Microwave-assisted three-component coupling-addition-SNAr (CASNAR) sequences to annelated 4H-thiopyran-4-ones†
Benjamin Willya, Walter Frank‡b and Thomas J. J. Müller*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr., 1, D-40225, Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@uni-duesseldorf.de; Fax: +492118114324
bLehrstuhl für Anorganische Chemie und Strukturchemie der Heinrich-Heine-Universität Düsseldorf, Universitätsstraße, 1, D-40225, Düsseldorf, Germany
First published on 23rd October 2009
Abstract
A whole family of annelated 4H-thiopyran-4-ones as the core structural unit was readily synthesized in good yields by a microwave-assisted coupling-addition-SNAr (CASNAR) sequence starting from readily available (het)aroyl chlorides, alkynes and sodium sulfide nonahydrate in a consecutive one-pot three-component reaction. All representatives display a pronounced halochromicity of the absorption bands upon protonation. According to DFT calculations, the electronic ground state of the annelated 4H-thiopyran-4-ones possess a considerable zwitterionic character.
Introduction
Thiopyranone is the thio homologue of pyranone, a core constituent of many natural products. In their own right, thiochromenones, benzo-annelated thiopyranones that are related to the class of flavones, are potent drug candidates and serve as key intermediates for the synthesis of biologically active compounds. As a consequence, many thiochromenones are known to exhibit antimicrobial and antifungal,1 antibacterial,2 antibiotic,3 and anticarcinogenic4 activity. Some derivatives are used as antimalaria agents,5 and oxidized representatives reversibly inhibit the human cytomegalovirus protease.6In general, 4H-thiochromen-4-ones are synthesized either by condensation of β-keto esters and thiophenols with polyphosphoric acid7 or by cyclization of β-substituted cinnamates derived from thiophenols and appropriate propiolates.5,8 Condensation and subsequent acid-mediated cyclization of lithiated intermediates derived from acetoacetanilides,9 C(α),N-benzoyl hydrazones or C(α),N-carboalkoxy hydrazones10 with methyl thiosalicylates also lead to the formation of 4H-thiochromen-4-ones. Just recently, a synthesis via intramolecular thiolester carbonyl olefination using N-phenyl(triphenylphosphorany1idene)ethenimine as a starting material was reported.11
However, most of these methods could not successfully be applied to the synthesis of methoxy-substituted thioflavones.12 Either the reported strategies require many steps or the starting materials are difficult to synthesize. Hence, due to the interesting pharmacological potential of 4H-thiopyran-4-one derivatives and as a part of our program to develop multi-component syntheses of heterocycles,13 we recently communicated a novel three-component synthesis of 4H-thiochromen-4-ones with a highly flexible substitution pattern.14 Here, we report the methodological extension of this innovative strategy to various classes of annelated 4H-thiopyran-4-ones and their optical properties.
Results and discussion
Synthesis
Alkynones are accessible by Sonogashira coupling15 of acid chlorides with terminal alkynes.16 However, a modified protocol with THF as the solvent using only one stoichiometrically necessary equivalent of triethylamine as the base has proven to be most favorable for successful coupling.17 In turn, the resulting alkynones are reactive Michael acceptors ready to react with various nucleophiles18 and dipolarophiles19 in a one-pot fashion to give a plethora of heterocycles.Retrosynthetically, the formation of annelated 4H-thiopyran-4-ones in a one-pot fashion can be perceived via a sequence of intramolecular nucleophilic aromatic substitutions (SNAr) and Michael addition of hydrosulfide yielding alkynones 5 (Scheme 1). These alkynones should be accessible by Sonogashira coupling of 2-halobenzoyl chlorides 1 and terminal alkynes 2. Prior to our studies, only a few examples of SNAr-Michael addition of hydrosulfide to alkynones had been reported.20 Hence, the remaining methodological challenge is the development of a general rapid one-pot three-component coupling-addition-SNAr (CASNAR) process.
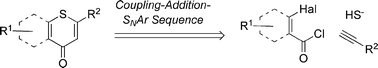 | ||
| Scheme 1 Mechanistic rationale of the coupling-addition-SNAr (CASNAR) sequence. | ||
Therefore, ortho-haloaroyl chloride 1a or 1b was reacted with ethynyl benzene (2a) under modified Sonogashira conditions for 1 h at room temperature to furnish the expected alkynones 5. Subsequently, sodium sulfide nonahydrate (3) and ethanol were added to the reaction mixture and heated in a microwave cavity to give 4H-thiochromen-4-one 4a (Scheme 2, Table 1). Mechanistically, it is likely that the rapid Michael addition of the hydrosulfide ion to the alkynone 5 to give adduct 6 precedes the cyclizing SNAr, assisted by Pd and/or Cu catalysis.
