Stereoselective synthesis and structure–affinity relationships of bicyclic κ receptor agonists†
Daniel Krachta, Elisabeth Racka, Dirk Schepmanna, Roland Fröhlichb and Bernhard Wünsch*a
aInstitut für Pharmazeutische und Medizinische Chemie der Universität Münster, Hittorfstraße 58-62, D-48149 Münster, Germany. E-mail: wuensch@uni-muenster.de; Fax: +49-251-8332144; Tel: +49-251-8333311
bOrganisch-Chemisches Institut der Westfälischen Wilhelms-Universität Münster, Corrensstr. 40, D-48149 Münster, Germany
First published on 12th November 2009
Abstract
Reductive amination of the bicyclic ketone 4 led diastereoselectively to endo-configured amines, which were transformed into the amides 7–10. The synthesis of the diastereomers 25 with an exo-configured amino moiety at position 6 was only successful after deactivation of both N-atoms of the 1,4-diazabicyclo[3.3.1]nonane system. The N-1-oxide 19 with an N-4-tosyl moiety was the crucial intermediate, which allows SN2 substitution with NaN3 under inversion of the configuration at position 6. Whereas the endo-configured pyrrolidine 7a (WMS-1302) revealed a κ receptor affinity of 73 nM, the exo-configured diastereomer 25a was almost inactive at the κ receptor (Ki > 1 μM). Replacement of the 3,4-dichlorophenylacetyl residue by other acyl and sulfonyl residues showed that it is essential for high κ affinity. The κ receptor affinities of the conformationally constrained pyrrolidines 7a and 25a were correlated with the dihedral angle N(pyrrolidine)–C–C–N(acetamide). A systematic conformational analysis of the potent but flexible κ agonist 2 showed that a dihedral angle of 168° (as in 25a) is energetically more disfavored than a dihedral angle of 58° (7a). However, even the conformation with a dihedral angle of 58° does not represent an energy minimum, which might explain the reduced κ affinity of 7a.
Introduction
The clinically used strong analgesics interact with the three classical opioid receptors, which are termed μ, κ and δ receptors. Whereas activation of all three opioid receptor subtypes leads to strong analgesia, the side effect profiles associated with the three subtypes differ considerably. In view of their side effect profile, κ agonists are of particular interest, because, in contrast to μ agonists, they cause minimal physical dependence, respiratory depression and inhibition of gastrointestinal motility. In addition to their analgesic effects in in vivo models, κ agonists have been shown to be potent neuroprotective and antihyperalgesic agents. However, sedation, dysphoria and strong diuresis are the most severe side effects associated with the application of κ agonists.1All three subtypes have already been cloned.2 However, X-ray crystal structures of the G-protein coupled membrane-bound receptors showing the exact three dimensional orientation of the ligand binding sites are not yet available. Therefore, for the development of novel selective κ agonists, lead compounds with high κ affinity were selected.
Most of the described κ agonists can be subdivided into four compound classes:1 peptides (e.g. the physiological agonist Dynorphine A1,3), benzomorphans (e.g. ethylketocyclazocine1,3), arylacetamides (e.g. the prototypical κ agonist U-50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4884) and the non-basic natural product salvinorin A.5,6 Monoacylated ethylenediamines derived from the prototypical κ agonist U-50488 are of particular interest for this project.
4884) and the non-basic natural product salvinorin A.5,6 Monoacylated ethylenediamines derived from the prototypical κ agonist U-50488 are of particular interest for this project.
During the last few years, potent κ agonists have been described, which are derived from the lead compound U-50488, including annulated compounds,7,8 simplified ligands,9,10 as well as heterocyclic analogues like 2-(aminomethyl)piperidines11,12 and 2-(aminomethyl)piperazines.13–15 The (2R)-configured 2-(aminomethyl)piperazine 1 (GR-89696) belongs to the most active κ agonists with an IC50-value of 0.018 nM13–15 (Ki = 0.36 nM,16Fig. 1). Very recently, we have shown that the κ receptor affinity of 1 was exceeded by the (S,S)-configured pyrrolidinylethyl derivative 2 (Ki = 0.31 nM), bearing an additional CH3-moiety at the C-2 side chain.16,17 In both compounds, the κ affinity is strongly dependent on the stereochemistry; in Fig. 1 the most active stereoisomers are shown.
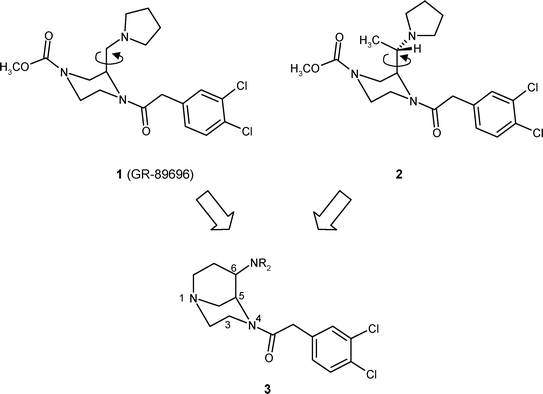 | ||
| Fig. 1 Design of conformationally constrained κ agonists 3, with a defined dihedral angle between the two pharmacophoric elements: pyrroline and phenylacetamide. | ||
The 1,4-diacylated piperazine system of 1 and 2 represents a rather rigid substructure. However, the axially oriented substituent in position 217 can rotate without hindrance around the indicated axial bond. In order to investigate the preferred orientation (bioactive conformation) of the pharmacophoric pyrrolidine moiety of 1 and 2, conformationally constrained κ receptor agonists were designed. Herein, we report on the stereoselective synthesis and pharmacological evaluation of bicyclic κ receptor agonists of type 3. The 1,4-diazabicyclo[3.3.1]nonane system of 3 results upon connection of the methyl residue of 2 with the second piperazine N-atom. The orientation of the basic amino group in position 6 can be adjusted by the stereochemistry.
