The stability of imine-containing dynamic [2]rotaxanes to hydrolysis†
Ken Cham-Fai Leung*ab, Wing-Yan Wonga, Fabio Aricób, Philip C. Haussmannb and J. Fraser Stoddart*bc
aCenter of Novel Functional Molecules, Department of Chemistry, The Chinese University of Hong Kong, Shatin, NT, Hong Kong SAR, P. R. China. E-mail: cfleung@cuhk.edu.hk; Fax: (+852) 2603-5057
bCalifornia NanoSystems Institute and Department of Chemistry and Biochemistry, The University of California, Los Angeles, 405 Hilgard Avenue, Los Angeles, California 90095, USA
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: stoddart@northwestern.edu; Fax: (+1) 847-491-1009
First published on 20th November 2009
Abstract
Large amounts (>100 mol equivalents) of water are required to effect by hydrolysis the partial disassembly of the rings from the dumbbell components of two dynamic [2]rotaxanes. The two dynamic [2]rotaxanes are comprised of [24]crown-8 rings—each of which incorporate two imine bonds—encircling a dumbbell component composed of a dibenzylammonium ion in which each of the two benzyl substituents carries two methoxyl groups attached to their 3- and 5-positions. A mechanism for the partial disassembly of the two dynamic [2]rotaxanes, involving the cleavage of the kinetically labile imine bonds by water molecules, is proposed. The most important experimental observation to be noted is the fact that the hydrolysis of the macrocyclic diimines, associated with the templating –CH2NH2+CH2–centres in the middle of their dumbbells, turns out to be an uphill task to perform in the face of the molecular recognition provided by strong [N+–H⋯O] hydrogen bonds and weaker, yet not insignificant, [C–H⋯O] interactions. The dynamic nature of the imine bond formation and hydrolysis is such that the acyclic components produced during hydrolysis of the imine bonds can be enticed to cyclise once again around the –CH2NH2+CH2–template, affording the [2]rotaxanes. The reluctance of imine bonds, present in substantial numbers in larger molecular and extended structures, is significant when it comes to exercising dynamic chemistry in compounds where multiple imine bonds are present.
Introduction
Recently, dynamic covalent chemistry1 (DCC) has attracted increasing attention on account of its usefulness in preparing complex products, such as molecular Borromean rings,2 dendrimers3 and other exotic compounds4 with high efficiencies using one-pot, templated self-assembly processes.5 The nearly quantitative formation of the (super)structures with mechanically intertwined and interlocked entities indicates that a template effect5a is highly efficient, yielding thermodynamically stable supramolecular intermediates on the way to the desired entities. The main advantage of the thermodynamic process over the kinetic one is that the former operates under equilibrium control such that the formation of undesired, kinetically competitive intermediates can eventually make their way back to the most energetically-favored product by means of error checking and proof-reading.1g DCC relies upon the use of dynamic covalent bonds that form in a reversible manner. Common dynamic equilibrating (reversible) reactions exploited in such syntheses include olefin metathesis,6 and disulfide7 and imine formation.8Generally, imines are relatively unstable since they are susceptible to rapid hydrolysis in the presence of water.9 Saggiomo and Lüning10 have reported recently, however, that imine-containing macrocycles possess remarkable stabilities in the presence of water. Other molecular systems,11 involving dative bonds between imines and transition metal ions,12 can also experience increased imine stabilities in water. It follows that a careful investigation of the dissociative mechanisms associated with the formation and hydrolysis of dynamic rotaxanes containing imine bonds is important in the context of the design of functional rotaxanes13 that can be obtained in high yields by DCC and possess potentially useful applications.14
In this paper, we report an investigation of the dissociation of dynamic, imine-containing rotaxanes8a in the presence of an excess of water. It transpires that not only is the hydrolysis far from complete, but also that the dissociated molecular components obtained at equilibrium can be assembled rather easily to afford the original [2]rotaxanes by removing the water from the aqueous solutions with appropriate drying agents.
