Facile P,N-heterocycle synthesis via tandem aminomethylation–cyclization of H-phosphinate building blocks†
Clémence Queffélec and Jean-Luc Montchamp*
Department of Chemistry, TCU Box 298860, Texas Christian University, Fort Worth, TX 76129, USA. E-mail: j.montchamp@tcu.edu; Fax: 1 817 257 5851; Tel: 1 817 257 6201
First published on 5th November 2009
Abstract
Various heterocycles containing phosphorus and nitrogen are easily synthesized from readily available H-phosphinate building blocks. Aminomethylation of these H-phosphinates is followed by in situ cyclization through substitution or cross-coupling to produce novel heterocycles in moderate to good yields.
Introduction
Perhaps surprisingly, few phosphorus–nitrogen heterocycles have been synthesized previously.1 In recent work, we reported the formation of such heterocycles starting from ω-amino-H-phosphinates (eqn (1)) through condensation with carbonyl compounds.2 The method was made possible by synthetic methodologies we developed to access the starting materials.3 Herein, we shifted the focus to precursors which do not contain an amino group, but instead possess a reactive halide.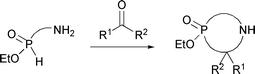 | (1) |
With the exception of our previous report (eqn (1)),2P,N-containing phosphinic heterocycles are rare in the literature. Since heterocycles have shown a variety of biological activities, and since cyclic P,N-phosphinates can be considered analogs of amino acids, we set out to expand the range of compounds accessible from simple building blocks. Below, we describe a general approach to P,N-heterocycles based on the Kabachnick–Fields aminomethylation4 of precursors, followed by in situ cyclization of the resulting amines (Scheme 1). Over the past several years, our laboratory has reported several general methods to prepare functionalized H-phosphinates.3 In the present work, such intermediates are prepared and their use in the synthesis of P,N-heterocycles is investigated.
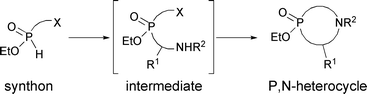 | ||
| Scheme 1 Strategy for the synthesis of P,N-heterocycles | ||
Results and discussion
Precursor synthesis
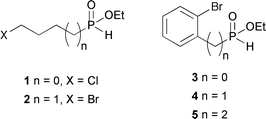
Several synthons were prepared through established reactions.3 Compounds 1, 2 and 5 were prepared via hydrophosphinylation,3d–j compound 3via Pd-catalyzed cross-coupling,3k and compound 4via base-promoted alkylation.3r The detailed conditions are shown in Schemes 2–4, and eqn (2) and (3). Synthon 1 (Scheme 2) was synthesized through our radical hydrophosphinylation using Et3B as the initiator, as reported previously.3h The resulting intermediate 6 was esterified5 directly to produce ester 1. The low yield of compound 1 was attributed to difficulties during the purification of this polar compound using chromatography on silica gel. Nonetheless, the preparation of 1 was easily accomplished.
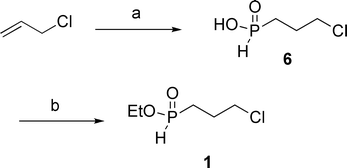 | ||
| Scheme 2 Synthesis of ethyl (3-chloropropyl)-H-phosphinate 1. (a) NaH2PO2, Et3B, rt, 2 h, 71%; (b) (EtO)4Si, toluene, reflux, 24 h, 40%. | ||
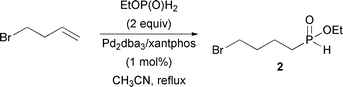
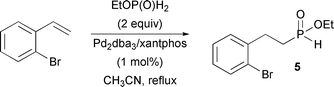
Homolog 2 was prepared in a single step through palladium-catalyzed hydrophosphinylation3d of 4-bromo-1-butene (eqn (2)). Synthon 2 was obtained in 67% yield. A similar reaction was used to prepare 5 from commercially available 2-bromostyrene (eqn (3)) in 64% yield.
 | (2) |
 | (3) |
Synthon 3 required the use of our palladium-catalyzed cross-coupling of anilinium hypophosphite3k with 1-bromo-2-iodobenzene. Esterification then proceeded in reasonable yield to form 3 (Scheme 3).