Results and discussion
Chemistry
The synthesis of the first series of κ agonists started with the bicyclic ketone 4, which has been prepared in four reaction steps from ethyl piperazine-2-carboxylate.18 Reductive amination of ketone 4 with pyrrolidine and NaBH(OAc)319 provided diastereoselectively the endo-configured pyrrolidine 5a in 59% yield. The corresponding dimethylamine 5c was prepared in the same manner (38% yield). Unfortunately, the diethylamine 5b, as a ring-opened pyrrolidine analogue, was not available by direct reductive amination of ketone 4 with the secondary amine diethylamine. Therefore, the diethylamine 5b was prepared stepwise. First, reductive amination of ketone 4 with the primary amine ethylamine and NaBH(OAc)3 led to the ethylamine 5d, which was reductively alkylated with acetaldehyde and NaBH(OAc)3 to afford the tertiary amine 5b (Scheme 1).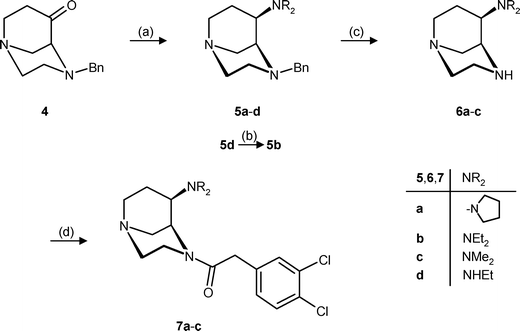 | ||
Scheme 1 The synthesized compounds are racemates; only one enantiomer is shown in this and the following schemes. Reagents and reaction conditions: (a) R2NH, NaBH(OAc)3, THF, rt, 5a: 59%; 5c: 38%; 5d: 47%. (b) CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NaBH(OAc)3, THF, 2 h, rt, 64%. (c) H2, Pd/C, CH3OH, rt. (d) 3,4-Dichlorophenylacetic acid, DCC, CH2Cl2, rt, 7a: 47%; 7b: 70%; 7c: 53%. O, NaBH(OAc)3, THF, 2 h, rt, 64%. (c) H2, Pd/C, CH3OH, rt. (d) 3,4-Dichlorophenylacetic acid, DCC, CH2Cl2, rt, 7a: 47%; 7b: 70%; 7c: 53%. | ||
Hydrogenolytic removal of the N-benzyl protecting group of 5a–c provided the secondary amines 6a–c, which were directly acylated, without purification, using 3,4-dichlorophenylacetic acid in the presence of dicyclohexylcarbodiimide (DCC), to form the desired dichlorophenylacetamides 7a–c.
In order to prove the endo-configuration of the final products 7 unequivocally, an X-ray crystal structure analysis was performed. For this purpose, pyrrolidine 7a was recrystallized from a mixture of diethyl ether and acetonitrile to obtain colorless crystals which were suitable for X-ray crystal structure analysis (Fig. 2). The X-ray crystal structure clearly shows the endo-configuration of the pyrrolidine substituent at position 6. Both six-membered heterocycles adopt the chair conformation and, moreover, the dihedral angle between the pyrrolidine N-atom and the acetamide N-atom could be determined to be 58.3° (see the Discussion of the dihedral angle section).
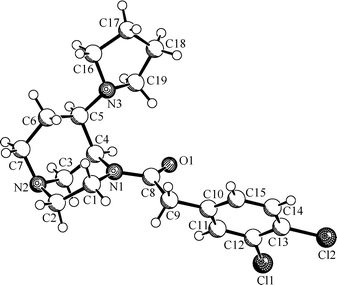 | ||
| Fig. 2 X-Ray crystal structure of 7a, proving the endo-configuration of the pyrrolidine substituent. | ||
In addition to the relative configuration of the final products, the X-ray crystal structure also proves the reaction path of the reductive amination. The reducing agent NaBH(OAc)3 can only attack the intermediate iminium ion from the exo-face, whereas the endo-face is shielded by the larger bridge.
Although it is known from the literature that the dichlorophenylacetyl residue is optimal for binding at κ receptors,1 it was replaced by some similar residues (Scheme 2). Thus, the N-benzyl moiety of pyrrolidine 5a was removed hydrogenolytically to form the secondary amine 6a, which was directly converted into the phenylacetyl (8a), benzoyl (9a) and tosyl (10a) derivatives. The diethylamine 10b with an N-tosyl group was also prepared by treatment of the benzyl derivative 5b with H2, Pd/C and subsequent tosylation.
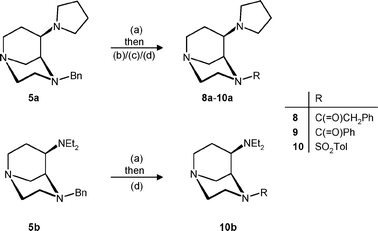 | ||
| Scheme 2 Reagents and reaction conditions: (a) H2, Pd/C, CH3OH, rt. (b) Phenylacetic acid, DCC, CH2Cl2, rt, 78%. (c) Benzoyl chloride, NEt3, CH2Cl2, rt, 48%. (d) Tosyl chloride, NEt3, CH2Cl2, rt, 10a: 35%; 10b: 80%. | ||
For the synthesis of the diastereomeric amines with exo-orientation of the amino group, ketone 4 should be reduced diastereomerically and the resulting endo-alcohol should be transformed into exo-amines by an inversion reaction. Indeed, reduction of ketone 4 with LiBH4 in THF provided exclusively the endo-alcohol 11 in 91% yield (Scheme 3). However, all attempts to activate the OH-moiety with mesyl chloride or tosyl chloride, even at low temperature, led to very fast decomposition of the alcohol 11. Also, the direct reaction of 11 in a Mitsunobu inversion20 (PPh3, diisopropyl azodicarboxylate (DIAD)) using different N-nucleophiles, like succinimide, phthalimide and Zn(N3)2·(pyridine)2 complex,21 failed due to fast decomposition. Moreover, it was shown that alcohol 11 itself was also very unstable. Storage in the refrigerator at 0 °C under N2 for three days led to a dark coloration of the compound and additional peaks in the HPLC.
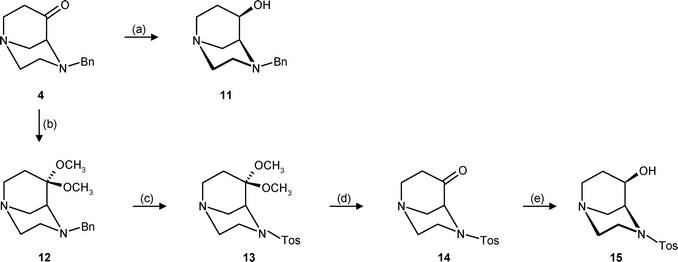 | ||
| Scheme 3 Reagents and reaction conditions: (a) LiBH4, THF, −20 °C, 91%. (b) CH3OH, HC(OCH3)3, p-TosOH, reflux, 79%. (c) H2, Pd/C, CH3OH, rt, then tosyl chloride, NEt3, CH2Cl2, rt, 80%. (d) 2 M HCl, H2O, reflux, 93%. (e) NaBH4, CH3OH, 0 °C, 91%. | ||
In order to stabilize alcohol 11, the N-benzyl group should be replaced by an N-tosyl moiety. Unfortunately, the direct synthesis of the sulfonamide 14 failed since the Dieckmann condensation of the tosyl-protected piperazine derivative did not give the corresponding bicyclic product.18 Therefore, an exchange of the N-protecting group was performed. First, ketone 4 was protected as dimethyl acetal 12. Hydrogenolytic removal of the N-benzyl group and subsequent reaction with tosyl chloride provided the sulfonamide 13, which was hydrolyzed with dilute HCl to form the N-tosyl-protected ketone 14. Reduction of 14 with NaBH4 in methanol provided diastereomerically the endo-configured alcohol 15 in 91% yield. In contrast to 11, alcohol 15 with the N-tosyl protecting group was considerably more stable, and could be stored at 0 °C for several months without decomposition.