Results and discussion
By employing DCC associated with reversible imine bond formation, we reported8c previously that the dynamic [2]rotaxane 1a–H·PF6 can be formed (Scheme 1) by condensation between three different components in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio—the diformylpyridine 2, the diamine 3a and the already stoppered dialkylammonium salt 4–H·PF6—in dry MeCN (60 mM) in over 90% yield in one pot. Similarly, another dynamic [2]rotaxane 1b–H·PF6 can also be formed using the same condensation method with 2, the diamine 3b and 4–H·PF6 in a 1:1:1 ratio. Moreover, we found out experimentally that the condensations occur effectively in both MeCN and MeNO2 (60 mM). The encirclement of a dialkylammonium ion by the imine-containing [24]crown-8 macrocycle can be regarded as a thermodynamically controlled template-directed self-assembly and amplification process. Since the dialkylammonium ion template 4–H·PF6 fits well into the macrocycle, which is generated from the [1 + 1] condensation between 2 and 3a/b, the [2]rotaxanes 1a/b–H·PF6 can be amplified as the major products in the dynamic competitive equilibrium mixture. Subsequently, the [2]rotaxanes 1a/b–H·PF6 were characterised by 1H NMR spectroscopy and matrix-assisted laser-desorption ionization mass spectrometry (HR-MALDI-MS) (mass spectra not shown). The 1H NMR spectra were obtained after 15 min of mixing between the free components. The 1H NMR spectra of the dynamic [2]rotaxanes reveal that the characteristic benzylic methylene proton signals, adjacent to the ammonium ion centre, observed at 3.8–3.9 ppm of 4–H·PF6 are shifted downfield to 4.5–4.6 ppm upon rotaxane formation. Moreover, the characteristic peaks for the protons on the ethyleneglycol chain are shifted upfield (see the ESI†). The [2]rotaxanes are stable entities on account of [N+–H⋯O] and [N+–C–H⋯X] (X
:1:1 ratio—the diformylpyridine 2, the diamine 3a and the already stoppered dialkylammonium salt 4–H·PF6—in dry MeCN (60 mM) in over 90% yield in one pot. Similarly, another dynamic [2]rotaxane 1b–H·PF6 can also be formed using the same condensation method with 2, the diamine 3b and 4–H·PF6 in a 1:1:1 ratio. Moreover, we found out experimentally that the condensations occur effectively in both MeCN and MeNO2 (60 mM). The encirclement of a dialkylammonium ion by the imine-containing [24]crown-8 macrocycle can be regarded as a thermodynamically controlled template-directed self-assembly and amplification process. Since the dialkylammonium ion template 4–H·PF6 fits well into the macrocycle, which is generated from the [1 + 1] condensation between 2 and 3a/b, the [2]rotaxanes 1a/b–H·PF6 can be amplified as the major products in the dynamic competitive equilibrium mixture. Subsequently, the [2]rotaxanes 1a/b–H·PF6 were characterised by 1H NMR spectroscopy and matrix-assisted laser-desorption ionization mass spectrometry (HR-MALDI-MS) (mass spectra not shown). The 1H NMR spectra were obtained after 15 min of mixing between the free components. The 1H NMR spectra of the dynamic [2]rotaxanes reveal that the characteristic benzylic methylene proton signals, adjacent to the ammonium ion centre, observed at 3.8–3.9 ppm of 4–H·PF6 are shifted downfield to 4.5–4.6 ppm upon rotaxane formation. Moreover, the characteristic peaks for the protons on the ethyleneglycol chain are shifted upfield (see the ESI†). The [2]rotaxanes are stable entities on account of [N+–H⋯O] and [N+–C–H⋯X] (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or N) hydrogen bonds, in addition to electrostatic and π–π stacking interactions.8c Thereafter, they were redissolved in MeCN prior to carrying out dissociation studies in the presence of different amounts of water.