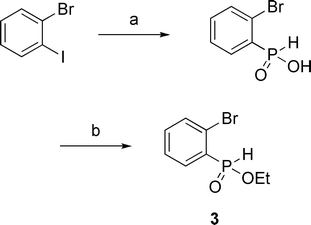 | ||
| Scheme 3 Synthesis of ethyl (2-bromophenyl)-H-phosphinate 3. (a) 1-bromo-2-iodobenzene, anilinium hypophosphite (3 equiv.), Pd(OAc)2 (2 mol%), dppp (2.2 mol%), CH3CN, reflux, 16 h, 65%; (b) (EtO)4Si, toluene, reflux, 24 h, 64%. | ||
Finally, synthon 4 was synthesized through the LiHMDS-promoted alkylation3r of 2-bromobenzyl bromide followed by deprotection of the acetal using TMSCl.6 The sequence produced 4 in 60% overall yield (Scheme 4).
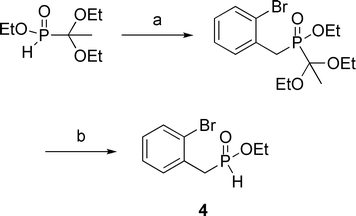 | ||
| Scheme 4 Synthesis of ethyl (2-bromobenzyl)-H-phosphinate 4. (a) 2-bromobenzyl bromide, LiHMDS, THF, −78 °C to rt, 3 h, 61%; (b) TMSCl, CH2Cl2, EtOH, rt, 16 h, 99%. | ||
Reactivity and cyclization
With the above precursors in hand, their reactions with imines were investigated. Table 1 shows the results obtained with synthons 1 and 2. Reaction of the H-phosphinates with imines proceeded smoothly and cyclization then took place uneventfully.| Entry | Synthon | Iminea | Product | Isolated yield (%)b |
|---|---|---|---|---|
| a Conditions: N-benzylideneaniline, toluene, reflux, 16 h; or 1,3,5-tribenzylhexahydro-1,3,5-triazine, toluene, reflux, 16 h.b Yield of pure compound after column chromatography. | ||||
| 1 | 1 | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh NPh |  | 61 |
| 2 | 1 | H2CNBn |  | 58 |
| 3 | 2 | PhCHNPh |  | 76 |
| 4 | 2 | H2CNBn |  | 45 |
The reactions of 2-bromophenyl-substituted H-phosphinate esters were investigated next (Table 2). Compounds 3 and 4 were subjected to similar reaction conditions, except that a base (Cs2CO3, 1.5 equiv.) and a palladium catalyst (Pd(PPh3)4, 2 mol%) were also added to the reaction mixtures. As expected, the aminomethylation took place easily, but, perhaps more surprisingly, the simplest Pd-catalyst delivered C–N bond formation in good yield, without the need for more sophisticated Buchwald–Hartwig-type cross-coupling catalysts.7 Thus, 5- and 6-membered P,N-heterocycles were obtained in a one-pot procedure (Table 2). Compound 3 reacted with a variety of imines (Table 2, entries 1–6). Interestingly, a reaction (entry 3) conducted with in situ formation of the imine (from paraformaldehyde and benzylamine) gave the expected product in only slightly lower yield than with the triazine precursor (entry 2). Compound 4 provided the 6-membered heterocycle in comparable yields (entries 7 and 8). Again, Pd(PPh3)4 successfully catalyzed the cross-coupling step.
