7, 110.7, 115.0, 126.6, 127.4, 128.0, 128.6, 129.1, 136.9, 158.5, 173.4; HRMS (ESI+) found 565.2580, C30H42NaO7Si (MNa+) requires 565.2592. Analogously, from 41b (28 mg, 55.1 μmol), diol 42b was obtained (25 mg, 84%). Rf 0.79 (dichloromethane/methanol, 95:5); νmax (thin film)/cm−1 3500–3100 br m, 2926 s, 1773 s, 1513 s, 1245 m, 1038 m; δH (400 MHz, CDCl3) 1.02–1.06 (21 H, m, TIPS), 2.29 and 2.50 (2 × 1 H, 2 × br s, 2 × OH), 2.85–2.95 (2 H, m, CH2CO), 3.63 (1 H, t, J 10.3, CHH′O), 3.75–3.85 (2 H, m, CHAr and CHH′O), 3.88–3.97 (2 H, m, 2 × CHOH), 4.08 (1 H, d, J 2.3, CHOTIPS), 5.05 (2 H, s, CH2Ph), 6.94 and 7.32 (2 × 2 H, 2 × d, J 8.7, Ar), 7.34–7.46 (5 H, m, Ph); δC (100 MHz, CDCl3) 12.1 (2 peaks) and 17.9 (3 peaks), 36.3, 48.2, 65.1, 67.8, 69.9, 73.0, 73.7, 106.8, 114.3, 127.5, 128.0, 128.6, 130.8, 136.9, 158.2, 174.3; HRMS (ESI+) found 543.2789, C30H43O7Si (MH+) requires 543.2773.(4S,5R,8R,9S,10S)-4-[4-(Benzyloxy)phenyl]-8,9,10-trihydroxy-1,6-dioxaspiro[4.5]decan-2-one (43-OBn)
To stirred a solution of diol 42a (35 mg, 64.6 μmol) in acetonitrile (2 mL) at RT was added H2SiF6 (28 μL, 22% by weight, 42.8 μmol) in three portions (8, 10 and 10 μL). The mixture was stirred for 24 h in total then the volatiles were evaporated and the residue purified by column chromatography (dichloromethane/methanol, 98:2) to give the title compound (11 mg, 44%) as an oil. [The same compound was obtained in similar yield by the analogous reaction of diol 42b (25 mg, 46.1 μmol).] Rf 0.36 (dichloromethane/methanol, 95:5); νmax (thin film)/cm−1 3700–3100 br m, 3079 s, 1784 m, 1616 s, 1543 s, 1229 s, 1035 m, 1012 m, 816 m, 682 m; δH (400 MHz, CD3OD) 2.90 (2 H, app. d, J 9.8, CH2CO), 3.49 (1 H, app. ddd, J 14.9, 7.4, 2.9, CHH′O), 3.66–3.81 (3 H, m, CHH′O and CH(OH)CH(OH)-spiro), 3.83 (1 H, t, J 9.8, CHAr), 3.95 (1 H, d, J 3.2, CH2CHOH), 5.07 (2 H, s, CH2Ph), 6.90 and 7.31 (2 × 2 H, 2 × d, J 8.6, Ar), 7.34–7.46 (5 H, m, Ph); δC (100 MHz, CD3OD) 36.2, 48.0, 65.3, 65.9, 69.9, 72.2, 73.9, 108.6, 114.2, 127.5, 127.8, 128.5, 129.6, 131.0, 137.9, 158.3, 175.6; HRMS (FI+) found 386.1359, C21H22O7 (M+) requires 386.1366.(4S,5R,8R,9S,10S)-8,9,10-Trihydroxy-4-(4-hydroxyphenyl)-1,6-dioxaspiro[4.5]decan-2-one (ent-sawaranospirolide D 43)
A stirred suspension of the triol obtained from TIPS deprotection of 42a,b (11 mg, 28.5 μmol) and 5% Pd/C (15 mg) in ethanol (2 mL) was purged with argon then stirred under hydrogen at RT for 29 h. The reaction vessel was then flushed with argon and the mixture filtered through Celite, washing through with more ethanol; the filtrate was concentrated in vacuo to give ent-sawaranospirolide D (8 mg, 95%) as a gum. Rf 0.17 (dichloromethane/methanol, 9:1); [α]23D +28.7 (c 0.4, MeOH), lit.1 (for enantiomer) −54 (c 0.74, MeOH); νmax (thin film)/cm−1 3650–3100 br s, 2925 m, 1771 s, 1615 m, 1519 s, 1260 s, 1024 m, 960 m; δH (400 MHz, d6-DMSO) 2.75 (1 H, dd, J 17.7, 9.8) and 2.90 (1 H, dd, J 17.7, 9.2, CH2CO), 3.25 (1 H, t, J 10.1, CHH′O), 3.47–3.61 (3 H, m, CHH′CH(OH)CH(OH)), 3.70–3.82 (2 H, m, CH(OH)-spiro and CHAr), 4.36, 4.88 and 5.01 (3 × 1 H, 3 × br s, 3 × OH), 6.62 and 7.10 (2 × 2 H, 2 × d, J 8.6, Ar); δC (100 MHz, d6-DMSO) 35.8, 46.3, 65.1, 65.4, 71.3, 72.9, 108.3, 114.2, 127.8, 130.7, 156.0, 174.3; HRMS (FI+) found 296.0888, C14H16O7 (M+) requires 296.0896.