O or N) hydrogen bonds, in addition to electrostatic and π–π stacking interactions.8c Thereafter, they were redissolved in MeCN prior to carrying out dissociation studies in the presence of different amounts of water.![Equilibrium for the self-assembly and dissociation of the dynamic [2]rotaxanes 1a/b–H·PF6 and their components 2, 3a/b and 4–H·PF6 depending on the concentration of water present in the reaction mixture.](/image/article/2010/OB/b915864b/b915864b-s1.gif) | ||
| Scheme 1 Equilibrium for the self-assembly and dissociation of the dynamic [2]rotaxanes 1a/b–H·PF6 and their components 2, 3a/b and 4–H·PF6 depending on the concentration of water present in the reaction mixture. | ||
Our investigations have shown that the dissociation of dynamic [2]rotaxanes 1a/b–H·PF6 requires large amounts of water. Thus, 1H NMR spectroscopy was employed for investigating the dissociation of 1a/b–H·PF6, following addition15 of 100, 200, 250 and 500 equivalents of D2O. Whether or not equilibration has been reached, sharp signals in the 1H NMR spectra of the [2]rotaxanes indicate that they are relatively stable kinetically on the NMR timescale at 500 MHz. The stability is presumably the result of the stabilizing multiple noncovalent bonding interactions. By way of an example, Fig. 1 shows the stacked 1H NMR spectra (500 MHz, CD3CN, 298 K, aromatic region, 16.6 mM, b = bound rotaxane and f = free components) of the [2]rotaxane 1a–H·PF6 in the presence of 200 equivalents of D2O. Initially, the NMR spectra of [2]rotaxane 1a–H·PF6 (at 0 min) reveal the proton signals at δ = 7.71 ppm and δ = 8.02 ppm for the bound (b) pyridine ring, at δ = 6.9–7.5 ppm for the bound (b) N,O-substituted aryl ring, and at δ = 6.10 ppm and δ = 6.45 ppm for the bound (b) dimethoxyaryl ring. With increasing time in the presence of water, the characteristic proton signals originating from the aldehyde (δ = 10.12 ppm), the free (f) pyridine ring (δ = 8.22 ppm) and the free (f) dimethoxyaryl ring (δ = 6.58 and 6.62 ppm) in separate cyclic (or acyclic) and dumbbell components were found to have their relative intensities increase, while those signals arising from the rotaxane decreased. The low intensity aldehyde signal at 10.10 ppm can be attributed to the intermediate monoaldehyde 5a/b formed during the hydrolysis of 1a/b–H·PF6. The results indicate that the dynamic [2]rotaxane dissociates slowly into its free components—2, 3a and 4–H·PF6—until the dissociation process has reached equilibrium after 24 h. Furthermore, the broad signal (δ = 9.98 ppm) for the NH2+ centre became diminished with time on account of proton/deuterium exchange. With particular reference to 1a–H·PF6, there were 52, 41 and 14% of the dynamic [2]rotaxane remaining in the mixture after 24 h following the additions, in turn, of 100, 200 and 500 equivalents of D2O. On the other hand, in the case of the dissociation of 1b–H·PF6, there were 19, 8 and 4% of the dynamic [2]rotaxane remaining in the mixture after 24 h following the additions, in turn, of 100, 250 and 500 equivalents of D2O. Moreover, the 1H NMR spectra reveal that the residual dynamic [2]rotaxanes 1a/b–H·PF6 are stable in the CD3CN–D2O mixture after 24 h, reflecting a situation that is able to maintain the integrity of the [2]rotaxane constitution, even in the presence of water.
![Partial 1H NMR spectra (aromatic region, 500 MHz, CD3CN, 298 K, 200 mol equivalents of D2O, total conc. = 16.6 mM) showing the dissociation of the dynamic [2]rotaxane 1a–H·PF6 with time (b = bound rotaxane and f = free components). With increasing time, the characteristic proton signals originating from the aldehyde (δ = 10.12 ppm), the free (f) pyridine ring (δ = 8.22 ppm) and the free (f) dimethoxyaryl ring (δ = 6.62 and 6.58 ppm) in separate cyclic (or acyclic) and dumbbell components were found to have their relative intensities increase while those signals arising from the rotaxane decreased. The results indicate that the dynamic [2]rotaxane dissociates slowly into its free components—2, 3a and 4–H·PF6—until the dissociation process has reached equilibrium after 24 h. With particular reference to 1a–H·PF6, there was 41% of the dynamic [2]rotaxane remaining in the mixture after 24 h.](/image/article/2010/OB/b915864b/b915864b-f1.gif) | ||