| Entry | Synthon | Iminea | Product | Isolated yield (%)b |
|---|---|---|---|---|
| a Conditions: imine or triazine (1 equiv.), Cs2CO3 (1.5 equiv.), Pd(PPh3)4 (2 mol%), toluene, reflux, 24 h.b Yield of pure compound after column chromatography.c 1,3,5-tribenzylhexahydro-1,3,5-triazine was used. | ||||
| 1 | 3 | PhCHNPh |  | 61 |
| 2 | 3 | H2CNBnc |  | 63 |
| 3 | 3 | (CH2O)n + BnNH2 | 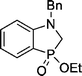 | 53 |
| 4 | 3 | PhCHNBn | 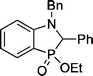 | 74 |
| 5 | 3 | PhCHN-tBu |  | 44 |
| 6 | 3 | PhCHNMe |  | 62 |
| 7 | 4 | PhCHNPh | 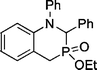 | 76 |
| 8 | 4 | H2CNBnc |  | 41 |
However, with synthon 5, although the Kabachnik–Fields reaction took place in good yield, the resulting intermediates did not cyclize to give the 7-membered ring under otherwise identical conditions. The aminomethylated products were obtained with N-benzylideneaniline or 1,3,5-tribenzylhexahydro-1,3,5-triazine in 51 and 67% isolated yields, respectively.
In a different (but related) approach, the heterocyclization of unsaturated ethyl cinnamyl-H-phosphinate was investigated through a tandem aminomethylation–Heck cyclization process (Scheme 5). Readily available cinnamyl-H-phosphinic acid87 was esterified5 to 8 using our typical conditions. Ester 8 was then reacted with 2-iodoaniline and paraformaldehyde to form aminomethylated iodide 9 in moderate isolated yield. Compound 9 was subjected to Heck-type9 reaction conditions (Pd/dppf, 2 mol%) in DMF to produce the desired 7-membered ring heterocycle 10 in 35% isolated yield. We have not optimized this reaction, other than the use of dppf10 instead of PPh3. While more work would be required to fully explore this strategy, Scheme 5 provides an interesting “proof of concept”, and the basis for future experiments.
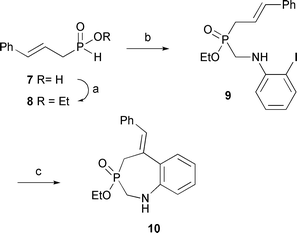 | ||
| Scheme 5 Tandem aminomethylation–Heck cyclization. (a) (EtO)4Si, toluene, reflux, 24 h, 92%; (b) 2-iodoaniline (1.2 equiv.), paraformaldehyde (1.2 equiv.), toluene, reflux, 16 h, 46%; (c) Pd(OAc)2 (2 mol%), dppf (2.2 mol%), i-Pr2NEt (2 equiv.), DMF, 110 °C, 35%. | ||
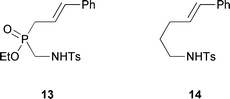
Along similar lines, we briefly attempted the Wacker-type cyclization11 of unsaturated amino-H-phosphinate 12 (Scheme 6). Unfortunately, attempts on 12a and 12b were unsuccessful. The use of ethyl cinnamyl-H-phosphinate 8via sulfonamide 13 similarly failed, although Liu and Stahl reported the successful cyclization of the all-carbon analog 14.11c
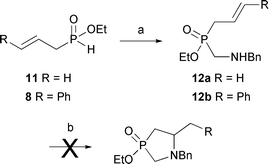 | ||
| Scheme 6 Attempted Pd(II)-catalyzed cyclization. (a) 1,3,5-tribenzylhexahydro-1,3,5-triazine (0.4 equiv.), toluene, reflux, 16 h, 12a 57%; 12b 83%. (b) Pd(OAc)2 (5 mol%), AcONa (2 equiv.), DMSO, O2, 80 °C, 60 h. | ||