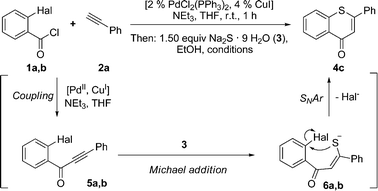 | ||
| Scheme 2 Mechanistic rationale of the coupling-addition-SNAr (CASNAR) sequence. | ||
| Entry | 1 (equiv.) | 2c (equiv.) | 3 (equiv.) | Addition-SNAr | 4a Yield (%)a |
|---|---|---|---|---|---|
| a Isolated yield after column chromatography on silica gel (ethyl acetate/hexanes).b Control experiment in an oil bath. | |||||
| 1 | 1a (1.00) | 1.00 | 1.10 | 90 °C, 120 minb | 52 |
| 2 | 1a (1.00) | 1.00 | 1.10 | 90 °C, 90 min | 58 |
| 3 | 1a (1.00) | 1.00 | 1.50 | 90 °C, 90 min | 59 |
| 4 | 1a (1.25) | 1.00 | 1.50 | 90 °C, 90 min | 73 |
| 5 | 1b (1.25) | 1.00 | 1.50 | 90 °C, 90 min | 40 |
Dielectric heating proved to be more efficient than conductive heating in an oil bath (entries 1 and 4), but only in the concluding addition-substitution step. Sonogashira coupling proceeded smoothly at room temperature and without formation of side products. Fluorine as a leaving group was superior to chlorine (entries 4 and 5). Hence, the stage was set for a novel microwave-assisted three-component coupling-addition-SNAr (CASNAR) synthesis of 4H-thiochromen-4-ones 4 in a one-pot fashion.
Therefore, the coupling of 2-halobenzoyl chlorides 1 with terminal alkynes 2 under modified Sonogashira conditions for 1 h at room temperature, and addition of sodium sulfide nonahydrate (3) and ethanol, with reaction for 90 min at 90 °C in a microwave cavity resulted in the formation of substituted 4H-thiochromen-4-ones 4 in moderate to good yields as bright yellow solids (Scheme 3, Table 2).
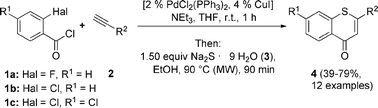 | ||
| Scheme 3 Coupling-addition-substitution (CASNAR) sequence to yield substituted 4H-thiochromen-4-ones 4. | ||
| Entry | Aroyl chloride 1 | Alkyne 2 | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 1a | 2a R2 = H [TMS] | 4a | 39 |
| 2 | 1a | 2b R2 = C6H5 | 4b | 73 |
| 3 | 1a | 2c R2 = 4-C6H4C(CH3)3 | 4c | 76 |
| 4 | 1a | 2d R2 = 4-C6H4OCH3 | 4d | 77 |
| 5 | 1a | 2e R2 = 3,4-C6H3(OCH3)2 | 4e | 73 |
| 6 | 1a | 2f R2 = 4-C6H4Cl | 4f | 52 |
| 7 | 1a | 2g R2 = nbutyl | 4g | 59 |
| 8 | 1a | 2h R2 = ferrocenyl | 4h | 63 |
| 9 | 1a | 2i R2 = 6-methoxynaphthalen-2-yl | 4i | 51 |
| 10 | 1c | 2a R2 = H [TMS] | 4j | 35 |
| 11 | 1c | 2b R2 = C6H5 | 4k | 61 |
| 12 | 1c | 2j R2 = 4-C6H4CH3 | 4l | 48 |
| 13 | 1c | 2c R2 = 4-C6H4C(CH3)3 | 4m | 66 |
| 14 | 1c | 2e R2 = 3,4-C6H3(OCH3)2 | 4n | 59 |
| 15 | 1c | 2k R2 = 4-C6H4F | 4o | 47 |
Interestingly, 2,4-dichlorobenzoyl chloride 1c selectively furnished 7-chloro-substituted products (Table 2, entries 10–15) without competing SNAr at the para-position. In comparison, existing protocols for the synthesis of 4H-thiochromen-4-one 4c generally require several steps, sometimes harsh reaction conditions, longer reaction times and give significantly lower overall yields (Table 3). With the method described herein, the synthesis and isolation of 4c is possible within 2 h, starting from commercially available starting materials.
