Identification and biosynthesis of tropone derivatives and sulfur volatiles produced by bacteria of the marine Roseobacter clade†
Verena
Thiel
a,
Thorsten
Brinkhoff
b,
Jeroen S.
Dickschat
a,
Susanne
Wickel
a,
Jörg
Grunenberg
a,
Irene
Wagner-Döbler
c,
Meinhard
Simon
b and
Stefan
Schulz
*a
aInstitut für Organische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: stefan.schulz@tu-bs.de; Fax: +49-531-391-5272; Tel: +49-531-391-5271
bInstitut für Chemie und Biologie des Meeres, Universität Oldenburg, Carl-von-Ossietzky-Str. 9-11, 26111 Oldenburg, Germany
cHelmholtz-Zentrum für Infektionsforschung, Inhoffenstr. 7, 38124 Braunschweig, Germany
First published on 12th November 2009
Abstract
Bacteria of the Roseobacter clade are abundant marine bacteria and are important contributors to the global sulfur cycle. The volatiles produced by two of its members, Phaeobacter gallaeciensis and Oceanibulbus indolifex, were analyzed to investigate whether the released compounds are derived from sulfur metabolism, and which biosynthetic pathways are involved in their formation. Both bacteria emitted different sulfides and thioesters, including new natural compounds such as S-methyl phenylethanethioate (16) and butyl methanesulfonate (21). The S-methyl alkanoates were identified by comparison with standards that were synthesized from the respective methyl alkanoates by a new method using an easily prepared aluminium/sulfur reagent. Phaeobacter gallaeciensis is also able to produce tropone (37) in large amounts. Its biosynthesis was investigated by various feeding experiments, showing that 37 is formed via a deviation of the phenylacetate catabolism. The unstable tropone hydrate 42 was identified as an intermediate of the tropone biosynthesis that was also released together with tropolone (38).
Introduction
The so-called Roseobacter clade within the Alphaproteobacteria is one of the most important groups of marine bacteria, comprising up to 25% of the marine microbial communities, especially in coastal regions and polar oceans.1 Members of this group exhibit diverse metabolic traits, like sulfur oxidation, aerobic anoxygenic photosynthesis (anaerobic photosynthesis for energy supply without production of oxygen under aerobic conditions), oxidation of carbon monoxide, dimethylsulfoniopropionate cleavage and demethylation, as well as the production of secondary metabolites.2–5 Some strains of the Roseobacter clade have been reported to produce the tropone antibiotic tropodithietic acid (TDA, 4).6–9 This antibiotic seems to be of ecological relevance, as in turbot larval farms Phaeobacter gallaeciensis and Ruegeria sp., both producing 4, were able to suppress growth of other bacteria such as Vibrio anguillarum.10Several natural tropones and tropolones produced by plants and fungi, such as colchicine from Colchicum autumnale, stipitatic acid (1) isolated from Penicillium stipitatum, as well as puberulonic (2) and puberulic acid (3) from Penicillium puberulum, are known,11–13 but they have been rarely described as bacterial metabolites. The first, and for a long time only, example was thiotropocin (5), isolated from Pseudomonas sp. CB-104 and Caulobacter PK 654.14,15 Thiotropocin and tropodithietic acid exhibit great structural similarity. Recent computational studies showed that both structures represent a pair of tautomers of similar energy that interconvert easily via a 1,5-hydrogen shift (Fig. 1).16 Therefore, structures 4 and 5 must be regarded as two tautomeric forms of the same compound. The biosynthesis of 5 in Pseudomonas sp. CB-104 has been shown to start from phenylacetate formed via the shikimate pathway.17
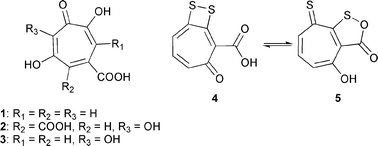 | ||
| Fig. 1 Stipitatic (1), puberulonic (2), puberulic (3), tropodithietic acids (4), and thiotropocin (5). | ||
Recently we became interested in the volatile compounds released by members of the Roseobacter clade,18,19 and investigated several strains for their ability to produce volatiles. The volatile bouquets of P. gallaeciensis and Oceanibulbus indolifex showed a high proportion of sulfur-containing metabolites, which is of interest because of the importance of the sulfur cycle in the ocean given the widespread occurrence of bacteria of the Roseobacter clade. In addition, P. gallaeciensis releases tropone (37) and the related compound tropolone (38). Tropone has previously only been reported by us as a minor volatile compound produced from the bacterium Loktanella sp. Bio-20418 and from a pacL mutant of Azoarcus evansii. In this mutant, the phenylacetate degradation pathway is impaired at the aromatic ring opening,20 leading to formation of 37 by an as yet unknown mechanism. The biosynthesis of the identified tropolonoids from L-phenylalanine and phenylacetate will be discussed based on results obtained from feeding experiments with labeled precursors.
Results
Identification of volatiles
Liquid cultures of P. gallaeciensis DSM17395 and O. indolifex HEL-45 grown in marine broth were connected to a closed-loop stripping apparatus (CLSA),21 which allowed sampling under continuous circulation of air. The volatiles released by the bacteria were trapped on activated charcoal, extracted with dichloromethane, and analyzed by gas chromatography-mass spectrometry (GC-MS). The compounds were identified by comparison of mass spectra and gas chromatographic retention times with those of authentic reference compounds either commercially available or synthesized in the course of other projects.The total ion chromatograms of the headspace extracts of both species are shown in Fig. 2 and the results of the analyses are summarized in Table 1.
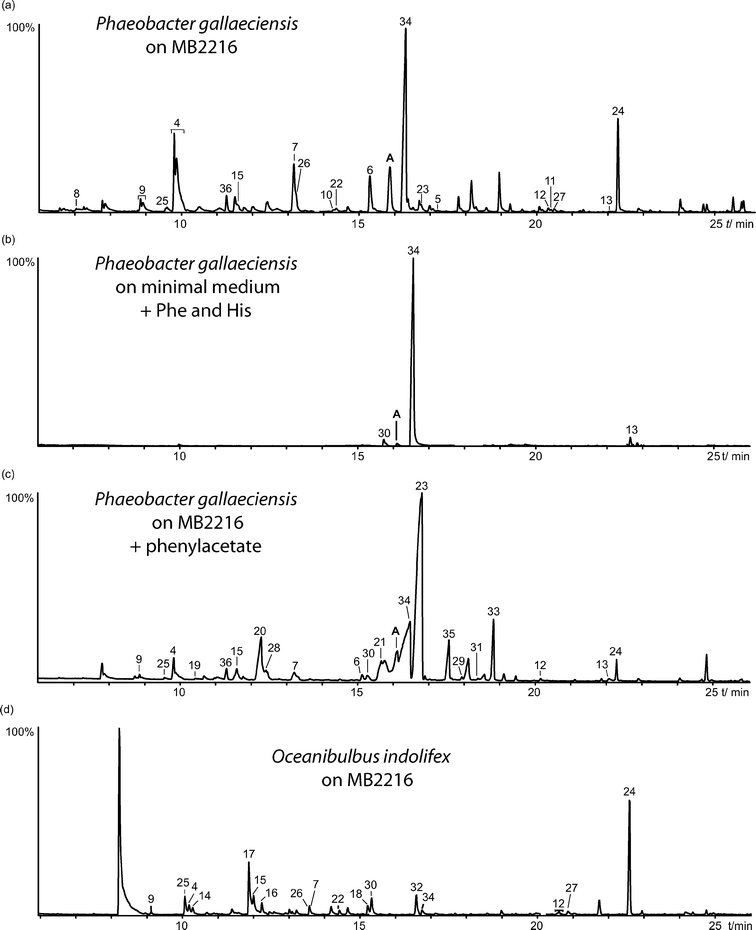 | ||
| Fig. 2 Total ion chromatogram of headspace extracts of P. gallaeciensis DSM17395 grown on MB2216 (a), P. gallaeciensis grown on a minimal medium containing L-phenylalanine and L-histidine (b), P. gallaeciensis grown on MB2216 containing phenylacetate (c), and of O. indolifex HEL-45 grown on MB2216 (d). Compound A proved to be identical to 42b. | ||
| Compound | I | Ident. | DSM17395 | DSM17395 (+ Phe, mi) | DSM17395 (+ PhAc, fu) | HEL-45 |
|---|---|---|---|---|---|---|
| a +++: Major component (>8%), ++: minor component (2–8%), +: trace component (<2%). The percentage refers to the peak area of the largest peak. b Compounds also present in medium alone, but in markedly lower amounts. | ||||||
| Dimethyl disulfide (6) | 775 | syn | +++ | +++ | +++ | |
| S-Methyl propanethioate | 807 | syn | +++ | ++ | ||
| S-Methyl butanethioate (11) | 902 | syn | + | |||
| 2,5-Dimethylpyrazineb | 927 | syn | + | + | +++ | |
| Ethyl 3-hydroxybutyrate | 937 | syn | + | |||
| S-Methyl 3-methylbutanethioate (12) | 952 | syn | + | + | + | |
| Benzaldehyde (28)b | 978 | syn | + | + | ++ | |
| Dimethyl trisulfide (7) | 981 | ms | +++ | + | ++ | |
| S,S′-Dimethyl carbonodithioate (17) | 984 | syn | ++ | |||
| Phenol (22) | 990 | syn | + | |||
| Trimethylpyrazineb | 1000 | syn | + | |||
| 2-Ethyl-3-methylpyrazine | 1015 | syn | + | + | ||
| Cyclohepta-3,5-dien-1-ol (39) | 1024 | syn | + | + | ||
| Butyl methylsulfone (20) | 1031 | ms | +++ | |||
| 2-Acetylthiazole (18) | 1035 | ms | + | + | ++ | |
| Methyl butyl disulfide (19) | 1042 | ms | ++ | |||
| Benzyl alcohol (23) | 1050 | syn | ++ | |||
| Salicylaldehyde (31) | 1055 | syn | + | |||