| Fig. 1 Partial 1H NMR spectra (aromatic region, 500 MHz, CD3CN, 298 K, 200 mol equivalents of D2O, total conc. = 16.6 mM) showing the dissociation of the dynamic [2]rotaxane 1a–H·PF6 with time (b = bound rotaxane and f = free components). With increasing time, the characteristic proton signals originating from the aldehyde (δ = 10.12 ppm), the free (f) pyridine ring (δ = 8.22 ppm) and the free (f) dimethoxyaryl ring (δ = 6.62 and 6.58 ppm) in separate cyclic (or acyclic) and dumbbell components were found to have their relative intensities increase while those signals arising from the rotaxane decreased. The results indicate that the dynamic [2]rotaxane dissociates slowly into its free components—2, 3a and 4–H·PF6—until the dissociation process has reached equilibrium after 24 h. With particular reference to 1a–H·PF6, there was 41% of the dynamic [2]rotaxane remaining in the mixture after 24 h. | ||
By comparing the integrations of characteristic signals arising from the dynamic rotaxanes 1a/b–H·PF6 and the dissociated free dumbbell 4–H·PF6 from the dissociation study, plots of the [Rotaxane]−1versus time can be obtained. The data, which were obtained (Fig. 2) from dissociations lasting 4–6 h, were best fitted to straight lines in agreement with second-order kinetics for both the a and b series. Rate constants (k2 and k−2), dissociation rates and half-lives (t1/2) for the breakdown of 1a/b–H·PF6 were calculated and are listed in Table 1. When comparing similar starting concentrations of the dynamic rotaxanes, in most cases, the stability to water dissociation of 1a–H·PF6 is higher than that of 1b–H·PF6. This observation may account for the enhanced template binding induced by translocational freedom involving the tetraethyleneglycol chain in 3a than that involving the more rigid 2,6-disubstituted pyridyl moiety in 3b.
![A plot of [Rotaxane]−1versus time, showing the dissociation profile of the dynamic [2]rotaxanes 1a/b–H·PF6 in the presence of different amounts of D2O. For the a series, the concentration of [2]rotaxane is 17.2, 16.6 and 15.2 mM when 100, 200 and 500 equivalents D2O were added, respectively. For the b series, the concentration of [2]rotaxane is 16.6, 15.9 and 14.9 mM when 100, 250 and 500 equivalents D2O were added, respectively. Data points were obtained from analyzing their 1H NMR spectra (500 MHz, CD3CN, 298 K). The data, which were obtained from dissociations lasting 4–6 h, were best fitted to straight lines in agreement with second-order kinetics for both the a and b series.](/image/article/2010/OB/b915864b/b915864b-f2.gif) | ||
| Fig. 2 A plot of [Rotaxane]−1versus time, showing the dissociation profile of the dynamic [2]rotaxanes 1a/b–H·PF6 in the presence of different amounts of D2O. For the a series, the concentration of [2]rotaxane is 17.2, 16.6 and 15.2 mM when 100, 200 and 500 equivalents D2O were added, respectively. For the b series, the concentration of [2]rotaxane is 16.6, 15.9 and 14.9 mM when 100, 250 and 500 equivalents D2O were added, respectively. Data points were obtained from analyzing their 1H NMR spectra (500 MHz, CD3CN, 298 K). The data, which were obtained from dissociations lasting 4–6 h, were best fitted to straight lines in agreement with second-order kinetics for both the a and b series. | ||
| 1a–H·PF6 | 1b–H·PF6 | |||||
|---|---|---|---|---|---|---|
| Equivalents of water (total concentration) | 100 (17.2 mM) | 200 (16.6 mM) | 500 (15.2 mM) | 100 (16.6 mM) | 250 (15.9 mM) | 500 (14.9 mM) |
| k2/M−1 h−1 | 247.4 ± 1.2 | 355.9 ± 1.8 | 1091.2 ± 5.4 | 606.7 ± 2.9 | 1298.5 ± 6.5 | 2354.2 ± 11.7 |
| k−2/M−1 h−1 | 268.0 ± 1.3 | 247.3 ± 1.2 | 177.6 ± 0.9 | 145.0 ± 0.7 | 115.4 ± 0.6 | 82.2 ± 0.4 |
| rate/mM h−1 | 73.2 | 98.1 | 252.1 | 179.5 | 357.8 | 543.9 |
| t1/2/min | 14.1 | 10.2 | 3.6 | 5.8 | 2.8 | 1.7 |
| % Remaining [2]rotaxane in the water–acetonitrile mixture | 52 | 41 | 14 | 19 | 8 | 4 |
From a mechanistic point of view, the ring associated with the [2]rotaxanes 1a/b–H·PF6 will eventually open up (Scheme 2) with only one equivalent of H2O present in the solution to give the monoaldehyde intermediates 5a/b, allowing the dumbbell 4–H·PF6 to dethread from these intermediates on account of the loss of the crown ether's integrity and the associated pre-organisation. Subsequently, the monoaldehyde intermediates 5a/b will be hydrolysed even further right down to the diformylpyridine 2, as well as into the diamines 3a/b with one additional equivalent of H2O. However, the template effect offered by the multiple noncovalent interactions and also the stopper effect may retain the relative stability and integrity of the rotaxanes. That is, at equilibrium, relatively large amounts of H2O are required to hydrolyse the imine bond(s) and to solubilise the cleaved components.