Furthermore, alcohol 15 could be reacted with mesyl chloride and tosyl chloride to obtain the rather stable mesylate 16 and tosylate 17 in 83 and 70% yields, respectively (Scheme 4).
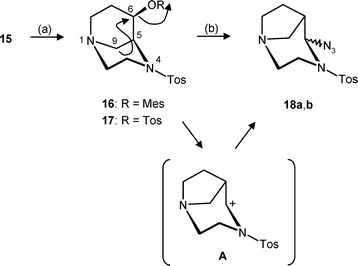 | ||
| Scheme 4 Reagents and reaction conditions: (a) Mesyl chloride or tosyl chloride, NEt3, CH2Cl2, rt, 16: 83%; 17: 70%. (b) NaN3, DMF, 80 °C, 18a: 22%; 18b: 32%. | ||
Treatment of the endo-mesylate 16 with NaN3 in DMF at 80 °C led to two new products, whose mass spectra were in agreement with the calculated molecular weights. Careful analysis of the 1H and 13C NMR spectra, however, revealed that the diastereomeric azides 18 had been formed instead of the desired exo-azide 21. Variation of the reaction conditions during the nucleophilic substitution of mesylate 16, starting with tosylate 17, and even the direct substitution of alcohol 15via a Mitsunobu inversion (PPh3, DIAD, Zn(N3)2·(pyridine)2 complex), always led to the rearranged azides 18.
This transformation is explained by a rearrangement of mesylate 16, which is outlined in Scheme 4. The rearrangement is driven by the exact antiperiplanar orientation of the C-9–C-5 bond and the C–O bond of the leaving group. The nitrogen atom at position 1, with its free electron pair, supports this rearrangement, and the nitrogen atom of the sulfonamide at position 4 stabilizes the intermediate carbenium ion A, which is finally trapped by the azide nucleophile. The intermediate carbenium ion A also explains nicely the formation of two diastereomeric azides 18a and 18b, since the planar carbenium ion A can be attacked from both sides.
After elucidation of this process, we postulated that a similar rearrangement is the reason for the instability of alcohol 11. Compared to the carbenium ion A, the analogous carbenium ion formed from the N-benzyl-protected alcohol 11 is much better stabilized by the N-benzyl moiety than the N-sulfonyl derivative A. Therefore, its formation, together with subsequent uncontrolled transformations, is generally favored.
The observation that the reduction of the electron donating properties of N-4 (compare the stability of alcohols 11 and 15) leads to an at least slightly reduced rearrangement tendency stimulated the idea to reduce the electron donating activity of N-1 as well, in order to suppress the unwanted rearrangement reaction completely. For this purpose, the N-oxide 19 was prepared by oxidation of tosylate 17 with m-chloroperbenzoic acid (MCPBA, Scheme 5).22 Indeed, the SN2 substitution of tosylate 19, bearing electron withdrawing groups at both N-atoms, with NaN3 in DMF at 150 °C successfully gave the exo-configured azides 20 and 21. Whereas azide 21 was isolated in 46% yield as the main product, the N-oxide 20 could not be separated completely from the starting tosylate 19. Performing this substitution reaction with microwave irradiation resulted in a considerable reduction of the reaction time from 20 h to 15 min. Obviously, during heating of the reaction mixture in DMF to 150 °C with an excess of NaN3, reduction of the N-oxide 20 occurred, directly forming azide 21 with a tertiary amino group. However, this side reaction is not detrimental, as in the next reaction step the azide moiety was reduced with H2 in the presence of Pd/C. Under these reaction conditions, both functional groups, the N-oxide and the azide, were reduced and, therefore, a mixture of 20 and 21 containing small amounts of the starting N-oxide 19 was usually employed for this reaction step (Scheme 5).
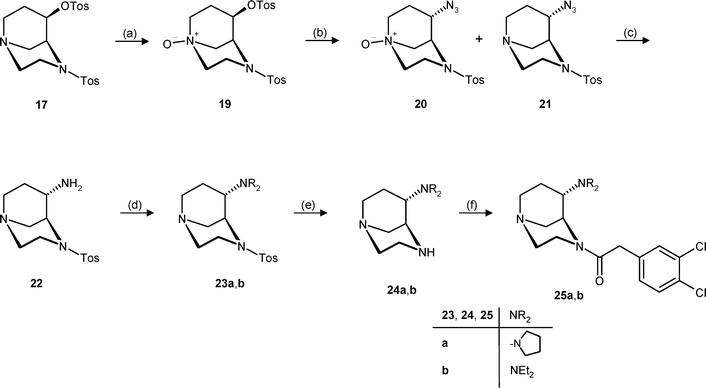 | ||
| Scheme 5 Reagents and reaction conditions: (a) MCPBA, K2CO3, CH2Cl2, 73%. (b) NaN3, DMF, 80 °C, 21: 46%. (c) H2, Pd/C, CH3OH, rt. (d) Succinaldehyde or CH3CHO, NaBH(OAc)3, THF, rt, 23a: 36%; 23b: 29%. (e) Mg, CH3OH, reflux. (f) 3,4-Dichlorophenylacetyl chloride, NEt3, CH2Cl2, rt, 25a: 25%; 25b: 45%. | ||
Hydrogenation of the mixture of azides 20 and 21 yielded the primary amine 22, which was reductively alkylated without further purification. Reductive amination is the preferred method in this reaction step, since alkyl halides react very fast with the bridgehead tertiary amine to form quaternary ammonium ions. For the synthesis of the diethylamine 23b, an excess of acetaldehyde in the presence of NaBH(OAc)3 was used. However, succinaldehyde, which was required for the synthesis of the pyrrolidine moiety, was not commercially available. Therefore, succinaldehyde was generated in situ upon hydrolysis of 2,5-dimethoxytetrahydrofuran with dilute HCl.23 This procedure provided the exo-configured pyrrolidine 23a in 36% yield, starting from the N-oxide 19.