The scope was then expanded to 2-chloronicotinoyl chloride (1d), 2,5-dichlorothiophene-3-carbonyl chloride (1e) and 3-chlorobenzo[b]thiophene-2-carbonyl chloride (1f) as heteroaroyl chlorides, giving rise to the formation of the class of 4H-thiopyrano[2,3-b]pyridin-4-ones 7 (Scheme 4, Table 4), 2-chloro-4H-thieno[2,3-b]thiopyran-4-ones 8 and 7H-benzo-[b]thieno[3,2-b]thiopyran-7-ones 9 (Scheme 4, Table 5) in modest to moderate yields.
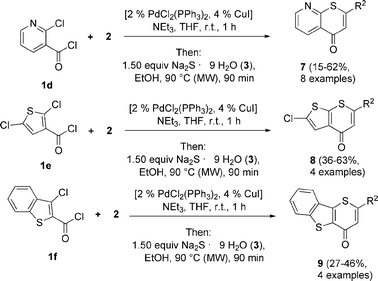 | ||
| Scheme 4 Coupling-addition-substitution (CASNAR) sequences to heteroaryl annelated 4H-thiopyran-4-ones 7, 8 and 9. | ||
| Entry | Alkyne 2 | Product | Yield (%) |
|---|---|---|---|
| 1 | 2a R2 = H [TMS] | 7a | 62 |
| 2 | 2b R2 = C6H5 | 7b | 55 |
| 3 | 2j R2 = 4-C6H4CH3 | 7c | 23 |
| 4 | 2c R2 = 4-C6H4C(CH3)3 | 7d | 15 |
| 5 | 2f R2 = 4-C6H4Cl | 7e | 31 |
| 6 | 2h R2 = ferrocenyl | 7f | 19 |
| 7 | 2g R2 = nbutyl | 7g | 17 |
| 8 | 2l R2 = cyclopropyl | 7h | 53 |
| Entry | Heteoaroyl chloride 1 | Alkyne 2 | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 1e | 2b R2 = C6H5 | 8a | 40 |
| 2 | 1e | 2c R2 = 4-C6H4C(CH3)3 | 8b | 63 |
| 3 | 1e | 2e R2 = 3,4-C6H3(OCH3)2 | 8c | 51 |
| 4 | 1e | 2g R2 = nbutyl | 8d | 36 |
| 5 | 1f | 2b R2 = C6H5 | 9a | 46 |
| 6 | 1f | 2c R2 = 4-C6H4C(CH3)3 | 9b | 41 |
| 7 | 1f | 2k R2 = 4-C6H4F | 9c | 27 |
| 8 | 1f | 2l R2 = cyclopropyl | 9d | 32 |
Since standard syntheses fail, to the best of our knowledge, only two examples of 4H-thiopyrano[2,3-b]pyridin-4-ones 7 have been reported in the literature prior to our studies.21 4H-thieno[2,3-b]thiopyran-4-ones 8 and 7H-benzo-[b]thieno[3,2-b]thiopyran-7-ones 9 are hitherto unknown heterocycles.
The structures of all annelated 4H-thiopyran-4-one derivatives 4, 7, 8 and 9 were unambiguously supported by spectroscopic (1H, 13C and DEPT NMR experiments, IR, UV/Vis, mass spectrometry) and combustion analyses, and later by X-ray structure analyses for the compounds 4i and 7h (Fig. 1 and 2§).
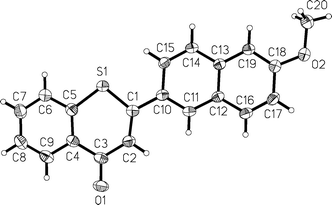 | ||
| Fig. 1 X-Ray structure of 4i. | ||
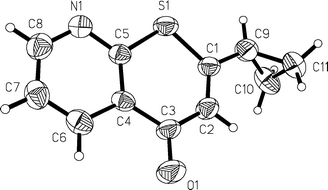 | ||
| Fig. 2 X-Ray structure of 7h. | ||
Methodologically, this novel one-pot three-component CASNAR synthesis of annelated 4H-thiopyran-4-ones 4, 7, 8 and 9 proceeds efficiently under mild reaction conditions and with a broad variety of structurally and electronically diverse alkynes. Aliphatic, aromatic, heteroaromatic, and even ferrocenyl- or heteroatom-substituted alkynes can be successfully transformed in this reaction. If trimethylsilyl acetylene is applied, the TMS group is presumably lost during the Michael addition and the resulting 4H-thiopyran-4-one then bears a hydrogen atom at the R2 position. Expectedly, free amino or hydroxy groups on the alkynes interfering with the electrophilic aroyl chloride reactivity should be protected. According to the substitution pattern of the compounds 4, 7, 8 and 9, it becomes apparent that the electronic structure of substituent R2 exerts the methodological limitation of the terminal Michael addition-SNAr steps. Unfortunately, alkynones resulting from the coupling with alkoxy, acetal and electron poor aromatic substituents on the alkyne 2 were not successfully transformed into annelated 4H-thiopyran-4-ones.