![Plausible mechanism for the dissociation of the dynamic [2]rotaxanes 1a/b–H·PF6 into their components 2, 3a/b, 4–H·PF6, 5a/b and 6a/b. The ring associated with the [2]rotaxanes 1a/b–H·PF6 eventually opens up with only one equivalent of H2O present in the solution to give the monoaldehyde intermediates 5a/b, allowing the dumbbell 4–H·PF6 to dethread from these intermediates on account of the loss of the crown ether's integrity and the associated pre-organisation. Subsequently, the monoaldehyde intermediates 5a/b will be hydrolysed even further right down to the diformylpyridine 2, as well as into the diamines 3a/b with one additional equivalent of H2O. On the other hand, on adding one equivalent of toluidine to the [2]rotaxane solutions, dissociation occurs on account of imine exchange, which results in the fast opening of the [24]crown-8 macrocycles to give the podand-like compounds 6a/b. Toluidine plays an important role in serving as a relatively bulky imine exchange partner with the macrocycle to give a disrupted open crown (podand) that cannot be easily converted back to the [24]crown-8 derivative.](/image/article/2010/OB/b915864b/b915864b-s2.gif) | ||
| Scheme 2 Plausible mechanism for the dissociation of the dynamic [2]rotaxanes 1a/b–H·PF6 into their components 2, 3a/b, 4–H·PF6, 5a/b and 6a/b. The ring associated with the [2]rotaxanes 1a/b–H·PF6 eventually opens up with only one equivalent of H2O present in the solution to give the monoaldehyde intermediates 5a/b, allowing the dumbbell 4–H·PF6 to dethread from these intermediates on account of the loss of the crown ether's integrity and the associated pre-organisation. Subsequently, the monoaldehyde intermediates 5a/b will be hydrolysed even further right down to the diformylpyridine 2, as well as into the diamines 3a/b with one additional equivalent of H2O. On the other hand, on adding one equivalent of toluidine to the [2]rotaxane solutions, dissociation occurs on account of imine exchange, which results in the fast opening of the [24]crown-8 macrocycles to give the podand-like compounds 6a/b. Toluidine plays an important role in serving as a relatively bulky imine exchange partner with the macrocycle to give a disrupted open crown (podand) that cannot be easily converted back to the [24]crown-8 derivative. | ||
Two control experiments were performed. Firstly, the dynamic [2]rotaxanes were dissolved separately in pre-dried CD3CN–DMF-d7 (9:1), CD3CN–CD3OD (2:1) and pure CD3CN. No significant changes were observed in the 1H NMR signals, from which it was concluded that dissociation of the dumbbell 4–H·PF6 does not occur even after standing for over 12 h in the presence of solvents that do not sustain hydrogen bonding. Secondly, since imine hydrolysis and formation can be catalysed by nucleophiles,8b as well as by acids,8f,16 the dissociations of the rotaxanes were investigated after adding a nucleophile—p-toluidine—as an imine exchange partner. On adding one equivalent of toluidine to the [2]rotaxane solutions, dissociation was found to occur on account of imine exchange, which results in the fast opening of the [24]crown-8 macrocycles to give the podand-like compounds 6a/b. Toluidine plays an important role in serving as a relatively bulky imine exchange partner with the macrocycle to give a disrupted open crown (podand), which cannot be easily converted back to the [24]crown-8 derivative. As a consequence, the [2]rotaxanes dissociate rapidly on simply adding one equivalent of toluidine.