In order to finalize the synthesis of the dichlorophenylacetamides 25, the tosyl protecting group was cleaved with Mg in refluxing methanol. The resulting secondary amines 24a and 24b were subsequently acylated with dichlorophenylacetyl chloride and NEt3, to give the exo-configured bicyclic amines 25a and 25b in 31% and 45% yield, respectively.
To broaden the structure–κ receptor affinity relationships, the tertiary amine (e.g. pyrrolidine) at position 6 was replaced by N-containing substituents with modified basicity. For this purpose N-heterocycles were annulated to the bicyclic framework. In particular, the weakly basic quinoline 28 and the non-basic indole 31 were envisaged, synthesized and pharmacologically evaluated. The key step in the synthesis of the weakly basic quinoline 28 was a Friedländer quinoline synthesis24 of the bicyclic ketone 4 with 2-aminoacetophenone in boiling glacial acetic acid to give the quinoline 26 in 73% yield (Scheme 6. ) Hydrogenolytic removal of the N-benzyl group led to the secondary amine 27, and attachment of the pharmacophoric dichlorophenylacetyl residue was performed by coupling with dichlorophenylacetic acid and DCC.
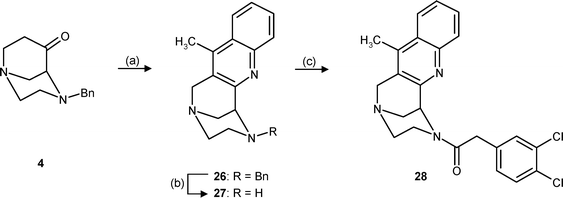 | ||
| Scheme 6 Reagents and reaction conditions: (a) o-Aminoacetophenone, HOAc, reflux, 73%. (b) NH4HCO2, Pd/C, CH3OH, reflux, 89%. (c) 3,4-Dichlorophenylacetic acid, DCC, CH2Cl2, rt, 74%. | ||
A Fischer indole synthesis25 of the bicyclic ketone 4 with p-methoxyphenylhydrazine in the presence of HCl provided the indole 29. After hydrogenolytic cleavage of the N-benzyl group, the resulting secondary amine 30 was acylated with dichlorophenylacetic acid and DCC to give indole 31 in 54% yield. (Scheme 7) In contrast to the other phenylacetamides, the indole N-atom of 31 does not possess basic properties.
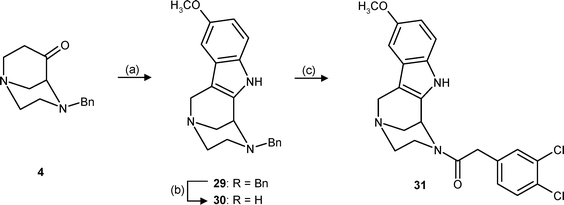 | ||
| Scheme 7 Reagents and reaction conditions: (a) p-Methoxyphenylhydrazine HCl, EtOH, HCl, reflux, 28%. (b) H2, Pd/C, CH3OH, rt. (c) 3,4-Dichlorophenylacetic acid, DCC, CH2Cl2, rt, 54%. | ||
Receptor binding studies
The κ receptor affinity of the bicyclic amines was determined in competition experiments with the radioligand [3H]-U-69593. In the assay, membrane preparations of guinea pig brains were used as receptor material. The non-specific binding was determined in the presence of a large excess of non-tritiated U-69593 (10 μM).16In Table 1, the κ receptor affinities of the synthesized amines, together with the κ affinities of some reference compounds, are summarized. In our assay, the pyrrolidine derivative 7a has a Ki-value of 73 nM. Whereas replacement of the pyrrolidine ring by a dimethylamino group (7c) did not influence the κ receptor affinity, the corresponding diethylamine shows a considerably reduced κ affinity. This result is not surprising, since the two ethyl residues on the N-atom can never adopt the same conformation as the pyrrolidine ring due to repulsion of the terminal methyl groups.
| Compound | κ Affinity [3H]-U-69593 Ki± SEM/nMa |
|---|---|
| a The Ki-values were determined in three independent experiments (n = 3) unless otherwise noted.b Percent inhibition at a concentration of 1 μM of the test compound. | |
| 7a (WMS-1302) | 73 ± 9.1 |
| 7b | 361 ± 65 |
| 7c | 65 ± 4.0 |
| 8a | 0%b |
| 9a | 0%b |
| 10a | 0%b |
| 10b | 0%b |
| 23a | 0%b |
| 23b | 0%b |
| 25a | 0%b |
| 25b | 0%b |
| 28 | 16%b |
| 31 | 1%b |
| 216 | 0.31 ± 0.1 |
| U-50488 | 0.31 ± 0.1 |
| U-69593 | 0.97 ± 0.4 |
| Naloxone | 7.3 ± 0.4 |
The data in Table 1 clearly show that the 3,4-dichlorophenylacetyl residue and the endo-configuration are essential for a strong interaction with the binding site of the κ receptor. Exchange of this residue for a phenylacetyl (8a), benzoyl (9a) or tosyl residue (10a, 10b) led to almost complete loss of the κ receptor affinity. The change of the configuration from endo-configured amines 7 to exo-configured amines 25 also led to inactive compounds. The quinoline and indole annulated bicyclic compounds 28 and 31 did not interact significantly with the κ receptor. It is assumed that the reduced (quinoline 28) or eliminated (indole 31) basicity together with the planar geometry around the N-atom are responsible for the reduced κ receptor affinity.
In addition to the κ receptor affinity, the affinity of the bicyclic amines towards the other two classical opioid receptors, μ and δ, as well as to the historically related σ1, σ2 and NMDA receptors were investigated in receptor binding studies with radioligands. At a concentration of 1 μM the test compounds did not compete significantly with the radioligands. Therefore the IC50-values are at least higher than 1 μM. This result indicates that the endo-configured pyrrolidine, 7a and its dimethylamine analogue 7c, are selective κ agonists with minimal affinity to the investigated receptor systems.
Discussion of the dihedral angle
Compared with the lead compound 2, the most potent κ agonist of this series, the endo-configured pyrrolidine 7a (WMS-1302), is about 200-fold less active (see Table 1). The reduction of the κ receptor affinity may be due to (1) the additional basic bridgehead N-atom in position 1 of the bicyclic system and/or (2) the fixation of the pyrrolidine ring in an unfavorable orientation.In addition to the basic pyrrolidine system, the known κ agonists with an ethylenediamine substructure generally do not have a further basic N-atom. Usually, additional N-atoms present in potential κ agonists are supplied with propionyl or methoxycarbonyl moieties.13–16 However, very recently we have demonstrated that an additional basic N-atom is indeed tolerated by the κ receptor protein. In the quinoxaline compound class of κ agonists, secondary amines as well as methyl and benzyl amines even exceed the corresponding propionyl and methoxycarbonyl derivatives with respect to κ receptor affinity.26 Therefore, we assume that the additional basic N-atom of 7a is at least not exclusively responsible for the reduced κ affinity.