Optical spectroscopy and electronic structure
Expectedly, UV/Vis absorption spectra of the annelated 4H-thiopyran-4-one derivatives 4, 7, 8 and 9 are quite similar (Table 6). For 4H-thiochromen-4-ones 4, three distinct maxima λmax,abs, appear in the spectra, whereas the longest wavelength band occurs at ∼345 nm causing the bright yellow color of the compounds (Fig. 3). Depending on the substitution pattern, the second absorption band is not pronounced (∼305 nm). The absorption band with the highest extinction coefficient is located at ∼275 nm. This feature is characteristic for aromatic cores and can be assigned to substituents R2.| Compound | λmax/nm (ε/mol−1 L cm−1) |
|---|---|
| 4a | 249 (7300), 287 (2300), 337 (4400) |
| 4b | 272 (30900), 301 (6900), 344 (11700) |
| 4c | 280 (17300), 306 (8200), 345 (7500) |
| 4d | 265 (33700), 319 (31900), 342 (27700) |
| 4e | 255 (23000), 284 (11400), 339 (17700) |
| 4f | 277 (42700), 302 (14400), 345 (15300) |
| 4g | 249 (9600), 286 (1400), 337 (5500) |
| 4h | 260 (20600), 285 (18400), 307 (11900), 347 (9800), 389 (2200), 486 (1800) |
| 4i | 249 (22000), 329 (14600), 337 (16000) |
| 4j | 255 (24100), 337 (10700) |
| 4k | 272 (31600), 304 (8200), 343 (11000) |
| 4l | 279 (20100), 309 (9700), 343 (8700) |
| 4m | 278 (23400), 307 (11800), 343 (9800) |
| 4n | 259 (23300), 282 (11300), 341 (17100) |
| 4o | 273 (41900), 305 (12000), 343 (15100) |
| 7a | 249 (22000), 329 (14600), 337 (16000) |
| 7b | 269 (18700), 297 (10000), 345 (8800) |
| 7c | 274 (12300), 306 (10400), 345 (7300) |
| 7d | 274 (23800), 306 (21900), 345 (15400) |
| 7e | 271 (21200), 299 (14800), 343 (10100) |
| 7f | 263 (17400), 310 (14600), 345 (10400), 395 (2300), 493 (2200) |
| 7g | 252 (7800), 285 (2000), 335 (5700) |
| 7h | 259 (18700), 329 (9600), 338 (10500) |
| 8a | 277 (24500), 317 (8500) |
| 8b | 284 (23700), 316 (10200) |
| 8c | 254 (20900), 299 (19300), 325 (19700) |
| 8d | 248 (21600), 309 (12800) |
| 9a | 260 (21300), 286 (27500), 298 (26700), 307 (26600), 351 (13300) |
| 9b | 261 (9000), 287 (12400), 299 (13400), 308 (13900), 350 (6400) |
| 9c | 259 (25100), 286 (31500), 298 (30600), 308 (30900), 351 (15400) |
| 9d | 250 (17000), 259 (18600), 266 (15300), 276 (17600), 295 (15400), 305 (18400), 334 (10500), 347 (12000) |
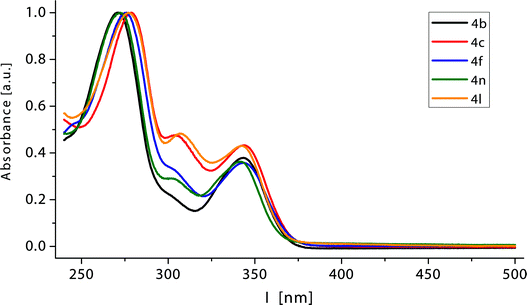 | ||
| Fig. 3 Normalized UV/Vis spectra of selected 4H-thiochromen-4-ones 4 (recorded in CH2Cl2, c0 = 10−3 M; T = 293 K). | ||
Interestingly, all annelated 4H-thiopyran-4-one derivatives can be easily protonated upon addition of acids. Upon adding aliquots of trifluoroacetic acid to a solution of 4, 7, 8 or 9 in dichloromethane, a significant change in the absorption behavior of the compound occurred (Fig. 4).