| Acetophenone (29) | 1082 | syn | ++ | + | ++ | |
| S-Methyl methanethiosulfonate (10) | 1084 | ms | ++ | + | + | |
| 2-Ethyl-3,6-dimethylpyrazine | 1088 | syn | + | + | ||
| S-Methyl hexanethioate (13) | 1102 | syn | + | |||
| Methyl benzoate (25) | 1108 | syn | + | + | ||
| Butyl methanesulfonate (21) | 1140 | syn | + | |||
| 1-Phenyl-2-propanone (33) | 1143 | syn | + | + | ++ | |
| 2,3,5-Trithiahexane (9) | 1144 | ms | ++ | + | ||
| Phenylacetonitrile (24) | 1160 | syn | ++ | |||
| Cycloheptene-1,3-dione (42) | 1171 | syn | ++ | + | ++ | |
| 1-Phenyl-1,2-propanedione (35) | 1177 | ms | ++ | |||
| Tropone (37) | 1182 | syn | +++ | +++ | +++ | + |
| Ethyl benzoate (26) | 1184 | syn | + | +++ | ||
| Tropolone (38) | 1212 | syn | ++ | |||
| 2-Aminobenzaldehyde (32) | 1225 | syn | + | |||
| Dimethyl tetrasulfide (8) | 1235 | ms | + | |||
| 1-Hydroxy-1-phenyl-2-propanone (34) | 1241 | ms | + | |||
| Ethyl phenylacetate (33) | 1256 | syn | ++ | |||
| S-Methyl octanethioate (14) | 1307 | syn | + | |||
| S-Methyl benzothioate (15) | 1317 | syn | + | + | + | |
| 2-Aminoacetophenone (30) | 1330 | syn | + | + | ||
| S-Methyl 2-phenylethanethioate (16) | 1379 | syn | + | + | + | |
| Butyl benzoate (27) | 1389 | syn | +++ | + | +++ | |
Several sulfur-containing compounds were identified in the headspace extracts of P. gallaeciensis and O. indolifex grown on MB2216 full medium, displayed in Fig. 3. Both species produced large amounts of dimethyl disulfide (6), dimethyl trisulfide (7), and S-methyl methanethiosulfonate (10), whereas dimethyl tetrasulfide (8) and 2,3,5-trithiahexane (9) were only found in P. gallaeciensis. Another important substance class emitted by P. gallaeciensis is a series of S-methyl alkanethioates including S-methyl butanethioate (11), S-methyl 3-methylbutanethioate (12), S-methyl hexanethioate (13), S-methyl octanethioate (14), S-methyl benzothioate (15) and S-methyl 2-phenylethanethioate (16). The esters 12, 15 and S,S′-dimethyl carbonodithioate (17) are released solely by O. indolifex. S-Methyl esters can be identified by the characteristic ions m/z 47 (CH3S+), 75 (CH3SCO+), [M − 47]+, and [M − 15]+ in their mass spectra (ESI, Fig. S1†). The proposed structures were confirmed by the synthesis of all thioesters 11–17 by the following strategy.
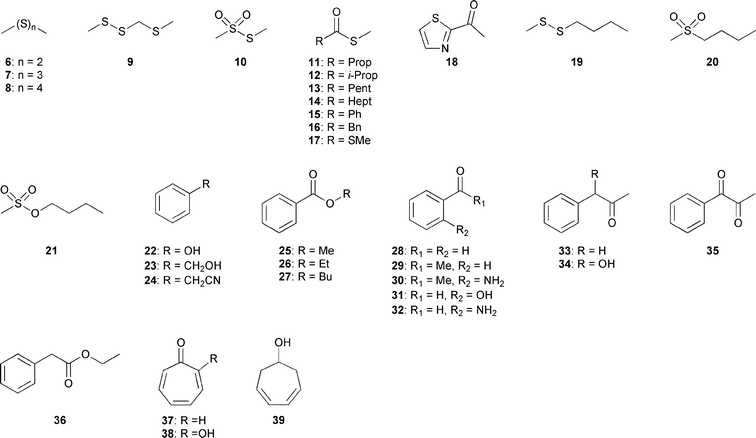 | ||
| Fig. 3 Volatiles identified in the headspace extracts of P. gallaeciensis and O. indolifex. | ||
Aluminium–sulfur reagents have frequently been used for the conversion of esters and lactones into their sulfur analogs, but these methods require thiols as starting materials.22,23 Kozikowski and Ames reported the preparation of seleno esters using dimethylaluminium methaneselenolate, a reagent that is easily obtained by heating trimethylaluminium in toluene with elemental selenium.24 We adopted this method for the preparation of thioesters by heating trimethylaluminium in toluene with elemental sulfur (Scheme 1). The putatively prepared dimethylaluminium methanethiolate was then used for the conversion of commercially available methyl esters into their respective S-methyl thiolates, the target compounds 11–17. The procedure is remarkably simple and high yielding, avoiding the handling of methanethiol that would be required if using other methods. The synthesized thioesters proved to be identical to the naturally occurring volatiles.
 | ||
| Scheme 1 Synthesis of S-methylthioates from methyl esters. | ||
The heterocyclic compound 2-acetylthiazole (18) and several butylated compounds such as the unusual methyl butyldisulfide (19) and butyl methylsulfone (20), present in large amounts, and butyl methanesulfonate (21) occurred in the headspace extract of O. indolifex. Another major component produced by this strain was butyl benzoate (27), also produced by P. gallaeciensis. Other aromatic compounds of both strains were represented by methyl benzoate (25), benzaldehyde (28), acetophenone (29) and 2-aminoacetophenone (30), whereas 1-phenyl-2-propanone (33) and 1-phenyl-1,2-propanedione (35) were found in O. indolifex only.
Tropone, tropolone and tropone hydrate
P. gallaeciensis also produced tropone (37) and some derivatives, but only within the first 72 h after inoculation. When grown on a nutrient-deficient medium charged with L-phenylalanine and L-histidine, the biosynthesis of the antibiotic TDA (4) was promoted in the liquid culture.25 The analysis of the headspace extract under these conditions showed striking changes in the production of volatile compounds. Compound 37 is produced almost exclusively, as well as small quantities of the ketone 33 and S-methyl 2-phenylethanethioate (16). In addition, an unknown compound eluting slightly earlier than tropone was present (peak A in Fig. 2a–c). The mass spectrum exhibited a molecular ion at m/z 124 corresponding, e.g., to the molecular formula C7H8O2 (Fig. 4e). On the basis of the fragmentation pattern with characteristic ions at m/z 96, 82, 54, and 39, this compound was suggested to be a cycloheptenedione or tropone hydrate, formally the product of water addition to 37. The loss of 28 amu pointed to the presence of a carbonyl group, while the loss of 42 amu and again 28 amu suggested a 1,3-dicarbonyl arrangement in the cycloheptene ring.![Mass spectra of tropone (37), its hydrate 42, and tropolone (38). (a) Unlabeled 37; (b) 37 obtained from [ring-2H5]phenylalanine; (c) 37 obtained from [2H8]phenylalanine; (d) unlabeled 38; (e) unlabeled 42; (f) 42 obtained from [ring-2H5]phenylalanine; (g) 42 obtained from [2H8]phenylalanine; (h) 38 obtained from [ring-2H5]phenylacetate. The mass spectra shown are those of the isotopomers with highest D-incorporation, eluting the earliest during GC.](/image/article/2010/OB/b909133e/b909133e-f4.gif) | ||
| Fig. 4 Mass spectra of tropone (37), its hydrate 42, and tropolone (38). (a) Unlabeled 37; (b) 37 obtained from [ring-2H5]phenylalanine; (c) 37 obtained from [2H8]phenylalanine; (d) unlabeled 38; (e) unlabeled 42; (f) 42 obtained from [ring-2H5]phenylalanine; (g) 42 obtained from [2H8]phenylalanine; (h) 38 obtained from [ring-2H5]phenylacetate. The mass spectra shown are those of the isotopomers with highest D-incorporation, eluting the earliest during GC. | ||
The most likely target compounds were therefore either 4- or 5-cycloheptene-1,3-dione (42a and 42b). Both structures can be transformed into the other via keto–enol tautomerism (see next section). The tautomer 5-cycloheptene-1,3-dione (42b) was synthesized from cyclohept-5-ene-1,3-diol (41), readily available via a three step synthesis from tropone according to Celestini et al. (Scheme 2).26 Oxidation of the diol with Cr(VI) oxide gave 5-cycloheptene-1,3-dione (42b), eluting at the same retention time and showing the same mass spectrum as compound A. Attempts to isomerize this compound to 4-cycloheptene-1,3-dione (42a) or to purify it by column chromatography failed because of its instability. The NMR spectrum of the crude product nevertheless indicated that the double bond is located at C-5. The Cr(VI) oxide oxidation is known to take place without isomerization of the double bond to the conjugated dione.27 Surprisingly, the double reduction product of tropone, cyclohepta-3,5-dien-1-ol (39), proved to be identical to a minor volatile produced by P. gallaeciensis.