In Fig. 3, the dihedral angles (N(pyrrolidine)–C–C–N(acetamide)) of the diastereomeric pyrrolidines 7a and 25a are compared. The dihedral angle of the energetically most favored conformer (MOE) of the almost inactive exo-configured pyrrolidine 25a is 168° (Fig. 3). Originally, we were very interested in this stereoisomer, since 25a is directly obtained by connecting the free methyl group with the carbamate N-atom of the very potent flexible κ agonist 2 (see Fig. 1).
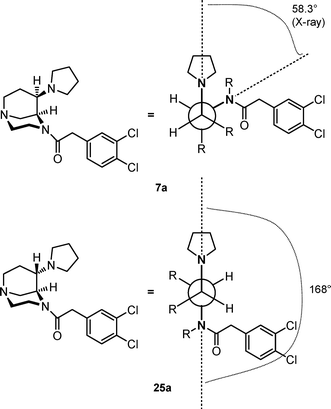 | ||
| Fig. 3 Dihedral angles of the diastereomeric pyrrolidines 7a and 25a. | ||
A systematic conformational analysis of N-protonated 2 was performed by rotation of the pyrrolidinylethyl residue in 10° intervals around the axially oriented C–C-bond (Fig. 4). Subsequently, the resulting conformations were energy minimized without changing the crucial dihedral angle N–C–C–N. The energy profile in Fig. 5 shows that a dihedral angle of 168° is energetically disfavored. Therefore, the potent κ agonist 2 probably does not adopt a conformation similar to 25a at the κ receptor.
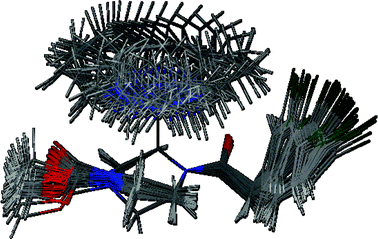 | ||
| Fig. 4 Superposition of the energy minimized conformations resulting upon 10° rotation of the axially oriented pyrrolidinylethyl moiety of 2. | ||
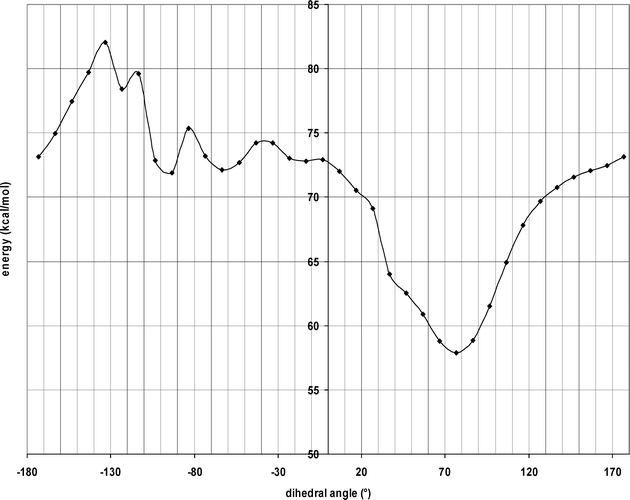 | ||
| Fig. 5 Energy profile resulting upon rotation of compound 2 with a proton at the pyrrolidine N-atom around the axially oriented C–C bond at position 3 (compare to Fig. 1). | ||
According to the X-ray crystal structure of the endo-configured pyrrolidine 7a (WMS-1302), both six-membered heterocycles adopt the chair conformation, which results in a dihedral angle of 58.3° (Fig. 3). Since the X-ray crystal structure is an energetically favored conformation, at least in the solid state, it is possible that 7a reacts in this conformation with the κ receptor protein. In comparison with the exo-diastereomer 25a, the κ affinity of 7a is increased, but reduced compared with the κ affinity of 2. So we assume that the dihedral angle of 58° is closer to the dihedral angle of the bioactive conformation of 2, but still is not optimal. This assumption is supported by the energy profile of 2 depicted in Fig. 5, which reveals that the dihedral angle of 58° is close to the energy minimum of 70°, but still differs slightly from it.
Conclusion
Starting from the very potent but flexible κ agonist 2, the conformationally constrained stereoisomeric bicyclic amines 7 and 25 have been synthesized. Compared with the lead compound 2 (Ki = 0.31 nM), the κ affinity of the endo-configured pyrrolidine 7a (WMS-1302) and the exo-configured pyrrolidine 25a is considerably reduced (Ki = 73 nM, Ki > 1 μM). It is assumed that the relative orientation of the pharmacophoric elements, the pyrrolidine ring and the dichlorophenylacetamide, determines the interaction of the bicyclic ligands with the κ receptor. Whereas a dihedral angle of 168° (25a) is not tolerated by the κ receptor, the dihedral angle of 58° of 7a is closer to the bioactive conformation of 2. The systematic conformational analysis of 2 revealed that the dihedral angle of 58° is energetically favored compared to 168°, but still differs from an energy minimum. This fact might explain the reduced κ receptor affinity of the bicyclic pyrrolidine 7a.Experimental
General chemistry
Unless otherwise noted, moisture sensitive reactions were conducted under dry nitrogen. THF was dried with sodium/benzophenone and was freshly distilled before use. Thin layer chromatography (tlc): silica gel 60 F254 plates (Merck). Flash chromatography (fc): silica gel 60, 40–64 μm (Merck); parentheses include: diameter of the column, height of SiO2 column, fraction size, eluent, Rf value. Melting point: melting point apparatus SMP 3 (Stuart Scientific), uncorrected. MS: MAT GCQ (Thermo-Finnigan); EI = electron impact, ESI = electrospray ionization. IR: IR spectrophotometer 480Plus FT-ATR-IR (Jasco). 