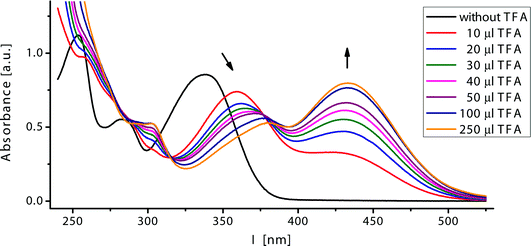 | ||
| Fig. 4 Titration of 4e with trifluoroacetic acid (recorded in CH2Cl2, T = 298 K). | ||
Upon protonation, the λmax,abs value shifted from 345 to 370–440 nm. Interestingly, this was not a static phenomenon. Increasing aliquots of acid increased the bathochromic shifts, even visible with the naked eye—the color changed from yellow to orange. In addition, protonation caused orange luminescence (λmax,em = 460 nm, see the ESI†) of the protonated heterocycle 4e-H+, with a fluorescence quantum yield lower than 1%, whereas the free base system 4e was essentially nonluminescent. This pronounced bathochromic halochromicity of the absorption band can be rationalized by the generation of a cyanine-type push-pull system upon protonation. According to the HSAB principle,22 protonation occurs preferentially at the harder carbonyl oxygen atom and not at the soft sulfur atom (Scheme 5).
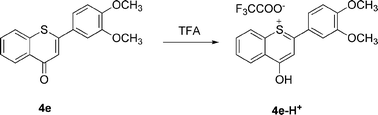 | ||
| Scheme 5 Protonation of compound 4e with trifluoroacetic acid. | ||
This qualitative rationalization is also supported by DFT calculations (B3LYP/6-311G++)23 on the thiopyranones and the corresponding related pyranones indicating a strongly zwitterionic character in the electronic ground state of the former (Fig. 5). The computations clearly show that the sulfur atom in the 4H-thiochromen-4-one structure possesses a significant positive partial charge of around +0.45 C, whereas the oxygen atom in the chromen-4-ones clearly has a negative partial charge of around −0.57 C. Compared to the natural product class of the flavones this connotes a complete inversion of the polarity of the ring system. The oxygen atoms of the carbonyl group in both systems expectedly display a negative partial charge of around −0.39 C.
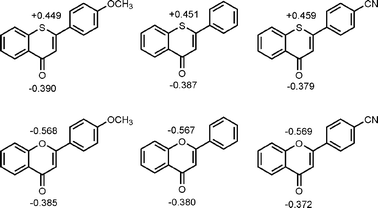 | ||
| Fig. 5 DFT-computed charge distribution in selected thiochromenones (top row) and their flavone analogues (bottom row) (atomic partial charges are given). | ||
Conclusions
The CASNAR sequence represents a straightforward and rapid method access to 4H-thiopyran-4-ones in the sense of a consecutive one-pot process and with a broad scope in the starting materials. Methoxy groups, which proved to be unfavorable in standard syntheses, are perfectly tolerated in this sequence. Furthermore, as a consequence of pronounced zwitterionic character of the computed electronic ground state, intense bathochromic halochromicity of the longest wavelength absorption bands with concomitant weak orange fluorescence can be observed upon protonation. This novel one-pot protocol now opens new avenues to related classes of annelated heterocycles. Studies addressing the detailed photophysics and the biological activity of 4H-thiopyran-4-ones are currently under investigation.Experimental