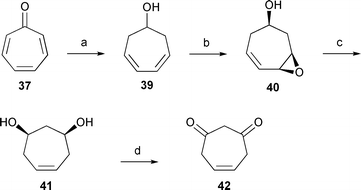 | ||
| Scheme 2 Synthesis of 5-cycloheptene-1,3-dione (42b). (a) NaBH4, MeOH–H2O; (b) m-CPBA, CH2Cl2–H2O, NaHCO3; (c) LiAlH4, THF; (d) CrO3, CH2Cl2–Et2O. | ||
Besides the two diketone forms discussed, tropone hydrate can occur in ten additional tautomeric forms. Therefore, a series of calculations at the density functional theory (DFT) level was performed in order to identify the low energy tautomers in the gas phase. We used Truhlar's hybrid meta exchange–correlation functional M05-2×28 in combination with a polarized triple zeta Pople basis set augmented with diffuse functions for carbon and oxygen atoms (6-311+G(d,p)). The relative energies of all 12 possible tautomers are shown in Fig. 5. Our calculations showed that the two 1,3-dicarbonyl tautomers of tropone hydrate are more stable compared to any enol form. The energy difference between 4- and 5-cycloheptene-1,3-dione (42a and 42b) is quite small (0.65 kcal mol−1). In contrast to open-chain mono-enol tautomers of 1,3-diketones, where intramolecular hydrogen bonds stabilize the mono-enol form, the conformational rigidity of the seven-membered ring does not allow such an interaction. Furthermore, the partly puckered ring poses considerable steric hindrance in some tautomers (ESI, Fig. S2†).
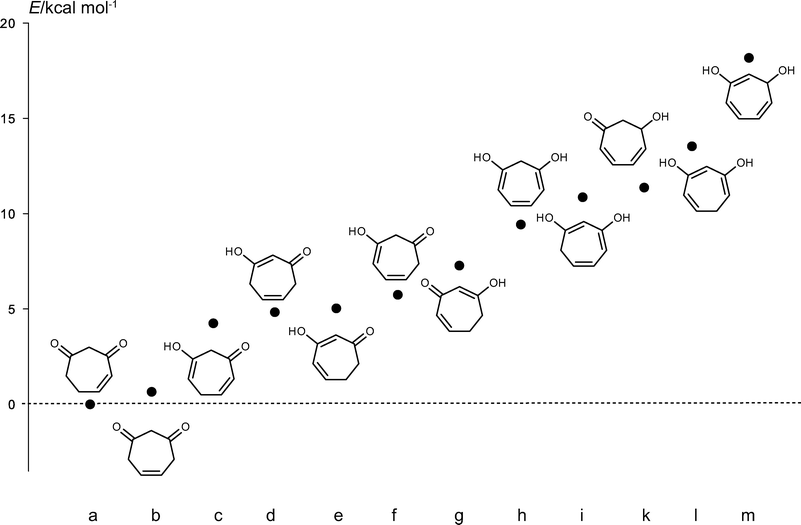 | ||
| Fig. 5 Relative energies of tropone hydrate tautomers 42a–m. | ||
The identity of the mass spectrum and retention index of the natural tropone hydrate as well as the synthesized material raised the suspicion that the high temperature in the injection port of the gas chromatograph might allow the interconversion of other tautomers to the most stable 42a. Therefore, GC-IR analyses were performed to determine the structure of the natural compound.29,30 The IR spectra of 42a, 42b, and 42c were calculated based on the DFT calculations. The gas phase IR spectra of the natural compound was most similar to the calculated spectrum of 42b, suggesting that the bacterial extract contains this form of tropone hydrate (ESI, Fig. S3†). Nevertheless, it cannot be ruled out that the bacteria initially produce a different tautomer. Furthermore, the observed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of 1730 cm−1 in the gas phase is typical for an isolated carbonyl group. A conjugated double bond as in 42a should have a frequency below 1710 cm−1 in the gas phase.31
O stretching frequency of 1730 cm−1 in the gas phase is typical for an isolated carbonyl group. A conjugated double bond as in 42a should have a frequency below 1710 cm−1 in the gas phase.31
P. gallaeciensis was also grown on a full medium spiked with phenylacetate, a proposed intermediate in the biosynthesis of 37 (see section below). This addition had only minor effects on the production of the released sulfur compounds (see Table 1). Again, large amounts of tropone (37) were produced, accompanied by 42b. Trace amounts of tropolone (38) were also detected. The volatile profile of the aromatic compounds shifted more profoundly and several additional aromatic compounds were found, which were not produced by P. gallaeciensis grown on MB2216 alone. While only traces of butyl benzoate (27) were released, ethyl benzoate (26) became the most abundant volatile in the headspace extracts. The additional compounds found were phenol (22), benzyl alcohol (23), benzaldehyde (28), salicylaldehyde (31), 2-aminobenzaldehyde (32), acetophenone (29), phenylacetonitrile (24), 1-phenyl-2-propanone (33), 1-hydroxy-1-phenyl-2-propanone (34), and ethyl phenylacetate (36).
Biosynthesis of tropone and tropolone
The large amounts of tropone released by P. gallaeciensis DSM17395 grown on a minimal medium containing L-phenylalanine and L-histidine suggested that L-phenylalanine is its biosynthetic precursor. Feeding experiments with differently [2H]-labeled phenylalanines were performed. Labeled [ring-2H5]phenylalanine offered to P. gallaeciensis was incorporated into tropone in high abundance (the incorporation rate was 92%, as determined from the peak areas of the molecular ions‡). The mass spectrum (Fig. 4b) showed the incorporation of four D-atoms, indicated by a characteristic shift of the molecular ion from m/z 106 to m/z 110, as well as a shift of the base peak, formed by loss of CO, from m/z 78 to m/z 82. Furthermore, minor amounts of triply labeled isotopomers also occurred (m/z 109), with a ratio of [2H3]-37/[2H4]-37 of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7, as determined from the peak areas of the molecular ions. Similarly, incorporation of [2H8]phenylalanine into 37 was observed, also resulting in the incorporation of three and four D-atoms (the incorporation rate was 92%), with a ratio of [2H3]-37/[2H4]-37 of 1:2 (Fig. 4c). The feeding experiments also showed incorporation of deuterium into the hydrate 42b. Feeding of [ring-2H5]- or [2H8]-phenylalanine resulted in an incorporation rate of 96 and 92%, respectively (Fig. 4f–g). Similarly to tropone, in both feeding experiments a shift of the molecular ion from m/z 124 to m/z 128 was observed, indicating the incorporation of four deuterium atoms. However, triply deuterated isotopomers occurred as well (m/z 127), while the most prominent isotopomer contained only two deuterium atoms (m/z 126). The isotopomer ratio obtained by feeding of [ring-2H5]phenylalanine was [2H2]-42b/[2H3]-42b/[2H4]-42b 1:0.4:0.2, while for [2H8]phenylalanine it was [2H2]-42b/[2H3]-42b/[2H4]-42b 1:0.6:0.2. The formation of the [2H2]- and [2H3]-isotopomers of 42b can be explained by tautomerization after release from the producing enzyme, leading to H/D-exchange. This cannot happen in tropone, so a higher 2H-content was observed in 37.
:1.7, as determined from the peak areas of the molecular ions. Similarly, incorporation of [2H8]phenylalanine into 37 was observed, also resulting in the incorporation of three and four D-atoms (the incorporation rate was 92%), with a ratio of [2H3]-37/[2H4]-37 of 1:2 (Fig. 4c). The feeding experiments also showed incorporation of deuterium into the hydrate 42b. Feeding of [ring-2H5]- or [2H8]-phenylalanine resulted in an incorporation rate of 96 and 92%, respectively (Fig. 4f–g). Similarly to tropone, in both feeding experiments a shift of the molecular ion from m/z 124 to m/z 128 was observed, indicating the incorporation of four deuterium atoms. However, triply deuterated isotopomers occurred as well (m/z 127), while the most prominent isotopomer contained only two deuterium atoms (m/z 126). The isotopomer ratio obtained by feeding of [ring-2H5]phenylalanine was [2H2]-42b/[2H3]-42b/[2H4]-42b 1:0.4:0.2, while for [2H8]phenylalanine it was [2H2]-42b/[2H3]-42b/[2H4]-42b 1:0.6:0.2. The formation of the [2H2]- and [2H3]-isotopomers of 42b can be explained by tautomerization after release from the producing enzyme, leading to H/D-exchange. This cannot happen in tropone, so a higher 2H-content was observed in 37.