1H NMR (400 MHz), 13C NMR (100 MHz): Mercury-400BB spectrometer (Varian); δ in ppm relative to tetramethylsilane; coupling constants are given with 0.5 Hz resolution. HPLC method 1: Merck Hitachi Equipment; UV detector: L-7400; autosampler: L-7200; pump: L-7100; degasser: L-7614; column: LiChrospher® 60 RP-select B (5 μm), 250-4 mm; flow rate: 1.00 mL/min; injection volume: 5.0 μL; detection at λ = 210 nm; solvents: A: water with 0.05% (v/v) trifluoroacetic acid; B: acetonitrile with 0.05% (v/v) trifluoroacetic acid: gradient elution: (A%): 0 min: 90%, 4 min: 90%, 29 min: 0%, 31 min: 0%, 31.5 min: 90%, 40 min: 90%. HPLC method 2: Equipment: pump: HPLC pump 64 (Knauer); UV-Detector: Variable Wavelength Monitor (Knauer); data acquisition: D-2500 Chromato-Integrator (Merck Hitachi); injection volume: 20.0 μL; stop time: 2 × tR; A: column: LiChroCART® 250-4 with Superspher® 100 RP-18; solvent: methanol/water = 75:25 + 0.1% triethylamine; flow rate: 0.6 mL/min; detection: wavelength: 235 nm; B: column: LiChroCART® 250-4 with Superspher® 100 RP-18; solvent: acetonitrile/water = 70:30 + 0.1% triethylamine; flow rate: 1.0 mL/min; detection: wavelength: 254 nm; C: column: phenomenex Gemini 5 μm C18 100A, 250–21.2 mm; solvent: methanol/water = 50:50 + 0.1% triethylamine; flow rate: 0.8 mL/min; detection: wavelength: 235 nm.![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) [cm−1] = 697 and 668 (m, arom. out of plane). MS (EI): m/z [%] = 285 (M, 100), 194 (M–PhCH2, 13), 91 (PhCH2, 31). [cm−1] = 735 and 700 (m, arom. out of plane). MS (EI): m/z [%]: 287 (M, 55), 91 (PhCH2, 86). [cm−1] = 701 and 648 (m, arom. out of plane). MS (EI): m/z [%] = 259 (M, 100), 91 (PhCH2, 50). [cm−1] = 3300 (br w, N-H), 737 and 695 (m, arom. out of plane). MS (EI): m/z [%] = 259 (M, 55), 168 (M–PhCH2, 5), 91 (PhCH2, 82). [cm−1] = 1632 (s, CO), 881 and 821 (w, arom. out of plane). MS (EI): m/z [%] = 381 (M, 2×35Cl, 100), 383 (M, 35Cl/37Cl, 63), 385 (M, 2×37Cl, 9). HPLC (Method 1): tR = 14.7 min, purity 98.9%. X-ray crystal structure analysis:27–30 see the ESI.† [cm−1] = 1634 (s, CO), 881 and 822 (w, arom. out of plane). MS (EI): m/z [%] = 383 (M, 2×35Cl, 97), 385 (M, 35Cl/37Cl, 60), 387 (M, 2×37Cl, 9). HPLC (Method 1): tR = 13.9 min, purity 96.5%. [cm−1] = 1631 (s, CO), 874 and 811 (w, arom. out of plane). MS (ESI): m/z [%] = 356 (MH, 2×35Cl, 100), 358 (MH, 35Cl/37Cl, 60), 360 (MH, 2×37Cl, 9). HPLC (Method 1): tR = 13.9 min, purity 97.2%. [cm−1] = 3317 (m br, O-H), 739 and 696(m, arom. out of plane). MS (EI): m/z [%] = 232 (M, 100), 141 (M–PhCH2, 97), 91 (PhCH2, 45). [cm−1] = 1715 (s, CO), 1160 (s, SO), 809 (m, arom. out of plane). MS (EI): m/z [%] = 294 (M, 9), 139 (M–SO2C6H4CH3, 100). HPLC (Method 1): tR = 11.5 min, purity 99.9%. [cm−1] = 3200 (br w, O-H), 1153 (s, SO), 822 (m, arom. out of plane). MS (EI): m/z [%] = 296 (M, 19), 141 (M–SO2C6H4CH3, 100). HPLC (Method 1): tR = 11.6 min, purity 99.1%. [cm−1] = 1343 (s, N-O), 1157 (s, SO), 812 (m, arom. out of plane). MS (ESI): m/z [%] = 467 (M + H, 23), 489 (M + Na, 24), 933 (2M + H, 100). HPLC (Method 1): tR = 18.8 min, purity 98.2%. [cm−1] = 2094 (s, NNN), 1159 (s, SO), 815 (m, arom. out of plane). MS (ESI): m/z [%] = 322 (MH, 27), 665 (M + Na, 100). HPLC (Method 1): tR = 15.7 min, purity 95.5%. [cm−1] = 1157 (s, SO), 815 (m, arom. out of plane). MS (ESI): m/z [%] = 350 (MH, 100). HPLC (Method 1): tR = 11.4 min, purity 96.6%. [cm−1] = 1157 (s, SO), 814 (m, arom. out of plane). MS (ESI): m/z [%] = 352 (MH, 100). HPLC (Method 1): tR = 11.4 min, purity 96.6%. [cm−1] = 1634 (s, CO), 877 and 824 (w, arom. out of plane). MS (EI): m/z [%] = 381 (M, 2×35Cl, 100), 383 (M, 35Cl/37Cl, 65), 385 (M, 2×37Cl, 11). HPLC (Method 1): tR = 13.6 min, purity 97.5%. [cm−1] = 1636 (s, CO), 870 and 824 (w, arom. out of plane). MS (EI): m/z [%] = 383 (M, 2×35Cl, 100), 385 (M, 35Cl/37Cl, 53), 387 (M, 2×37Cl, 8). HPLC (Method 1): tR = 14.1 min, purity 95.6%. [cm−1] = 1634 (s, CO), 1499 (m, CC), 756 and 737 (m, arom. out of plane). MS (ESI): m/z [%] = 426 (MH, 2×35Cl, 86), 428 (MH, 35Cl/37Cl, 55), 873 (2M + Na, 4×35Cl, 81), 875 (2M + Na, 3×35Cl/1×37Cl, 100), 877 (2M + Na, 2×35Cl/2×37Cl, 48). HPLC (Method 1): tR = 18.9 min, purity 97.6%. [cm−1] = 3205 (m broad, N-H), 1632 (s, CO), 786, 777 and 740 (m, arom. out of plane). MS (EI): m/z [%] = 429 (M, 2×35Cl, 30), 431 (M, 35Cl/37Cl, 16), 433 (M, 2×37Cl, 3). HPLC (Method 1): tR = 19.4 min, purity 98.5%.