The reversible formation of the [2]rotaxanes 1a/b–H·PF6 from their separate components has been demonstrated (Fig. 3) by 1H NMR spectroscopy. For example, from the dissociated mixture of the dynamic [2]rotaxane 1a/b–H·PF6 achieved on the addition of 200 equivalents of H2O (i.e., with 41% of the [2]rotaxane remaining in the mixture), after treatment with anhydrous MgSO4, the percentage of the corresponding dynamic [2]rotaxane 1a/b–H·PF6 is restored (Fig. 3a) to ∼70%. Somewhat surprisingly, molecular sieves (4 Å) are not useful for drying these systems, since the dynamic [2]rotaxanes 1a/b are unstable in their presence. Moreover, treatment with anhydrous Na2SO4, followed by evaporation and the redissolving of the dry MeCN (three consecutive cycles), gives (Fig. 3b) the corresponding dynamic [2]rotaxane 1a–H·PF6. This reversibility of the equilibrium was found to be adjustable many times over, without destroying either the individual components or the dynamic [2]rotaxanes.
![Partial 1H NMR spectra (500 MHz, CD3CN, 298 K, aromatic region) of the dissociated dynamic [2]rotaxane 1a–H·PF6 (b = bound and f = free) after treatment with (a) anhydrous MgSO4 and (b) anhydrous Na2SO4, evaporated and redissolved in MeCN through three consecutive cycles.](/image/article/2010/OB/b915864b/b915864b-f3.gif) | ||
| Fig. 3 Partial 1H NMR spectra (500 MHz, CD3CN, 298 K, aromatic region) of the dissociated dynamic [2]rotaxane 1a–H·PF6 (b = bound and f = free) after treatment with (a) anhydrous MgSO4 and (b) anhydrous Na2SO4, evaporated and redissolved in MeCN through three consecutive cycles. | ||
Conclusions
The separation of the dynamic [2]rotaxanes 1a/b into their dumbbell and cleaved ring components in the presence of different concentrations of water has been demonstrated. Large amounts (>100 equivalents) of water are required for the degradation of half of the molecules of the [2]rotaxanes. The stability of the [2]rotaxane 1a–H·PF6 towards the hydrolysis is higher than that of 1b–H·PF6. A mechanism for the hydrolysis, involving the hydrolytic cleavage of one of the two imine bonds in the [24]crown-8 ring, has been proposed. It involves the formation of an acyclic intermediate following hydrolysis of one of the kinetically labile bonds in the ring. The separate components are also relatively stable in the presence of large amounts of water and can even be re-assembled to afford the dynamic [2]rotaxanes 1a/b–H·PF6, once again by eliminating water from their aqueous solutions. In principle, the concentrations of the dynamic [2]rotaxanes in the mixture can be controlled by the addition and subtraction of appropriate amounts of water. It is possible that by covalent attachment with fluorescent probes (reporter groups), such as anthracene or naphthalene moieties, detailed hydrolysis mechanisms for the degradation of these dynamic rotaxanes in the presence of various stimuli—e.g., water, acids, nucleophiles, etc.—could be investigated by monitoring the fluorescence quenching effects in the rotaxanes.Experimental
Materials and methods
All reagents were purchased from Aldrich or Cambridge Isotopes Laboratories, Inc. 2,6-Pyridinedicarboxaldehyde (2) is commercially available, while tetraethyleneglycol bis(2-aminophenyl)ether (3a) and bis-3,5-dimethoxybenzyl ammonium hexafluorophosphate (4–H·PF6) were prepared according to literature procedures.8c The dynamic [2]rotaxanes were prepared according to the literature procedure,8c and were then dried in vacuo and isolated. Compound 3b was synthesized as illustrated in Scheme 3. Acetonitrile-d3 (99.8% D), N,N-dimethylformamide-d7 (99.8% D) and methanol-d4 (99.8% D) were dried with activated molecular sieves (4 Å). Deuterium oxide (99.9% D) was used. Thin-layer chromatography (TLC) was carried out using aluminium sheets, precoated with silica gel 60F (Merck 5554). The plates were inspected by UV light, prior