In another feeding experiment, [ring-2H5]phenylacetic acid (46) synthesized from [2H5]bromobenzene (43, Scheme 3) was applied to P. gallaeciensis. The reaction of the corresponding Grignard reagent with formaldehyde yielded [ring-2H5]benzyl alcohol (44) that was transformed into [ring-2H5]benzyl bromide (45). A subsequent Grignard reaction with carbon dioxide furnished the acid 46.
![Synthesis of [ring-2H5]phenylacetate (46). (a) 1. Mg, Et2O, 2. H2CO; (b) PPh3, Br2, CH2Cl2; (c) 1. Mg, Et2O, 2. CO2.](/image/article/2010/OB/b909133e/b909133e-s3.gif) | ||
| Scheme 3 Synthesis of [ring-2H5]phenylacetate (46). (a) 1. Mg, Et2O, 2. H2CO; (b) PPh3, Br2, CH2Cl2; (c) 1. Mg, Et2O, 2. CO2. | ||
Feeding of this deuterated acid resulted in the incorporation of three to four D-atoms into 37 (the incorporation rate was 67%), with a ratio for [2H3]-28/[2H4]-28 of 1:2.2 determined from the peak areas of the ions at m/z 81 and 82 (mass spectrum not shown). In addition, the incorporation of 46 into tropolone (38) was indicated by a shift of the molecular ion from m/z 122 to 126, (the incorporation rate was 63%) (Fig. 4h). The incorporation of three deuterium atoms was also observed (m/z 125 and 97), with a [2H4]-38/[2H3]-38 ratio of 1:1.7.
These data are in accordance with the biosynthetic pathway shown in Fig. 6. It is closely related to the previously published biosynthetic pathway to TDA (4/5).17L-Phenylalanine (47) is first converted by transamination and oxidative decarboxylation into phenylacetate (46) and then into phenylacetyl-CoA (48),32 which is oxidized to form the 1,2-dihydrobenzenediol derivative 49. A second oxidation in the side chain introduces another hydroxy group to give 50, which sets the stage for a following ring-expansion, forming the seven-membered tropone core. The resulting CoA-derivative 51 is hydrolyzed to the key intermediate, the corresponding free acid 52. This acid can either be transformed into 4/5 after sulfuration,17 or it can undergo decarboxylation to tropone hydrate 42. Which tautomer of 42 is initially formed cannot be clarified, and several tautomers seem to be possible. Elimination of water finally yields tropone (37), which can be subsequently oxidized to tropolone (38).
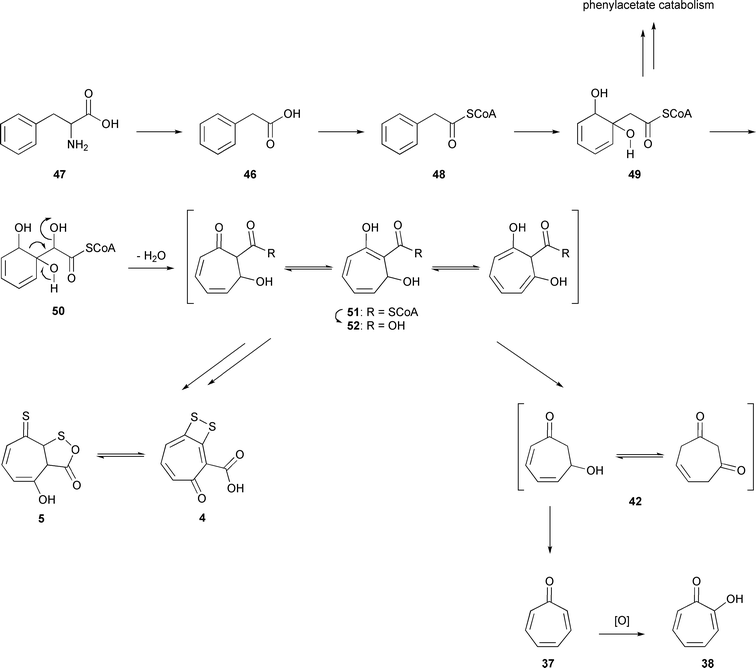 | ||
| Fig. 6 Biosynthesis of tropone (37), tropone hydrate (42), and tropolone (38) in P. gallaeciensis DSM17395. | ||
The described pathway yields [2H5]-52 and [2H6]-52 from [ring-2H5]phenylalanine and [2H8]phenylalanine, respectively (Fig. 7). At this stage, one deuterium of [2H5]-52 from [ring-2H5]phenylalanine and two deuterium atoms of [2H6]-52 from [2H8]phenylalanine can be washed out by keto–enol tautomerism (Fig. 6), resulting in [2H4]-52 in both cases, the precursor for [2H4]tropone ([2H4]-37) and [2H4]tropone hydrate ([2H4]-42). Additional D-atoms of [2H4]-42 can be lost by the interconversion of tautomers discussed before.
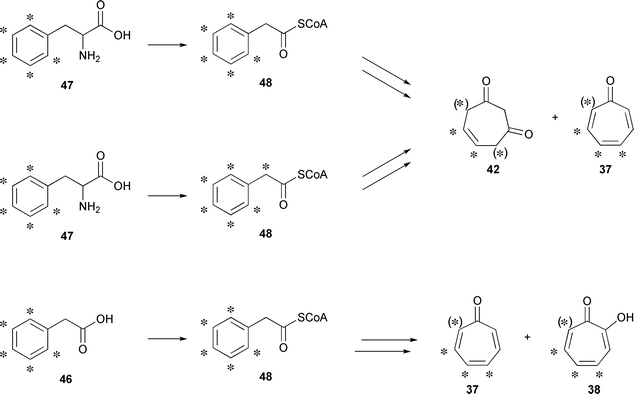 | ||
| Fig. 7 Position of deuterium labels after feeding of labeled precursors. Asterisks indicate the positions of D-atoms. | ||
Discussion
Sulfur volatiles
The headspace extracts of P. gallaeciensis and O. indolifex contain several volatile sulfur compounds, reflecting the important role of bacteria of the Roseobacter clade in the sulfur turnover and metabolism in marine ecosystems.1,2 The released sulfides, dimethyl disulfide (6), dimethyl trisulfide (7) and dimethyl tetrasulfide (8), can be formed by oxidation of methanethiol and H2S.33 Dimethyl disulfide and dimethyl trisulfide are emitted by a wide variety of bacteria,34 whereas higher homologs have less often been described as bacterial volatiles; e.g., dimethyl tetrasulfide (8) is also produced by Streptomyces griseus and Myxococcus xanthus.35,36 An oxidation product of dimethyl disulfide found in both species investigated, S-methyl methanethiosulfonate (10), was already observed in other Roseobacters such as Dinoroseobacter shibae and Loktanella sp., and the myxobacterium Stigmatella aurantiaca.18,37 Another sulfur compound released by P. gallaeciensis is 2,3,5-trithiahexane (9), known to be formed by photolysis of dimethyl disulfide.38 Several thioesters (11–16) are produced by P. gallaeciensis, whereas O. indolifex produces only 12 and 15. These compounds have rarely been described as bacterial volatiles. Some cheese-ripening microorganisms like coryneform bacteria, Micrococcaceae, Staphylococcaceae and lactic acid bacteria are reported to generate thioesters when the appropriate precursors methanethiol and fatty acids are available.39 While S-methyl benzothioate (15) was previously found in a strain of the marine bacterium Streptomyces caviscabies,40S-methyl 2-phenylethanethioate (16) was identified as a bacterial volatile for the first time. O. indolifex releases the unusual sulfur volatile S,S′-dimethyl carbonodithioate (17), also known as an aroma compound of white truffle (Tuber magnatum), and as a volatile of the offensively smelling plant Paederia foetida,41,42 but has never been reported from bacteria before.In conclusion, all these sulfur volatiles originate from methanethiol, and the question arises how it is formed in Phaeobacter and Oceanibulbus. Bacteria of the Roseobacter clade can be associated to dinoflagellates, coccolithophores and various macroalgae that produce the organic sulfur compound dimethylsulfoniopropanoate (DMSP) in substantial amounts as a cryo- and osmoprotectant. DMSP can be metabolised by bacteria in a cleavage reaction catalyzed by the DMSP lyase to form dimethylsulfide (DMS).43,44 Two different mechanisms are known for this cleavage reaction. In the first, DMSP is converted into its CoA thioester which is lytically degraded to DMS and 3-hydroxypropionate. The dddD gene first identified from Marinomonas is involved in this process and encodes an acyl CoA transferase.45 In contrast, the dddL gene from Sulfitobacter encodes a DMSP lyase that catalyses the direct cleavage of DMSP to DMS and acrylate.46 DMSP can also be demethylated to S-methyl 3-mercaptopropanethioate (MMPA) which is lytically cleaved to release methanethiol.47 The responsible gene, dmdA, for the demethylation reaction has been found in Ruegeria pomeroyi.48 These reactions make bacteria of the Roseobacter clade important players in the global sulfur cycle, because their production of DMS contributes to an estimated 13–45 Tg of sulfur efflux from the oceans per year.49 Atmospheric DMS is rapidly oxidised, resulting in the formation of a sulfate aerosol causing cloud formation with significant impact on the global climate.50,51 However, although homologs of the dmdA gene encoding the DMSP demethylase are present in the genomes of P. gallaeciensis DSM17395 and O. indolifex HEL-45, the described reactions cannot serve to explain the origin of methanethiol as a precursor for the identified sulfur volatiles, because the laboratory culture medium does not contain DMSP. Instead, other mechanisms of sulfate reduction to sulfide and its subsequent methylation can be assumed.