[cm−1] = 697 and 668 (m, arom. out of plane). MS (EI): m/z [%] = 285 (M, 100), 194 (M–PhCH2, 13), 91 (PhCH2, 31). [cm−1] = 735 and 700 (m, arom. out of plane). MS (EI): m/z [%]: 287 (M, 55), 91 (PhCH2, 86). [cm−1] = 701 and 648 (m, arom. out of plane). MS (EI): m/z [%] = 259 (M, 100), 91 (PhCH2, 50). [cm−1] = 3300 (br w, N-H), 737 and 695 (m, arom. out of plane). MS (EI): m/z [%] = 259 (M, 55), 168 (M–PhCH2, 5), 91 (PhCH2, 82). [cm−1] = 1632 (s, CO), 881 and 821 (w, arom. out of plane). MS (EI): m/z [%] = 381 (M, 2×35Cl, 100), 383 (M, 35Cl/37Cl, 63), 385 (M, 2×37Cl, 9). HPLC (Method 1): tR = 14.7 min, purity 98.9%. X-ray crystal structure analysis:27–30 see the ESI.† [cm−1] = 1634 (s, CO), 881 and 822 (w, arom. out of plane). MS (EI): m/z [%] = 383 (M, 2×35Cl, 97), 385 (M, 35Cl/37Cl, 60), 387 (M, 2×37Cl, 9). HPLC (Method 1): tR = 13.9 min, purity 96.5%. [cm−1] = 1631 (s, CO), 874 and 811 (w, arom. out of plane). MS (ESI): m/z [%] = 356 (MH, 2×35Cl, 100), 358 (MH, 35Cl/37Cl, 60), 360 (MH, 2×37Cl, 9). HPLC (Method 1): tR = 13.9 min, purity 97.2%. [cm−1] = 3317 (m br, O-H), 739 and 696(m, arom. out of plane). MS (EI): m/z [%] = 232 (M, 100), 141 (M–PhCH2, 97), 91 (PhCH2, 45). [cm−1] = 1715 (s, CO), 1160 (s, SO), 809 (m, arom. out of plane). MS (EI): m/z [%] = 294 (M, 9), 139 (M–SO2C6H4CH3, 100). HPLC (Method 1): tR = 11.5 min, purity 99.9%. [cm−1] = 3200 (br w, O-H), 1153 (s, SO), 822 (m, arom. out of plane). MS (EI): m/z [%] = 296 (M, 19), 141 (M–SO2C6H4CH3, 100). HPLC (Method 1): tR = 11.6 min, purity 99.1%. [cm−1] = 1343 (s, N-O), 1157 (s, SO), 812 (m, arom. out of plane). MS (ESI): m/z [%] = 467 (M + H, 23), 489 (M + Na, 24), 933 (2M + H, 100). HPLC (Method 1): tR = 18.8 min, purity 98.2%. [cm−1] = 2094 (s, NNN), 1159 (s, SO), 815 (m, arom. out of plane). MS (ESI): m/z [%] = 322 (MH, 27), 665 (M + Na, 100). HPLC (Method 1): tR = 15.7 min, purity 95.5%. [cm−1] = 1157 (s, SO), 815 (m, arom. out of plane). MS (ESI): m/z [%] = 350 (MH, 100). HPLC (Method 1): tR = 11.4 min, purity 96.6%. [cm−1] = 1157 (s, SO), 814 (m, arom. out of plane). MS (ESI): m/z [%] = 352 (MH, 100). HPLC (Method 1): tR = 11.4 min, purity 96.6%. [cm−1] = 1634 (s, CO), 877 and 824 (w, arom. out of plane). MS (EI): m/z [%] = 381 (M, 2×35Cl, 100), 383 (M, 35Cl/37Cl, 65), 385 (M, 2×37Cl, 11). HPLC (Method 1): tR = 13.6 min, purity 97.5%. [cm−1] = 1636 (s, CO), 870 and 824 (w, arom. out of plane). MS (EI): m/z [%] = 383 (M, 2×35Cl, 100), 385 (M, 35Cl/37Cl, 53), 387 (M, 2×37Cl, 8). HPLC (Method 1): tR = 14.1 min, purity 95.6%. [cm−1] = 1634 (s, CO), 1499 (m, CC), 756 and 737 (m, arom. out of plane). MS (ESI): m/z [%] = 426 (MH, 2×35Cl, 86), 428 (MH, 35Cl/37Cl, 55), 873 (2M + Na, 4×35Cl, 81), 875 (2M + Na, 3×35Cl/1×37Cl, 100), 877 (2M + Na, 2×35Cl/2×37Cl, 48). HPLC (Method 1): tR = 18.9 min, purity 97.6%. [cm−1] = 3205 (m broad, N-H), 1632 (s, CO), 786, 777 and 740 (m, arom. out of plane). MS (EI): m/z [%] = 429 (M, 2×35Cl, 30), 431 (M, 35Cl/37Cl, 16), 433 (M, 2×37Cl, 3). HPLC (Method 1): tR = 19.4 min, purity 98.5%.Receptor binding studies
500 ×g for 20 min at 4 °C. The pellet was resuspended in 5–6 volumes of buffer (50 mM TRIS, pH 7.4), and centrifuged again at 23500 ×g (20 min, 4 °C). This procedure was repeated twice. The final pellet was resuspended in 5–6 volumes of buffer, the protein concentration was determined according to the method of Bradford31, using bovine serum albumin as the standard, and subsequently the preparation was frozen (−80 °C) in 1.5 mL portions containing about 1.5 mg protein mL−1.593 (55 Ci mmol−1, Amersham, Little Chalfont, UK). The thawed membrane preparation (about 75 μg of the protein) was incubated with various concentrations of test compounds, 1 nM [3H]-U-69‚593, and TRIS-MgCl2-buffer (50 mM, 8 mM MgCl2, pH 7.4) in a total volume of 200 μL for 150 min at 37 °C. The incubation was terminated by rapid filtration through the pre-soaked filtermats using a cell harvester. After washing each well five times with 300 μL of water, the filtermats were dried at 95 °C. Subsequently, the solid scintillator was placed on the filtermat and melted at 95 °C. After 5 min, the solid scintillator was allowed to solidify at room temperature. The bound radioactivity trapped on the filters was counted in the scintillation analyzer. The non-specific binding was determined with 10 μM unlabelled U-69593. The Kd-value of U-69593 is 0.69 nM.32500 ×g for 20 min at 4 °C. The pellet was resuspended in 5–6 volumes of buffer (50 mM TRIS, pH 7.4), and centrifuged again at 23500 ×g (20 min, 4 °C). This procedure was repeated twice. The final pellet was resuspended in 5–6 volumes of buffer, the protein concentration was determined according to the method of Bradford31 using bovine serum albumin as the standard, and subsequently the preparation was frozen (−80 °C) in 1.5 mL portions containing about 1.5 mg protein mL−1.Acknowledgements
This work was performed within the International Research Training Group “Complex Functional Systems in Chemistry: Design, Synthesis and Applications” in collaboration with the University of Nagoya. Financial support of this IRTG by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. Thanks are also due to Dr W. Englberger, Grünenthal GmbH, Aachen, for the determination of the δ receptor affinity of some bicyclic amines.References
- (a) D. I. C. Scopes, Drugs Future, 1993, 18, 933–947; (b) U. Holzgrabe, C. Nachtsheim, T. Siener, S. Drohsin and W. Brandt, Pharmazie, 1997, 52, 4–22.