to their development with iodine vapor. Melting points were determined on an Electrothermal 9200 apparatus and are uncorrected. Column chromatography was performed on silica gel 60F (Merck 9385, 0.040–0.063 mm). All nuclear magnetic resonance (NMR) spectra were recorded on a Brüker Advance 500 (1H at 500 MHz and 13C at 126 MHz). Chemical shifts are reported in parts per million (ppm) downfield from the Me4Si resonance as the internal standard for recording the 1H and 13C NMR spectra. High-resolution matrix-assisted laser-desorption ionization mass spectra (HR-MALDI-MS) were measured on an IonSpec Fourier Transform mass spectrometer. The reported molecular mass (m/z) values are for the most abundant monoisotopic mass.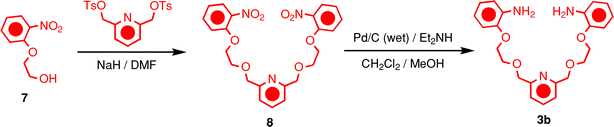 :1) as the eluent to yield the bisnitro compound 8 as an off-white solid (0.32 g, 30% yield); m.p. 90.2–92.4 °C. 1H NMR (500 MHz, CDCl3): δ = 3.94–3.96, (m, 4H), 4.30–4.32 (m, 4H), 4.72 (s, 4H), 7.02 (t, J = 7.7 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 7.7 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.76 (t, J = 7.7 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H); 13C NMR (126 MHz, CDCl3): δ = 68.7, 69.1, 74.1, 114.8, 120.2, 120.5, 125.5, 133.9, 137.6, 139.9, 152.1, 157.4; HRMS (HR-MALDI): calcd for C23H24N3O8m/z = 470.1563 and C23H23N3O8Na m/z = 492.1383; found m/z = 470.1560 [M + H]+ and 492.1383 [M + Na]+.
:1) as the eluent to yield the bisnitro compound 8 as an off-white solid (0.32 g, 30% yield); m.p. 90.2–92.4 °C. 1H NMR (500 MHz, CDCl3): δ = 3.94–3.96, (m, 4H), 4.30–4.32 (m, 4H), 4.72 (s, 4H), 7.02 (t, J = 7.7 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 7.7 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.76 (t, J = 7.7 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H); 13C NMR (126 MHz, CDCl3): δ = 68.7, 69.1, 74.1, 114.8, 120.2, 120.5, 125.5, 133.9, 137.6, 139.9, 152.1, 157.4; HRMS (HR-MALDI): calcd for C23H24N3O8m/z = 470.1563 and C23H23N3O8Na m/z = 492.1383; found m/z = 470.1560 [M + H]+ and 492.1383 [M + Na]+.Acknowledgements
This material is based upon work supported by the National Science Foundation under CHE0924620 in the United States and by the RGC-GRF (CUHK401808) of Hong Kong SAR.Notes and References
- (a) P. A. Brady, R. P. Bonar-Law, S. J. Rowan, C. J. Suckling and J. K. M. Sanders, Chem. Commun., 1996, 319–320 RSC; (b) P. A. Brady and J. K. M. Sanders, Chem. Soc. Rev., 1997, 26, 327–336 RSC; (c) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (d) J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265–2274 CrossRef CAS; (e) L. M. Greig and D. Philp, Chem. Soc. Rev., 2001, 30, 287–302 RSC; (f) R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 CrossRef CAS; (g) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (h) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS; (i) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- (a) K. S. Chichak, S. J. Cantrill, A. R. Pease, S. H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS; (b) K. S. Chichak, S. J. Cantrill and J. F. Stoddart, Chem. Commun., 2005, 3391–3393 RSC; (c) A. J. Peters, K. S. Chichak, S. J. Cantrill and J. F. Stoddart, Chem. Commun., 2005, 3394–3396 RSC; (d) C. D. Pentecost, K. S. Chichak, A. J. Peters, G. W. V. Cave, S. J. Cantrill and J. F. Stoddart, Angew. Chem., Int. Ed., 2007, 46, 218–222 CrossRef CAS; (e) C. D. Pentecost, N. Tangshaivang, S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, J. Chem. Educ., 2007, 84, 855–859 CrossRef CAS.
- (a) K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 5808–5810 CrossRef CAS; (b) K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, Macromolecules, 2007, 40, 3951–3959 CrossRef CAS; (c) K. C.-F. Leung, Macromol. Theory Simul., 2009, 18, 328–335 CrossRef CAS; (d) K. C.-F. Leung, S. Xuan and C.-M. Lo, ACS Appl. Mater. Interfaces, 2009, 1, 2005–2012 Search PubMed.