All reactions involving water-sensitive compounds were carried out in flame-dried glassware under an argon atmosphere. Reagents and catalysts were purchased reagent grade and used without further purification. Solvents were dried by a solvent purification system. Flash column chromatography: silica gel 60, mesh 230–400. TLC: silica gel plates (60 F254). 1H, 13C, DEPT, NOESY, COSY, HMQC and HMBC spectra were recorded with a 500 MHz NMR spectrometer using CDCl3 as the solvent. The assignments of Cquat, CH, CH2 and CH3 were made on the basis of DEPT spectra. Mass spectra were recorded with a quadruple spectrometer. The melting points are uncorrected. IR spectra were taken on a FT-IR-spectrometer in KBr pellets and are reported in cm−1. Elemental analyses were carried out in the microanalytical laboratory of the Pharmazeutisches Institut of the Heinrich-Heine-Universität Düsseldorf. Dielectric heating was performed in a single-mode microwave cavity (Discover Labmate, CEM GmbH, Kamp-Lintfort) producing continuous irradiation at 2450 MHz. The applied temperatures for microwave heating were held constant over the indicated reaction time by the implemented regulation system. Temperature and pressure as well as the cooling curve were monitored by the integrated controlling device.General procedure for the synthesis of annelated 4H-thiochromen-4-ones 4, 7, 8 and 9
In a 10 ml microwave tube, PdCl2(PPh3)2 (15 mg, 0.02 mmol) and CuI (8 mg, 0.04 mmol) were dissolved in degassed THF (4 mL). Then, to this orange solution, acid chloride 1 (1.25 mmol), alkyne 2 (1.00 mmol) and triethylamine (1.05 mmol) were added. The reaction mixture was stirred at room temperature for 1 h. Finally, sodium sulfide nonahydrate (3) followed by ethanol (1 mL) were added to this suspension and the reaction mixture was heated at 90 °C in the microwave cavity for 90 min. After cooling to room temperature, the solvent was removed under reduced pressure and the crude products were purified by silica gel flash column chromatography (hexane/ethyl acetate) to afford the analytically pure 4H-thiochromen-4-ones 4, 7, 8 and 9 (for experimental details see the ESI, Tables 1–3†).2-(4-Methoxyphenyl)-4H-thiochromen-4-one (4d)
Yellow solid, mp 97 °C. 1H NMR (500 MHz, CDCl3): δ 3.88 (s, 3 H), 7.01 (d, 3J = 8.8 Hz, 2 H), 7.20 (s, 1 H), 7.54 (ddd, 3J = 8.2 Hz, 3J = 6.9 Hz, 4J = 1.3 Hz, 1 H), 7.61 (ddd, 3J = 8.2 Hz, 3J = 6.9 Hz, 4J = 1.3 Hz, 1 H), 7.64-7.67 (m, 3 H), 8.54 (dd, 3J = 8.1 Hz, 4J = 1.3 Hz, 1 H). 13C NMR (125 MHz, CDCl3): δ 55.5 (CH3), 114.7 (2 CH), 122.2 (CH), 126.4 (CH), 127.6 (CH), 128.3 (2 CH), 128.5 (CH), 128.8 (Cquat), 130.9 (Cquat), 131.5 (CH), 137.6 (Cquat), 152.7 (Cquat), 161.9 (Cquat), 180.9 (Cquat). EI MS (Rf = 20.3 min, 70 eV, m/z (%)): 269 (19), 268 (M+, 100), 267 (17), 240 (39), 225 (25), 197 (11), 136 (34), 132 (56), 120 (10), 117 (14), 108 (29), 89 (22), 69 (10), 63 (13). IR (KBr): ṽ = 1628 cm−1 (s), 1605 (s), 1551 (w), 1509 (s), 1438 (w), 1336 (m), 1311 (w), 1269 (s), 1246 (w), 1184 (m), 1130 (w), 1117 (w), 1103 (w), 1020 (m), 862 (w), 831 (s), 798 (m), 774 (m), 732 (m), 666 (w), 623 (w), 568 (w), 517 (m). Anal. calcd for C16H12O2S (268.3): C 71.62, H 4.51; found: C 71.66, H 4.21%.4H-Thiopyrano[2,3-b]pyridin-4-one (7a)
Yellow solid, mp 135 °C. 1H NMR (500 MHz, CDCl3): δ 7.04 (d, 3J = 10.6 Hz, 1 H), 7.50 (dd, 3J = 8.1 Hz, 3J = 4.5 Hz, 1 H), 7.93 (d, 3J = 10.6 Hz, 1 H), 8.77 (dd, 3J = 8.1 Hz, 4J = 1.8 Hz, 1 H), 8.80 (dd, 3J = 4.5 Hz, 4J = 1.8 Hz, 1 H). 13C NMR (125 MHz, CDCl3): δ 123.0 (CH), 126.1 (CH), 129.7 (Cquat), 136.9 (CH), 139.5 (CH), 152.7 (CH), 158.8 (Cquat), 180.6 (Cquat). EI MS (70 eV, m/z (%)): 164 (11), 163 (M+, 100), 137 (18), 135 (28), 109 (11). IR (KBr): ṽ = 1625 cm−1 (s), 1579 (m), 1509 (w), 1450 (w), 1398 (s), 1366 (m), 1305 (w), 1265 (w), 1157 (w), 1072 (w), 842 (w), 791 (m), 717 (w), 670 (w). Anal. calcd for C8H5NOS (163.2): C 58.88, H 3.09, N 8.58; found: C 58.81, H 3.18, N 8.45%.6-(4-tButylphenyl)-2-chloro-4H-thieno[2,3-b]thiopyran-4-one (8b)