Acknowledgements
We thank the University of Oxford and the Engineering and Physical Sciences Research Council for funding. We also thank Drs Tim Claridge and Barbara Odell, University of Oxford, for supporting NMR studies.Notes and references
- S. Hasegawa, H. Koyanagi and Y. Hirose, Phytochemistry, 1990, 29, 261–266 CrossRef CAS.
- This is based on the proposed biosynthesis described in ref. 1, and references cited therein.
- Spirolactones of this type show a strong thermodynamic preference for an axially-disposed acyl oxygen–carbon bond. For a general discussion of the axial preference of anomeric acyloxy functionality see: A. J. Kirby, The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, Springer-Verlag, Berlin, 1983, pp. 60–61 Search PubMed.
- For our recent applications of this approach in the context of natural product synthesis see: (a) J. Robertson, P. Meo, J. W. P. Dallimore, B. M. Doyle and C. Hoarau, Org. Lett., 2004, 6, 3861–3863 CrossRef CAS; (b) J. Robertson and J. W. P. Dallimore, Org. Lett., 2005, 7, 5007–5010 CrossRef CAS; (c) J. Robertson, K. Stevens and S. Naud, Synlett, 2008, 2083–2086 CrossRef CAS; see also: (d) J. Robertson and S. Naud, Org. Lett., 2008, 10, 5445–5448 CrossRef CAS.
- H. Fukuda, M. Takeda, Y. Sato and O. Mitsunobu, Synthesis, 1979, 368–370 CrossRef CAS.
- P. Angehrn, P. Hebeisen, I. Heinze-Krauss, M. Page, Eur. Pat. Appl., 1998, EP0831093 (A1) Search PubMed.
- Products of the type 8 show a tendency to fragment by retro-aldol reaction under the dihydroxylation conditions. For this reason, the reaction was terminated before complete consumption of the starting alkene and the latter could be recycled.
- Although reduction with L-Selectride favoured the formation of isomer 10α, benzoyl migration complicated product isolation which resulted in a lower overall yield.
- The stereochemistry of 10α/10β was assigned retrospectively following spirocyclisation.
- D. Woollaston, D.Phil. Thesis, Oxford, 2007. Search PubMed.
- In another approach (see ref. 10) we prepared the 3,5-dibromofuryl variant of substrate 12 in order to provide a handle for introducing the p-hydroxyphenyl group by cross-coupling. However, the two bromine substituents deactivated the furan and we were unable to effect oxidation.
- B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771–806 CrossRef CAS.
- R. M. Boyce and R. M. Kennedy, Tetrahedron Lett., 1994, 35, 5133–5136 CrossRef CAS.
- S. J. Danishefsky and W. H. Pearson, J. Org. Chem., 1983, 48, 3865–3866 CrossRef CAS.
- A. I. Meyers and M. Shipman, J. Org. Chem., 1991, 56, 7098–7102 CrossRef CAS.
- J. M. Withey, Chemistry Part II Thesis, Oxford, 2000 Search PubMed.
- T. Fowler, Chemistry Part II Thesis, Oxford, 2001 Search PubMed.
- L. A. Paquette, M. J. Kinney and U. Dullweber, J. Org. Chem., 1997, 62, 1713–1722 CrossRef CAS.
- A. Chesney, M. R. Bryce, A. S. Batsanov, J. A. K. Howard and L. M. Goldenberg, Chem. Commun., 1998, 677–678 RSC.
- We note that this represents the stereochemically matched case; repetition of this sequence starting with ent-31 gave a mixture of diastereomers in the conjugate addition. Thus, although the cheaper valinol-derived auxiliary serves for some purposes, the tert-leucinol-analogue (ref. 15) still remains the most generally useful.
- This amide was examined as an oxidative cyclisation substrate but under a variety of conditions satisfactory results were not achieved; instead, recovered starting material, eliminated material, and over-oxidised products were obtained.
- A. K. Dilger, V. Gopalsamuthiram and S. D. Burke, J. Am. Chem. Soc., 2007, 129, 16273–16277 CrossRef CAS.
- The use of BF3·OEt2 (1.1 equiv.) in THF at RT gave a similar yield of spirocycles 41a,b, essentially free of the simple acid-catalysed spirocyclisation products.
- M. J. Kurth and O. H. W. Decker, J. Org. Chem., 1985, 50, 5769–5775 CrossRef CAS.
- This coupling pattern suggests that a significant proportion of the conformers bear a 1,2-diaxial arrangement of the silyloxy substituents, which was supported by preliminary molecular mechanics calculations (Monte Carlo conformational search, MMFF).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for Schemes 3, 4 and 6; copies of selected 1H and 13C NMR spectra for the compounds in Schemes 7 and 8. See DOI:10.1039/b918091e |
| This journal is © The Royal Society of Chemistry 2010 |