In addition, butylated sulfur compounds such as butyl methyl disulfide (19), butyl methyl sulfone (20), and butyl methanesulfonate (21) were identified that might be formed by related biosynthetic pathways. Compound 19 is known as a volatile constituent from Allium spp.,52 but has never been reported from bacteria, whereas 20 and 21 have not been reported from nature before.
Aromatic volatiles
A second class of important volatiles from O. indolifex are aromatic compounds. Benzaldehyde (28) and acetophenone (29) are commonly emitted by marine bacteria.34 2-Aminoacetophenone (30) is the character component of Pseudomonas aeruginosa,53 but occurs also in other bacteria such as Myxococcus xanthus.35 The aromatic ketone 1-phenyl-2-propanone (33) was found in the volatile profiles of Streptomyces40 and Klebsiella54 strains, while 1-phenyl-1,2-propanedione (35) was previously only identified in a Loktanella sp. together with 33;18 the headspace extract also contained aromatic esters. The major component butyl benzoate (27) is also released by Dinoroseobacter shibae18 and P. gallaeciensis, whereas methyl benzoate (25) is a common bacterial volatile.34In contrast to O. indolifex, P. gallaeciensis only released a complex mixture of aromatic compounds during growth on a full medium with added phenylacetate. Although L-phenylalanine is a precursor for phenylacetate by transamination and oxidative decarboxylation, a similar pattern of aromatic volatiles did not occur during growth on L-phenylalanine and L-histidine, possibly because this experiment was based on a minimal medium and not a full medium. The principal compound produced by P. gallaeciensis from phenylacetate, ethyl benzoate (26), is a typical plant volatile, but has never been reported from bacteria. The higher homolog ethyl phenylacetate (33) was also identified from bacteria for the first time, but the related butyl phenylacetate is produced by Streptomyces.40 Phenol (22) is found in the enterobacteria Klebsiella pneumoniae and Citrobacter freundii,54,55 whereas benzyl alcohol (23) is widespread among bacteria.34 Two related aromatic aldehydes, salicylaldehyde (31) and 2-aminobenzaldehyde (32), are also produced from phenylacetate by Phaeobacter. Chondromyces crocatus also emits 32 and methyl salicylate,21 but no 31. However, 31 is produced by different bacteria from naphthalene.56 Phenylacetonitrile (24) is also reported from Loktanella and Dinoroseobacter from the Roseobacter clade,18 and from Serratia and Pseudomonas, where it might contribute to bacterial antagonism against fungi.57 Interestingly, tropone (37) and tropolone (38) have antibacterial activities against a wide range of bacteria.58,59 While tropone has been reported earlier, the quite unstable tropone hydrate (42) as well as cyclohepta-3,5-dien-1-ol (39) have not been reported before from nature. Tropolone (38) has been reported from Pseudomonas ATCC 3109960 and the rice seedling blight-inducing bacterium P. plantarii ATCC 43733.61
Tropone biosynthesis
The biosynthesis of tropone and tropolone was investigated in feeding experiments with isotopically labeled precursors, and proceeds from L-phenylalanine via phenylacetate and phenylacetyl-CoA. The downstream part of the biosynthesis is linked to the phenylacetate catabolism and the biosynthesis of TDA (4/5).The phenylacetate degradation pathway has been investigated in detail in E. coli,62 and the available genomic information revealed that this pathway is a common feature within bacteria of the Roseobacter clade.63 The first steps of this degradation pathway are the conversion of phenylacetate to phenylacetyl-CoA and oxidation of the aromatic ring to form the diol 49 (Fig. 6). Further processing by the ring-opening enzyme PaaN and additional enzymes finally lead to succinyl-CoA in the investigated E. coli system. Tropone has previously been identified in an Azoarcus evansii mutant with a disruption in the paaN homolog (pacL),20 suggesting that the biosynthesis of tropone in P. gallaeciensis is linked to the phenylacetate degradation pathway.
The biosynthetic pathway leading to TDA (4/5)9,17 and tropone requires an additional oxidation at the benzylic carbon of phenylacetyl-CoA to attach a hydroxyl group that later serves as a leaving group, forming compound 50. Originally the biosynthesis of 4 was proposed to proceed via free phenylacetic acid,17 but in view of the phenylacetate degradation pathway, a bound phenylacetate-CoA substrate seems more likely. After ring expansion, an oxidation of the free acid 52 leads to further processing culminating in the TDA production, while decarboxylation favors the formation of tropone and related compounds. Therefore, both tropone and TDA biosynthesis are tightly linked. The genes of the TDA biosynthetic gene cluster are not present in the genome of O. indolifex, and accordingly, no production of tropone is found in this species.
Tropone (37) is likely to be the precursor of tropolone (38) requiring one oxidation step. This oxidation occurs in the position next to the carbonyl group of 37. A similar oxidation of 52 is needed in the biosynthesis of TDA, and this reaction might be carried out by the same enzyme encoded in the TDA biosynthetic gene cluster. The feeding of [ring-2H5]phenylacetate (46) resulted in the formation of [2H3]- and [2H4]-38 in a ratio of 1:1.7, whereas [2H3]- and [2H4]-37 are produced in a ratio of 1:2.2. Due to the C2v symmetry of 37 the oxidation of [2H4]-37 can take place in two equal positions, one of which is deuterated and one is not. If both positions were oxidized at a similar rate, a significant loss of deuterium labeling would be expected. However, the majority of 38 is still labeled with four deuterium atoms, suggesting that the replacement of a proton by OH is preferred due to a kinetic isotope effect. Furthermore, almost no [2H2]-38 is present, indicating that in [2H3]-37, both α-positions carry a proton and not a deuterium atom.
Interestingly, TDA (4/5) and tropone are formed from phenylacetate, while related tropolonoids as the fungal acids 1–3 are of polyketide origin.
Experimental
Strains, culture conditions and feeding experiments
Phaeobacter gallaeciensis DSM17395 was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany), Oceanibulbus indolifex HEL-45T (DSM 14862) was isolated from the North Sea.64Precultures were grown on a basis medium containing MgCl2·6 H2O (9.98 g L−1), NaCl (21.74 g L−1), Na2SO4 (3.62 g L−1), NaHCO3 (0.27 g L−1), CaCl2·2 H2O (1.37 g L−1), KCl (0.63 g L−1). The basis medium was supplemented with peptone (5 g L−1) and yeast extract (1 g L−1). Incubation was carried out for 8 h at 20 °C with shaking (100 rpm). Cultivation in liquid cultures was carried out in 500 mL Erlenmeyer flasks containing 100 mL marine broth (MB2216, Roth) medium and 100 μL of a preculture. In addition, experiments with P. gallaeciensis DSM17395 were performed using basis medium (as described above) supplemented with 5 mM L-phenylalanine and 50 μM L-histidine. Racemic labeled [ring-2H5]phenylalanine (10 mM, Sigma-Aldrich) and [2H8]phenylalanine (10 mM, Sigma-Aldrich) were used for feeding experiments under identical conditions. For inoculation of each of these cultures, 4 mL of a preculture were used. Cells of the preculture were sedimented by centrifugation, the supernatant was discarded and the cells resuspended in the medium used for further cultivation. Incubation of the liquid cultures was carried out overnight at 15 °C with shaking (100 rpm). Experiments with phenylacetic acid were performed with MB2216 liquid medium spiked with 1 mM [ring-2H5]phenylacetic acid or 1 mM phenylacetic acid, respectively, inoculated with 1 mL of the preculture. The resulting liquid cultures were incubated overnight with shaking at 28 °C (180 rpm) and directly subjected to headspace analysis.