Brown solid, mp. 157 °C. 1H NMR (500 MHz, CDCl3): δ 1.35 (s, 9 H), 7.16 (s, 1 H), 7.51 (d, 3J = 8.8 Hz, 2 H), 7.55 (d, 3J = 8.8 Hz, 2 H), 7.57 (s, 1 H). 13C NMR (125 MHz, CDCl3): δ 31.1 (3 CH3), 34.9 (Cquat), 124.1 (CH), 124.4 (CH), 126.4 (2 CH), 126.6 (2 CH), 131.0 (Cquat), 133.0 (Cquat), 137.2 (Cquat), 142.6 (Cquat), 151.5 (Cquat), 154.6 (Cquat), 175.7 (Cquat). EI MS (Rf = 32.0 min, 70 eV, m/z (%)): 336 (37Cl-M+, 32), 335 (12), 334 (35Cl-M+, 62), 321 (40), 320 (19), 319 (100), 290 (11), 263 (16), 207 (11), 179 (13), 178 (13), 177 (25), 176 (20), 148 (18), 147 (15), 146 (27), 128 (15), 127 (11), 115 (27), 69 (19). IR (KBr): ṽ = 2961 cm−1 (w), 1607 (s), 1508 (w), 1436 (w), 1414 (w), 1361 (w), 1271 (w), 1240 (w), 1114 (w), 1001 (w), 902 (w), 879 (w), 849 (w), 822 (w), 714 (w), 663 (w), 587 (w), 522 (w). Anal. calcd for C17H15ClOS2 (334.9): C 60.97, H 4.51; found: C 60.80, H 4.60%.6-Phenyl-4H-benzothieno[2,3-b]thiopyran-4-one (9a)
Beige solid, mp 157 °C. 1H NMR (500 MHz, CDCl3): δ 7.30 (s, 1 H), 7.49-7.53 (m, 4 H), 7.55–7.59 (m, 1 H), 7.68–7.70 (m, 2 H), 7.96–7.98 (m, 2 H). 13C NMR (125 MHz, CDCl3): δ 122.4 (CH), 132.7 (CH), 124.6 (CH), 125.2 (CH), 127.2 (2 CH), 128.6 (CH), 129.4 (2 CH), 130.8 (CH), 135.3 (Cquat), 136.1 (Cquat), 136.3 (Cquat), 137.3 (Cquat), 140.7 (Cquat), 152.1 (Cquat), 176.9 (Cquat). EI MS (70 eV, m/z (%)): 296 (12), 295 (20), 294 (M+, 100), 266 (33), 192 (13), 164 (18), 133 (20), 120 (21), 40 (56). IR (KBr): ṽ = 1620 cm−1 (s), 1593 (s), 1544 (m), 1528 (w), 1499 (m), 1443 (m), 1345 (m), 1300 (m), 1242 (w), 1085 (m), 1052 (m), 951 (m), 864 (m), 753 (s), 727 (m), 684 (s), 578 (s). Anal. calcd for C17H10OS2·1/8 CH2Cl2 (294.4 + 10.6): C 67.44, H 3.49; found: C 67.49, H 3.79%.Acknowledgements
The authors gratefully acknowledge the Fonds der Chemischen Industrie and CEM for a research corporation.Notes and references
- H. Nakazumi, T. Ueyama and T. Kitao, J. Het. Chem., 1985, 22, 1593 Search PubMed.
- H. Nakazumi, T. Ueyama and T. Kitao, J. Het. Chem., 1984, 21, 193 Search PubMed.
- J. Couquelet, P. Tronche, P. Niviere and G. Andraud, Trav. Soc. Pharm. Montpellier, 1963, 23, 214 Search PubMed.
- M. H. Holshouser, L. J. Loeffler and I. H. Hall, J. Med. Chem., 1981, 24, 853 CrossRef CAS.
- R. K. Razdan, R. J. Bruni, A. C. Mehta, K. K. Weinhardt and Z. B. Papanastassiou, J. Med. Chem., 1978, 21, 643 CrossRef CAS.
- D. Dhanak, R. M. Keenan, G. Burton, A. Kaura, M. G. Darcy, D. H. Shah, L. H. Ridgers, A. Breen, P. Lavery, D. G. Tew and A. West, Bioorg. Med. Chem. Lett., 1998, 8, 3677 CrossRef CAS.
- F. Bossert, Liebigs Ann. Chem., 1964, 40, 680; S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59 CAS; H. Nakazumi, S. Wanatabe, T. Kitaguchi and T. Kitao, Bull. Chem. Soc. Jpn., 1990, 63, 847 CAS.
- W. E. Truce and D. L. Goldhamer, J. Am. Chem. Soc., 1959, 81, 5795 CrossRef CAS; K. Buggle, J. J. Delahunty, E. M. Philbin and N. D. Ryan, J. Chem. Soc. C, 1971, 3168 RSC.