- (a) A. R. Williams, B. H. Northrop, T. Chang, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2006, 45, 6665–6669 CrossRef CAS; (b) B. H. Northrop, N. Tangchiavang, J. D. Badjic and J. F. Stoddart, Org. Lett., 2006, 8, 3899–3902 CrossRef CAS; (c) J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266–17271 CrossRef CAS; (d) L. M. Klivansky, G. Koshkakavyan, D. Cao and Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 4185–4189 CrossRef CAS.
- (a) F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 CAS; (b) K. E. Griffiths and J. F. Stoddart, Pure Appl. Chem., 2008, 80, 485–506 CrossRef CAS.
- For examples of rotaxane syntheses with ring closing metathesis, see: (a) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS; (b) A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281–3285 CrossRef CAS; (c) F. Aricó, P. Mobian, J.-M. Kern and J.-P. Sauvage, Org. Lett., 2003, 5, 1887–1890 CrossRef CAS; (d) J. D. Badjic, S. J. Cantrill, R. H. Grubbs, E. N. Guidry, R. Orenes and J. F. Stoddart, Angew. Chem., Int. Ed., 2004, 43, 3273–3278 CrossRef CAS; (e) E. N. Guidry, S. J. Cantrill, J. F. Stoddart and R. H. Grubbs, Org. Lett., 2005, 7, 2129–2132 CrossRef CAS; (f) H. Hou, K. C.-F. Leung, D. Lanari, A. Nelson, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 15358–15359 CrossRef CAS.
- For rotaxane-containing disulfide units in their dumbbell-shaped components that are formed by riveting and stoppering approaches, see: (a) A. G. Kolchinski, N. W. Alcock, R. A. Roesner and D. H. Busch, Chem. Commun., 1998, 1437–1438 RSC; (b) Y. Furusho, T. Hasegawa, A. Tsuboi, N. Kihara and T. Takata, Chem. Lett., 2000, 18–19 CrossRef CAS; (c) T. Oku, T. Furush and T. Takata, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 119–123 CrossRef CAS; (d) Y. Furusho, T. Oku, G. A. Rajkumar and T. Takata, Chem. Lett., 2004, 33, 52–53 CrossRef CAS.
- (a) S. J. Cantrill, S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1363–1366 CrossRef CAS; (b) S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1913–1916 CrossRef CAS; (c) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875 CrossRef CAS; (d) M. Horn, J. Ihringer, P. T. Glink and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 4046–4054 CrossRef CAS; (e) F. Aricó, T. Chang, S. J. Cantrill, A. I. Khan and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 4655–4666 CrossRef; (f) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (g) P. C. Haussmann, S. I. Khan and J. F. Stoddart, J. Org. Chem., 2007, 72, 6708–6713 CrossRef CAS; (h) P. C. Haussmann and J. F. Stoddart, Chem. Rec., 2009, 9, 136–154 CrossRef CAS.
- (a) C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef CAS; (b) W. Lu and T. H. Chang, J. Org. Chem., 2001, 66, 3467–3473 CrossRef CAS; (c) T. Hirashita, Y. Hayashi, K. Mitsui and S. Araki, J. Org. Chem., 2003, 68, 1309–1313 CrossRef CAS.
- V. Saggiomo and U. Lüning, Eur. J. Org. Chem., 2008, 4329–4333 CrossRef CAS.
- A. Simion, C. Simion, T. Kanda, S. Nagashima, Y. Mitoma, T. Yamada, K. Mimura and M. Tashiro, J. Chem. Soc., Perkin Trans. 1, 2001, 2071–2078 RSC.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem.–Eur. J., 2006, 12, 4069–4076 CrossRef CAS.
- S. Nygaard, K. C.-F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S.-Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 960–970 CrossRef CAS.
- S. Saha, K. C.-F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS.
- D2O was employed instead of H2O because of the ease for 1H NMR spectroscopic characterization. Notice that the dissociation of the [2]rotaxanes in H2O would proceed faster than those in D2O because of the isotope effect.
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS.
- T. Ventrice, E. M. Campi, W. R. Jackson and A. F. Patti, Tetrahedron, 2001, 57, 7557–7574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectra. See DOI: 10.1039/b915864b |
| This journal is © The Royal Society of Chemistry 2010 |