Extraction of volatiles
Extraction of volatile organic compounds emitted by cultures of the Roseobacter strains DSM17395 and HEL-45 grown in marine broth was performed in a commercial CLSA apparatus (Brechbühler, Zurich, Switzerland). The samples were stirred by a magnetic stirrer; the adsorbent trap was a 5 mg filter of activated charcoal (Chromtech Gesellschaft für analytische Messtechnik mbH, Idstein, Germany). The filters were extracted with 30 μL dichloromethane after 18 h of sampling. The extracts were analyzed by GC-MS.GC-MS analyses
GC-MS analyses were carried out on a HP 6890 Series GC System connected to a HP 5973 Mass Selective Detector (Hewlett-Packard Company, Wilmington, USA) fitted with a BPX5 fused-silica capillary column (25 m × 0.22 mm i.d., 0.25 mm film, SGE Inc., Melbourne, Australia). Conditions were as follows: splitless injection (60 s valve time), inlet pressure: 77.1 kPa; injection volume: 1 μL; transfer line: 300 °C; electron energy: 70 eV. The GC was programmed as follows: 5 min at 50 °C then with 5 °C min−1 to 320 °C. The carrier gas was He at 1 mL min−1 flow rate.Computational details
All computations were performed using the Gaussian03 program. Truhlar's hybrid meta exchange–correlation functional M05-2×28 was used in combination with a polarized triple zeta Pople basis set augmented with diffuse functions for carbon and oxygen atoms (6-311+G(d,p)). The force constants and the dipole moment derivatives were calculated using analytical second derivatives. Normal coordinate analysis was carried out using Wilsons GF Method.65 In order to make the theoretical line spectra comparable to the experiment, the calculated line peaks are broadened by Lorentzian lines with a band width at half maximum of 40 cm−1.General methods
Chemicals were purchased from Fluka Chemie GmbH (Buchs, Switzerland) or Sigma-Aldrich Chemie GmbH (Steinheim, Germany) and used without further purification. Racemic [ring-2H5]phenylalanine and [2H8]phenylalanine for feeding experiments were purchased from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). NMR spectra were obtained using a Bruker AMX400 spectrometer (operating at 400 MHz for 1H and 100 MHz for 13C) using CDCl3 as the solvent. Tetramethylsilane (0.00 ppm) served as the internal standard in 1H NMR and CDCl3 (77.0 ppm) in 13C NMR. Coupling constants (J values) are given in Hertz (Hz). Chemical shifts are expressed in parts per million (ppm). IR spectra were recorded on a Bruker Tensor 27 ATR spectrometer. The FT-IR spectrum of 42 was recorded on a HP 6890 series GC system connected to a HP 5965B IR detector (Hewlett Packard) fitted with a HP-5 fused-silica cap. column (30 m × 0.32 mm i.d., 0.25 μm film thickness; Hewlett-Packard) with He as carrier gas (2 mL m−1). UV spectra were obtained with a Varian Cary 100 Bio spectrometer.Column chromatography was carried out using Merck Kieselgel 60. Thin layer chromatography was carried out using 0.2 mm pre-coated plastic sheets Polygram Sil G/UV254 (Marcherey-Nagel, Düren, Germany). Solvents were purified by distillation and dried according to standard methods.
S-Methyl thioates
A mixture of Me3Al (20 mL, 2 M in toluene, 40 mmol) and elemental sulfur (1.34 g, 42 mmol) was heated to reflux for 2 h resulting in complete consumption of the sulfur and the formation of a translucent pale yellow solution. After cooling to 0 °C the methyl ester (38 mmol) or dimethyl carbonate (19 mmol) dissolved in dry CH2Cl2 (20 mL) was added. The mixture was stirred for 1 h at 0 °C and for 1 h at room temperature. Cautious hydrolysis of the remaining organometallic reactant was performed at 0 °C by the dropwise addition of water, saturated NH4Cl, and 2 N HCl (Caution! The hydrolysis reaction was slow in the beginning, but became very violent after an initiation period.) The aqueous phase was separated and extracted with diethyl ether (3 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated. The pure S-methyl thioates were obtained by column chromatography on silica gel with pentane–diethyl ether (20:1) as a colourless oils.
:1): RF 0.48; GC: I 897; νmax(film)/cm−1 2965, 2932, 2876, 1688, 1458, 1417, 1261, 1114, 996, 885, 800, 761, 695 and 598; λmax(CH2Cl2)/nm 232 (ε/dm3 mol−1 cm−1 3810); δH(400 MHz; CDCl3; Me4Si) 0.96 (3 H, t, J 7.4, 1JC,H 125.7, CH3), 1.70 (2 H, sextet, J 7.4, 1JC,H 128.8, CH2), 2.29 (3 H, s, 1JC,H 141.4, CH3) and 2.54 (2 H, t, J 7.4, 1JC,H 128.4, CH2); δC(100 MHz; CDCl3; Me4Si) 11.5 (CH3), 13.5 (CH3), 19.2 (CH2), 45.7 (CH2) and 199.9 (CO); m/z (EI) 118 (7%), 103 (11), 75 (10), 71 (45) and 43 (100).
:1): RF 0.57; GC: I 951; νmax(film)/cm−1 2959, 2931, 2872, 1688, 1466, 1134, 1018, 940 and 757; λmax(CH2Cl2)/nm 232 (ε/dm3 mol−1 cm−1 3190); δH(400 MHz; CDCl3; Me4Si) 0.96 (6 H, d, J 6.7, 1JC,H 125.3, 2 × CH3), 2.17 (1 H, septet, J 6.8, CH), 2.30 (3 H, s, CH3) and 2.44 (2 H, d, J 7.1, CH2); δC(100 MHz; CDCl3; Me4Si) 11.5 (CH3), 22.3 (2 × CH3), 26.4 (CH), 52.7 (CH2) and 199.4 (CO); m/z (EI) 132 (4%), 117 (6), 85 (55), 75 (20), 57 (100) and 41 (74).
:1): RF 0.68; GC: I 1105; νmax(film)/cm−1 2957, 2929, 2861, 1690, 1462, 1121, 1032, 922, 736, 696 and 599; λmax(CH2Cl2)/nm 232 (ε/dm3 mol−1 cm−1 4060); δH(400 MHz; CDCl3; Me4Si) 0.89 (3 H, t, J 7.0, 1JC,H 124.7, CH3), 1.26–1.37 (4 H, m, 2 × CH2), 1.63–1.71 (2 H, m, CH2), 2.29 (3 H, s, 1JC,H 141.4, CH3) and 2.55 (2 H, t, J 7.5, 1JC,H 128.2, CH2); δC(100 MHz; CDCl3; Me4Si) 11.5 (CH3), 13.8 (CH3), 22.3 (CH2), 25.4 (CH2), 31.1 (CH2), 43.9 (CH2) and 200.0 (CO); m/z (EI) 131 (17%), 99 (52), 75 (16), 71 (48), 55 (18) and 43 (100).
:1): RF 0.79; GC: I 1312; νmax(film)/cm−1 2954, 2926, 2856, 1691, 1462, 1416, 1124, 1041, 999, 760, 726, 696 and 599; λmax(CH2Cl2)/nm 232 (ε/dm3 mol−1 cm−1 4190); δH(400 MHz; CDCl3; Me4Si) 0.88 (3 H, t, J 6.9, 1JC,H 124.5, CH3), 1.22–1.33 (8 H, m, 4 × CH2), 1.66 (2 H, quintet, J 7.4, CH2), 2.29 (3 H, s, 1JC,H 141.4, CH3) and 2.55 (2 H, t, J 7.5, 1JC,H 128.3, CH2); δC(100 MHz; CDCl3; Me4Si) 11.5 (CH3), 14.0 (CH3), 22.6 (CH2), 25.7 (CH2), 28.9 (CH2), 31.6 (CH2), 43.9 (CH2) and 200.0 (CO); m/z (EI) 159 (12%), 127 (57), 109 (7), 75 (14), 57 (100) and 41 (44).
:1): RF 0.29; GC: I 1317; νmax(film)/cm−1 3062, 2928, 1659, 1580, 1447, 1312, 1204, 1174, 967, 906, 771, 684 and 645; λmax(CH2Cl2)/nm 222 (ε/dm3 mol−1 cm−1 7850), 238 (10300) and 268 (7380); δH(400 MHz; CDCl3; Me4Si) 2.47 (3 H, s, 1JC,H 141.9, CH3), 7.41–7.47 (2 H, m, 2 × CH), 7.56 (1 H, tt, J 7.4, 4JH,H 1.5, CH) and 7.95–7.98 (2 H, m, 2 × CH); δC(100 MHz; CDCl3; Me4Si) 11.7 (CH3), 127.1 (2 × CH), 128.6 (2 × CH), 133.2 (CH), 137.1 (Cquart.) and 192.4 (CO); m/z (EI) 152 (7%), 105 (100), 77 (69) and 51 (30).
:1): RF 0.41; GC: I 1389; νmax(film)/cm−1 3063, 3030, 2928, 1682, 1495, 1453, 1420, 1309, 1182, 1074, 1018, 998, 912, 774, 723, 699 and 596; λmax(CH2Cl2)/nm 234 (ε/dm3 mol−1 cm−1 4580); δH(400 MHz; CDCl3; Me4Si) 2.27 (3 H, s, 1JC,H 141.7, CH3), 3.82 (2 H, s, 1JC,H 130.2, CH2) and 7.26–7.35 (5 H, m, 5 × CH); δC(100 MHz; CDCl3; Me4Si) 11.9 (CH3), 50.3 (CH2), 127.4 (CH), 128.6 (2 × CH), 129.5 (2 × CH), 133.7 (Cquart.) and 197.7 (CO); m/z (EI) 166 (17%), 138 (1), 119 (16), 91 (100), 75 (5), 65 (22) and 39 (10).
:1): RF 0.62; GC: I 981; νmax(film)/cm−1 2929, 1745, 1642 (CO), 1422, 1310, 1043, 966, 856 and 573; λmax(CH2Cl2)/nm 227 (ε/dm3 mol−1 cm−1 3270) and 249 (5230); δH(400 MHz; CDCl3; Me4Si) 2.44 (6 H, s, 1JC,H 142.4, 2 × CH3); δC(100 MHz; CDCl3; Me4Si) 13.0 (2 × CH3) and 190.3 (CO); m/z (EI) 122 (50%), 94 (61), 75 (100) and 45 (73).