- A. J. Angel, A. E. Finefrock, K. L. French, D. R. Hurst, A. R. Williams, M. E. Rampey, S. L. Studer-Martinez and C. F. Beam, Can. J. Chem., 1999, 77, 94 CrossRef CAS.
- K. L. French, A. J. Angel, A. R. Williams, D. R. Hurst and C. F. Beam, J. Heterocycl. Chem., 1998, 35, 45 CrossRef CAS.
- P. Kumar, A. T. Rao and B. Pandey, J. Chem. Soc., Chem. Commun., 1992, 1580 RSC; P. Kumar and M. S. Bodas, Tetrahedron, 2001, 57, 9755 CrossRef CAS.
- D. H. Wadsworth and M. R. Detty, J. Org. Chem., 1980, 45, 4611 CrossRef CAS.
- For reviews, see: B. Willy and T. J. J. Müller, ARKIVOC, 2008, Part (i), 195 Search PubMed; B. Willy and T. J. J. Müller, Curr. Org. Chem., 2009, 13, 1777, DOI:10.2174/138527209789630479.
- B. Willy and T. J. J. Müller, Synlett, 2009, 1255 CAS.
- For lead reviews on Sonogashira couplings, see e.g.: S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 Search PubMed; K. Sonogashira, in Metal catalyzed cross-coupling reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 203 CrossRef CAS; H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 CrossRef CAS; L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 Search PubMed.
- Y. Toda, K. Sonogashira and N. Hagihara, Synthesis, 1977, 777 CrossRef CAS.
- A. S. Karpov and T. J. J. Müller, Org. Lett., 2003, 5, 3451 CrossRef CAS; D. M. D'Souza and T. J. J. Müller, Nat. Protoc., 2008, 3, 1660 Search PubMed.
- T. J. J. Müller, Chimica Oggi/Chemistry Today, 2007, 25, 70 Search PubMed; T. J. J. Müller, Targets in Heterocyclic Systems, 2006, 10, 54 Search PubMed; B. Willy and T. J. J. Müller, Eur. J. Org. Chem., 2008, 4157 CrossRef CAS.
- B. Willy, F. Rominger and T. J. J. Müller, Synthesis, 2008, 293 CAS; B. Willy, W. Frank, F. Rominger and T. J. J. Müller, J. Organomet. Chem., 2009, 694, 942 CrossRef CAS; A. V. Rotaru, I. D. Druta, T. Oeser and T. J. J. Müller, Helv. Chim. Acta, 2005, 88, 1798 CrossRef.
- M. S. Shvartsberg and I. D. Ivanchikova, ARKIVOC, 2003,(13), 87 CAS; I. D. Ivanchikova and M. S. Shvartsberg, Russ. Chem. Bull., 2004, 53, 2303 Search PubMed.
- A. Couture, P. Grandclaudon and E. Huguerre, Synthesis, 1989, 456 CrossRef CAS; J. Becher, M. C. Christensen, M. C. Möller and J. Winckelmann, Sulfur Lett., 1982, 1, 43.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B.3), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray coordinates, molecular modeling coordinates and fluorescence spectrum of 4e-H+. CCDC reference numbers 754614–754615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b917627f |
| ‡ X-Ray structure analyses. |
| § CCDC 745614 (4i) and CCDC 745615 (7h) contain supplementary crystallographic data for these structures. For crystallographic date in CIF or other electronic format see DOI: 10.1039/b917627f. Data were collected on a Bruker APEX diffractometer. Mo Kα radiation (λ = 0.71073 Å) was used in all cases, and the intensities were corrected for absorption effects using SADABS based on the laue symmetry of the reciprocal space. The structures were solved by direct methods and refined against F2 with a full matrix least square algorithm using the SHELXTL software package. Hydrogen atoms were considered at calculated positions and refined using appropriate riding models. Relevant crystal and data collection parameters for the individual structures are given below. Crystal data: 4i: C20H14O2S, M = 318.38, monoclinic, space group P21/n, a = 13.6458(10), b = 5.7353(3), c = 19.9386(14) Å, β = 106.548(8)°, V = 1495.82(18) Å3, T = 291(2) K, Z = 4, ρ = 1.414 g cm−3, dimensions 0.60 × 0.35 × 0.12 mm3, μ = 0.223 mm−1, 0.3° omega-scans, 18 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 476 reflections measured, 2629 unique [
476 reflections measured, 2629 unique [| This journal is © The Royal Society of Chemistry 2010 |