- (a) K. Yasuda, K. Raymor, H. Kong, C. D. Breder, J. Takeda, T. Reisine and G. I. Bell, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6736–6740 CrossRef CAS; (b) Y. Chen, A. Mestek, J. Liu, J. A. Hurley and L. Yu, Mol. Pharmacol., 1993, 44, 8–12 CAS.
- D. C. Horwell, Drugs Future, 1988, 13, 1061–1071.
- B. R. de Costa, W. D. Bowen, S. B. Hellewell, C. George, R. B. Rothman, A. A. Reid, J. M. Walker, A. E. Jacobson and K. C. Rice, J. Med. Chem., 1989, 32, 1996–2002 CrossRef CAS.
- B. L. Roth, K. Baner, R. Westkaemper, D. Siebert, K. C. Rice, S. Steinberg, P. Ernsberger and R. B. Rothman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11934–11939 CrossRef CAS.
- D. Siebert, J. Pharmacol., 1994, 43, 53–56 Search PubMed.
- J. P. Freeman, E. T. Michalson, S. V. D'Andrea, L. Baczynskyj, P. F. VonVoigtländer, R. A. Lahti, M. W. Smith, C. F. Lawson, T. A. Scahill, S. A. Mizsak and J. Szmuszkovicz, J. Med. Chem., 1991, 34, 1891–1896 CrossRef CAS.
- V. Vecchietti, G. D. Clarke, R. Colle, G. Dondio, G. Giardina, G. Petrone and M. Sbacchi, J. Med. Chem., 1992, 35, 2970–2978 CrossRef CAS.
- G. J. Barlow, T. P. Blackburn, G. F. Costello, R. James, D. J. Le Count, B. G. Main, R. J. Pearce, K. Russell and J. S. Shaw, J. Med. Chem., 1991, 34, 3149–3158 CrossRef CAS.
- A.-C. Chang, A. E. Takemori, W. H. Ojala, W. B. Gleason and P. S. Portoghese, J. Med. Chem., 1994, 37, 4490–4498 CrossRef CAS.
- V. Vecchietti, A. Giordani, G. Giardina, R. Colle and G. D. Clarke, J. Med. Chem., 1991, 34, 397–403 CrossRef CAS.
- V. Vecchietti and A. Giordani, Eur. Pat. Appl., EP-275,696, 1988 Search PubMed.
- P. J. Birch, H. Rogers, A. G. Hayes, N. J. Hayward, M. B. Tyers, D. I. C. Scopes, A. Naylor and D. B. Judd, Br. J. Pharmacol., 1991, 103, 1829–1823.
- A. Naylor, D. B. Judd, J. E. Lloyd, D. I. C. Scopes, A. G. Hayes and P. Birch, J. Med. Chem., 1993, 36, 2075–2083 CrossRef CAS.
- A. Naylor, D. B. Judd and D. S. Brown, Eur. Pat. Appl., EP-343,900, 1989 Search PubMed.
- S. Soukara, C. A. Maier, U. Predoiu, A. Ehret, R. Jackisch and B. Wünsch, J. Med. Chem., 2001, 44, 2814–2828 CrossRef CAS.
- C. Röhr, S. Soukara and B. Wünsch, Eur. J. Med. Chem., 2001, 36, 211–214 CrossRef CAS.
- D. Kracht, S. Saito and B. Wünsch, Austr. J. Chem., 2009 DOI:10.1071/CH09206.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
- M. C. Viaud and P. Rollin, Synthesis, 1990, 130–132 CrossRef CAS.
- H. G. Aurich, M. Soeberdt and K. Harms, Eur. J. Org. Chem., 1999, 1249–1252 CrossRef.
- (a) G. F. Costello, R. James, J. S. Shaw, A. M. Staer and N. C. Stutchbury, J. Med. Chem., 1991, 34, 181–189 CrossRef CAS; (b) G. Höfner, B. Sttreicher and B. Wünsch, Arch. Pharm., 2001, 334, 284–290 CrossRef CAS.
- C. C. Cheng and S. J. Yan, Org. React., 1982, 28, 37–201 CAS.
- B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef.
- (a) C. Bourgeois, Synthese von kappa-Opioid-Rezeptor-Agonisten mit Chinoxalin-Struktur und Untersuchung der pharamkologischen Eigenschaften, dissertation, University of Münster, Münster, 2007. Search PubMed; (b) B. Wünsch, D. Schepmann, C. Bourgeois, DE 10 2007 062 550.4, appl. 20.12.2007; PCT/EP2008/068000.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- Z. Otwinowski, D. Borek, W. Majewski and W. Minor, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 228–234 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- S. Soukara, 2-[1-(Pyrrolidin-1-yl)ethyl]-substituierte Piperazine: Stereoselektive Synthesen und Rezeptorbindungsstudien zur Prüfung der κ-Rezeptoraffinität und–selektivität, dissertation, University of Freiburg, Freiburg, 2000 Search PubMed.
- K. Raynor, H. Kong, Y. Chen, K. Yasuda, L. Yu, G. I. Bell and T. Reisine, Mol. Pharmacol., 1994, 45, 330–334 CAS.
- (a) B. Jung, W. Englberger, R. Fröhlich, D. Schepmann, K. Lehmkuhl and B. Wünsch, Bioorg. Med. Chem., 2008, 16, 2870–2885 CrossRef CAS; (b) R. Holl, B. Jung, D. Schepmann, W. Englberger, R. Grünert, H.-U. Humpf, P. Bednarski and B. Wünsch, ChemMedChem DOI:10.1002/cmdc.200900358.
- (a) C. A. Maier and B. Wünsch, J. Med. Chem., 2002, 45, 438–448 CrossRef CAS; (b) C. Oberdorf, D. Schepmann, J. M. Vela, J. L. Diaz and B. Wünsch, J. Med. Chem., 2008, 51, 6531–6537 CrossRef CAS.
- U. Wirt, D. Schepmann and B. Wünsch, Eur. J. Org. Chem., 2007, 462–475 CrossRef CAS.
- Y.-C. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details. CCDC reference number 748563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b915180j |
| This journal is © The Royal Society of Chemistry 2010 |