Tropone hydrate 42b
The diol 41 was prepared according to the method of Celestini et al.26 This diol (0.1 g, 0.78 mmol) was dissolved in a mixture of diethyl ether (15 mL) and dichloromethane (5 mL), and Celite (0.31 g) was added. The mixture was cooled to 0 °C, and CrO3 (0.31 g, 3.1 mmol) was added in small portions.27 After stirring for 30 min, the reaction mixture was diluted with diethyl ether and then filtered through a Celite pad. Drying over MgSO4 and removal of the solvent gave the desired product in 2:1 mixture with the starting material. Purification was not possible because it started to decompose directly after synthesis. GC: I 1171; νmax/cm−1 1012, 1131, 1244, 1405, 1539, 1730, 2990, 3045; δH(300 MHz, CDCl3, Me4Si) 3.29 (4 H, dd, 3JH,H 3.5, 4JH,H 1.1, 2 × CH2CH), 3.77 (2 H, s, CH2(CO)), 5.93 (2 H, tt, 3JH,H 3.5, 4JH,H 1.2, 2 × CH), δC(75 MHz, CDCl3, Me4Si) 43.9 (2 × CH2), 59.4 (CH2), 128.2 (2 × CH), 200.8 (2 × CO);; m/z (EI) 124 (33%), 96 (12), 82 (43), 54 (100), 42 (13) and 39 (41).
[ring-2H5]Benzyl alcohol 44
A solution of the Grignard reagent [2H5]phenylmagnesium bromide in diethyl ether (40 mL) was prepared from 43 (5 g, 30.9 mmol) and magnesium (0.75 g, 30.9 mmol). Paraformaldehyde (1.11 g, 37 mmol) was added, the mixture was heated to reflux for 3 h, and quenched by the addition of a saturated NH4Cl solution. The aqueous layer was extracted with diethyl ether (3 × 50 mL). The combined extracts were dried (MgSO4) and concentrated. Purification of the crude product by flash chromatography on silica gel with hexane–ethyl acetate (3:1) yielded 44 (2.16 g, 62%). TLC (pentane–diethyl ether = 3:1): RF 0.30; GC: I 1140; νmax(film)/cm−1 3314br (OH), 2930, 2873, 2275 (CD), 1373, 1213, 1148, 1056, 1004, 843, 820 and 535; λmax(CH2Cl2)/nm 253sh (ε/dm3 mol−1 cm−1 2070) and 258 (2250); δH(400 MHz; CDCl3; Me4Si) 1.83 (1 H, br s, OH) and 4.67 (2 H, d, J 1.3, 1JC,H 142.5, CH2); δC(100 MHz; CDCl3; Me4Si) 65.2 (CH2), 126.5 (t, 1JC,D 24.4, 2 × CD), 127.1 (t, 1JC,D 24.8, CD), 128.0 (t, 1JC,D 24.5, 2 × CD) and 140.7 (Cquart.); m/z (EI) 113 (100%), 96 (20), 84 (68) and 54 (19).
[ring-2H5]Benzyl bromide 45
Bromine (3.82 g, 1.25 mL, 23.9 mmol) was added dropwise to a cooled (0 °C) solution of triphenylphosphane (6.26 g, 23.9 mmol) in dry CH2Cl2 until the orange color persisted. The mixture was stirred for 30 min, and then 44 (2.16 g, 19.1 mmol) was added. The reaction mixture was stirred at 0 °C for 3 h, and then subsequently washed with saturated Na2SO3 and Na2CO3 solutions. The organic layer was dried (MgSO4) and concentrated to ca. 20 mL. Triphenylphosphane oxide was precipitated by the addition of pentane and removed by filtration. The filtrate was concentrated, and the crude product was purified by column chromatography with hexane–ethyl acetate (20:1) to give 45 (2.54 g, 76%). TLC (pentane–diethyl ether = 20:1): RF 0.54; GC: I 1235; νmax(film)/cm−1 2968, 2276 (CD), 1436, 1335, 1213, 1150, 871, 844, 821, 747, 654, 582 and 538; λmax(CH2Cl2)/nm 230 (ε/dm3 mol−1 cm−1 7090); δH(400 MHz; CDCl3; Me4Si) 4.44–4.48 (2 H, s, 1JC,H 152.7, CH2); δC(100 MHz; CDCl3; Me4Si) 33.5 (CH2), 127.9 (t, 1JC,D 24.8, CD), 128.2 (t, 1JC,D 24.6, 2 × CD), 128.6 (t, 1JC,D 24.3, 2 × CD); m/z (EI) 175 (6%), 96 (100), 68 (8), 52 (3) and 41 (4).
[ring-2H5]Phenylacetic acid 46
A solution of the Grignard reagent [ring-2H5]benzylmagnesium bromide in diethyl ether (30 mL) was prepared from 45 (1.22 g, 6.9 mmol) and magnesium (168 mg, 6.9 mmol). A stream of carbon dioxide was passed through the solution for 30 min, resulting in the precipitation of a colorless solid. The reaction mixture was quenched by the addition of 2 N HCl and extracted with diethyl ether (3 × 50 mL). The combined organic layers were dried (MgSO4) and concentrated. The residue was purified by flash chromatography on silica gel with hexane–ethyl acetate (3:1) to yield 46 (450 mg, 46%). A small sample (ca. 1 mg) was treated with MSTFA for 1 h at 60 °C for GC-MS analysis. TLC (pentane–diethyl ether = 3:1): RF 0.25; GC (MSTFA): I 1314; νmax(film)/cm−1 3020, 2952, 2921, 2726, 2639, 2531, 2277 (CD), 1691 (CO), 1407, 1328, 1213, 1189, 912, 880, 852, 821, 796, 684, 620, 586 and 541; λmax(CH2Cl2)/nm 228 (ε/dm3 mol−1 cm−1 450), 247sh (120), 252 (150), 258 (170) and 263 (130); δH(400 MHz; CDCl3; Me4Si) 3.64 (2 H, s, 1JC,H 129.4, CH2) and 11.09 (1 H, br s, CO2H); δC(100 MHz; CDCl3; Me4Si) 41.0 (CH2), 126.8 (t, 1JC,D 25.1, CD), 128.1 (t, 1JC,D 24.4, 2 × CD), 128.9 (t, 1JC,D 24.0, 2 × CD), 133.1 (Cquart.) and 177.9 (CO); m/z (EI, MSTFA) 213 (1%), 198 (20), 169 (26), 142 (5), 131 (2), 117 (3), 96 (18), 73 (100), 61 (3) and 45 (6).
Conclusions
The two bacterial strains Phaeobacter gallaeciensis DSM17395 and Oceanibulbus indolifex HEL 45 of the Roseobacter clade produce a variable blend of volatiles originating from sulfur and phenylacetate metabolism. The bouquet of sulfur volatiles is derived from methanethiol, likely to be formed by degradation from DMSP supplied by other organisms in the ocean. A major metabolite of P. gallaeciensis is tropone (37), so far not known to be produced by wild-type bacteria outside the Roseobacter clade. Under defined culture conditions, the production of sulfur metabolites can be suppressed, leading to the production of almost solely tropone, accompanied by related components. The elucidation of its biosynthesis shows that this compound may be an indicator for strains able to produce the antibiotic tropodithietic acid (4/5). These bacteria elaborate two different biosynthetic pools for volatile production. In the case of tropone an unusual biosynthesis occurs, probably indicative for a specific function of this compound for the bacterium, e.g. communication or defense.Acknowledgements
We thank R. Gahl-Janssen and B. Elxnat for technical assistance. S.S. thanks the Fonds der Chemischen Industrie for financial support.References
- T. Brinkhoff, H.-A. Giebel and M. Simon, Arch. Microbiol., 2008, 189, 531–539 CrossRef CAS.
- I. Wagner-Döbler and H. Biebl, Annu. Rev. Microbiol., 2006, 60, 255–280 CrossRef.
- L. Gram, H.-P. Grossart, A. Schlingloff and T. Kiørboe, Appl. Environ. Microbiol., 2002, 68, 4111–4116 CrossRef CAS.
- B. Lafay, R. Ruimy, C. Rausch de Traubenberg, V. Breittmayer, M. J. Gauthier and R. Christen, Int. J. Syst. Bacteriol., 1995, 55, 290–296 Search PubMed.
- K. Böröczky, H. Laatsch, I. Wagner-Döbler, K. Stritzke and S. Schulz, Chem. Biodiversity, 2006, 3, 622–634 CrossRef.
- T. Brinkhoff, G. Bach, T. Heidorn, L. Liang, A. Schlingloff and M. Simon, Appl. Environ. Microbiol., 2004, 70, 2560–2565 CrossRef CAS.
- J. B. Bruhn, L. Gram and R. Belas, Appl. Environ. Microbiol., 2007, 73, 442–450 CAS.
- J. B. Bruhn, K. F. Nielsen, M. Hjelm, M. Hansen, J. Bresciani, S. Schulz and L. Gram, Appl. Environ. Microbiol., 2005, 71, 7263–7270 CrossRef CAS.
- H. Geng, J. B. Bruhn, K. F. Nielsen, L. Gram and R. Belas, Appl. Environ. Microbiol., 2008, 74, 1535–1545 CrossRef CAS.
- C. H. Porsby, K. F. Nielsen and L. Gram, Appl. Environ. Microbiol., 2008, 74, 7356–7364 CrossRef CAS.
- J. H. Birkinshaw and H. Raistrick, Biochem. J., 1932, 26, 441–453 CAS.
- R. Bentley, Nat. Prod. Rep., 2008, 25, 118–138 RSC.
- J. H. Birkinshaw, A. R. Chambers and H. Raistrick, Biochem. J., 1942, 36, 242–251 CAS.
- K. Kintaka, H. Ono, S. Tsubotani, S. Harada and H. Okazaki, J. Antibiot., 1984, 37, 1294–1300 CAS.
- S. Tsubotani, Y. Wada, K. Kamiya, H. Okazaki and S. Harada, Tetrahedron Lett., 1984, 25, 419–422 CrossRef.
- E. M. Greer, D. Aebisher, A. Greer and R. Bentley, J. Org. Chem., 2008, 73, 280–283 CrossRef CAS.
- D. E. Cane, Z. Wu and J. E. van Epp, J. Am. Chem. Soc., 1992, 114, 8479–8483 CrossRef CAS.
- J. S. Dickschat, I. Wagner-Döbler and S. Schulz, J. Chem. Ecol., 2005, 31, 925–947 CrossRef CAS.
- J. S. Dickschat, H. Reichenbach, I. Wagner-Döbler and S. Schulz, Eur. J. Org. Chem., 2005, 4141–4153 CrossRef CAS.
- R. Rost, S. Haas, E. Hammer, H. Herrmann and G. Burchhardt, Mol. Genet. Genomics, 2002, 267, 656–663 CrossRef CAS.
- S. Schulz, J. Fuhlendorff and H. Reichenbach, Tetrahedron, 2004, 60, 3863–3872 CrossRef CAS.
- R. P. Hatch and S. M. Weinreb, J. Org. Chem., 1977, 42, 3960–3961 CrossRef CAS.
- E. J. Corey and D. J. Beames, J. Am. Chem. Soc., 1973, 95, 5829–5831 CrossRef CAS.
- A. P. Kozikowski and A. Ames, Tetrahedron, 1985, 41, 4821–4834 CrossRef CAS.
- T. Heidorn, Ph.D. Thesis, Universität Hannover, 2002 Search PubMed.
- P. Celestini, B. Danieli, G. Lesma, A. Sacchetti, A. Silvani, D. Passarella and A. Virdis, Org. Lett., 2002, 4, 1367–1370 CrossRef CAS.
- S. J. Flatt, G. W. Fleet and B. J. Taylor, Synthesis, 1979, 815–817 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- S. Franke, J. Grunenberg and J. Schwarzbauer, Int. J. Environ. Anal. Chem., 2007, 87, 437–448 CrossRef CAS.
- J. Grunenberg, H. Hopf, M. Bahadir, A. Pieper, R. Vogt and H. Wichmann, Chem. Phys. Lett., 2002, 366, 1–8 CrossRef CAS.
- G. Socrates, Infrared and Raman characteristic group frequencies, Wiley, Chichester, 2001 Search PubMed.
- H. Martinez-Blanco, A. Reglero, L. B. Rodriguez-Aporicio and J. M. Luengo, J. Biol. Chem., 1990, 265, 7084–7090 CAS.
- H.-W. Chin and R. C. Lindsay, Food Chem., 1994, 49, 387–392 CrossRef CAS.
- S. Schulz and J. Dickschat, Nat. Prod. Rep., 2007, 24, 814–842 RSC.
- K. Wilkins, Chemosphere, 1996, 32, 1427–1434 CrossRef CAS.
- J. S. Dickschat, S. C. Wenzel, H. B. Bode, R. Müller and S. Schulz, ChemBioChem, 2004, 5, 778–787 CrossRef CAS.
- J. S. Dickschat, H. B. Bode, S. C. Wenzel, R. Müller and S. Schulz, ChemBioChem, 2005, 6, 2023–2033 CrossRef CAS.
- R. G. Buttery and R. M. Seifert, J. Agric. Food Chem., 1977, 25, 434–434 CrossRef CAS.
- G. Lamberet, B. Auberger and J. L. Bergère, Appl. Microbiol. Biotechnol., 1997, 48, 393–397 CrossRef CAS.
- J. S. Dickschat, T. Martens, T. Brinkhoff, M. Simon and S. Schulz, Chem. Biodiversity, 2005, 2, 837–865 CrossRef CAS.
- A. M. Gioacchini, M. Menotta, M. Guescini, R. Saltarelli, P. Ceccaroli, A. Amicucci, E. Barbieri, G. Giomaro and V. Stocchi, Rapid Commun. Mass Spectrom., 2008, 22, 3147–3153 CrossRef CAS.
- K. C. Wong and G. L. Tan, Flavour Fragrance J., 1994, 9, 25–28 CrossRef CAS.
- T. R. Miller and R. Belas, Appl. Environ. Microbiol., 2004, 70, 3383–3391 CrossRef CAS.
- D. C. Yoch, Appl. Environ. Microbiol., 2002, 68, 5804–5815 CrossRef CAS.
- J. D. Todd, R. Rogers, Y. Gou Li, M. Wexler, P. L. Bond, L. Sun, A. R. J. Curson, G. Malin, M. Steinke and A. W. B. Johnston, Science, 2007, 315, 666–669 CrossRef CAS.
- A. R. J. Curson, R. Rogers, J. D. Todd, C. A. Brearley and A. W. B. Johnston, Environ. Microbiol., 2008, 10, 757–767 CrossRef CAS.
- B. F. Taylor and D. C. Gilchrist, Appl. Environ. Microbiol., 1991, 57, 3581–3584 CAS.
- E. C. Howard, J. R. Henriksen, A. Buchan, C. R. Reisch, H. Bürgmann, R. Welsh, W. Ye, J. M. Gonzalez, K. Mace, S. B. Joye, R. P. Kiene, W. B. Whitman and M. A. Moran, Science, 2006, 314, 649–652 CrossRef CAS.
- A. J. Kettle and M. O. Andreae, J. Geophys. Res., 2000, 105, 26793–26808 CrossRef CAS.
- R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Nature, 1987, 326, 655–661 CrossRef CAS.
- T. S. Bates, R. J. Carlson and R. H. Gammon, Nature, 1987, 329, 319–321 CrossRef CAS.
- J. A. Pino, V. Fuentes and M. T. Correa, J. Agric. Food Chem., 2001, 49, 1328–1330 CrossRef CAS.
- S. Mann, Arch. Microbiol., 1966, 54, 184–190 CrossRef CAS.
- D. C. Robacker and R. J. Bartelt, J. Chem. Ecol., 1997, 23, 2897–2915 CrossRef CAS.
- C.-J. Lee, A. B. DeMilo, D. S. Moreno and A. J. Martinez, J. Agric. Food Chem., 1995, 43, 1348–1351 CrossRef CAS.
- G. M. Pumphrey and E. L. Madsen, Microbiology, 2007, 153, 3730–3738 CrossRef CAS.
- M. Kai, U. Effmert, G. Berg and B. Piechulla, Arch. Microbiol., 2007, 187, 351–360 CrossRef CAS.
- T. J. Trust and K. H. Bartlett, Antimicrob. Agents Chemother., 1975, 8, 381–383 CAS.
- T. J. Trust, Antimicrob. Agents Chemother., 1975, 7, 500–506 CAS.
- G. D. Lindberg, J. M. Larkin and H. A. Whaley, J. Nat. Prod., 1980, 43, 592–594 CrossRef CAS.
- K. Azegami, K. Nishiyama and H. Kato, Appl. Environ. Microbiol, 1988, 54, 844–847 CAS.
- W. Ismail, M. E. Mohamed, B. L. Wanner, K. A. Datsenko, W. Eisenreich, F. Rohdich, A. Bacher and G. Fuchs, Eur. J. Biochem., 2003, 270, 3047–3054 CrossRef CAS.
- I. Wagner-Döbler, B. Ballhausen, M. Baumgart, T. Brinkhoff, I. Buchholz, B. Bunk, H. Cypionka, R. Daniel, T. Drepper, G. Gerdts, S. Hahnke, C. Han, D. Jahn, D. Kalhoefer, H. Kiss, H.-P. Klenk, N. Kyrpides, W. Liebl, H. Liesegang, L. Meincke, A. Pati, J. Petersen, T. Piekarski, C. Pommerenke, S. Pradella, R. Pukall, R. Rabus, E. Stackebrandt, S. Thole, L. Thompson, P. Tielen, J. Tomasch, M. von Jan, N. Wanphrut, A. Wichels, H. Zech and M. Simon, ISME J. DOI:10.1038/ismej.2009.94.
- M. Allgaier, H. Uphoff, A. Felske and I. Wagner-Döbler, Appl. Environ. Microbiol., 2003, 69, 5051–5059 CrossRef CAS.
- E. B. Wilson, J.C. Decius, and P. C. Cross, Molecular Vibrations, Dover, New York, 1989 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectra of compounds 12, 14 and 16, and the gas phase structures and FT-IR spectra of the most stable tautomers of compound 42. See DOI: 10.1039/b909133e |
| ‡ The incorporation rates were determined by peak integration over all isotopomers. The mass spectra shown in Fig. 4 represent only the earliest eluting isotopomers for better visualization of the D-incorporation. Therefore, the peak ratios reported in the text may differ from those observed in the figures. |
| This journal is © The Royal Society of Chemistry 2010 |