Admittedly, we have not fully investigated the palladium-catalyzed heterocyclizations of precursors 9, 12 and 13. In spite of the above failed experiments, tremendous flexibility remains available to synthesize phosphorus heterocycles from simple precursors. For example, Wolfe-type carboaminations12 could also be tried on aminophosphinate precursors 12 and 13. Similarly, the use of allyl amine in the place of 2-iodoaniline (Scheme 5) could lead to a ring-closing metathesis precursor (the reaction of 8 with paraformaldehyde and allyl amine gives the corresponding diene in 53% yield). Our methodologies have made available a wide range of compounds through hydrophosphinylation, cross-coupling (halides, carboxylates, alcohols), alkylation, etc.,3 so that novel P-containing precursors could lead to facile syntheses of heterocyclic products. The preparation of P,O-heterocycles through the well-known addition of H-phosphinates to carbonyl compounds, using the precursors described in the present work, was not investigated because P,N-heterocycles are likely to be more interesting as amino acid analogs.13
Conclusions
The facile synthesis of P,N-heterocycles (substituted 3-hydroxy-1,3-azaphospholane and 3-hydroxy-1,3-azaphosphorinane-3-oxides) is described. With aryl bromide precursors, the cyclization proceeded well using Pd(PPh3)4 (2 mol%) as the cross-coupling catalyst. The availability of simple H-phosphinate building blocks opens up the possibility for the synthesis of various phosphorus heterocycles. Catalytic methods for the cyclization of other phosphinate precursors for the preparation of P,N- as well as other P-heterocycles will be the object of future studies.Experimental
For general chemistry information, and additional details on the synthesis of precursors and intermediates, spectral data, and a copy of the 31P, 1H and 13C NMR spectra, see the ESI.†Ethyl (3-chloropropyl)-H-phosphinate (Scheme 2, compound 1)
To (3-chloropropyl)-H-phosphinic acid3g (2.56 g, 18 mmol) in toluene (60 mL) was added tetraethyl orthosilicate (1.5 equiv., 5.63 g) under N2, and the mixture was refluxed for 24 h. The solvent was removed in vacuo and the resulting oil was purified by column chromatography (silica, EtOAc 100%) to afford the desired product as a yellow oil (1.22 g, 40%): 1H NMR (CDCl3, 300 MHz) δ 7.17 (d, J = 533 Hz, 1H, H–P), 4.00-4.30 (m, 2H, –CH2–O–), 3.64 (t, J = 5 Hz, 2H, –CH2–Cl), 1.85-2.10 (m, 4H, 2 x –CH2–), 1.39 (t, J = 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 62.8 (d, JPOC = 6 Hz), 44.9 (d, JPCCCCl = 17 Hz), 26.3 (d, JPC = 95 Hz), 24.2 (d, JPCC = 2 Hz), 16.4 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 37.7 (d, J = 533 Hz); HRMS (EI+) calc. for C5H12ClO2P 171.0342, found 171.0346.Ethyl (3-bromobutyl)-H-phosphinate (eqn (2), compound 2)3e
To a solution of EtOP(O)H2 in CH3CN (2 equiv., 60 mmol, 120 mL) and 4-bromo-1-butene (30 mmol, 4.05 g, 3 mL) were added Pd2dba3 (0.5 mol%, 137 mg) and xantphos (1.1 mol%, 191 mg). After 16 h of reflux, the mixture was concentrated and the resulting oil was diluted in EtOAc (60 mL) and washed with brine (1 × 20 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc 100%) to afford the desired product as a yellow oil (4.6 g, 67%): 1H NMR (CDCl3, 300 MHz) δ 7.13 (d, J = 530 Hz, 1H, H–P), 4.00-4.35 (m, 2H, –CH2–O), 3.43 (t, J = 7 Hz, 2H, –CH2–Br), 1.95-2.10 (m, 2H, –CH2–), 1.65-1.90 (m, 4H, 2 x –CH2–), 1.37 (t, J = 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 62.6 (d, JPOC = 7 Hz), 33.1 (d, JPCC = 15 Hz), 32.7, 27.9 (d, JPC = 94 Hz), 19.6, 16.4 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 38.8 (d, J = 530 Hz); HRMS (EI+) calc. for C6H14BrO2P 228.9993, found 228.9990.Ethyl 2-(2-bromophenyl)ethyl-H-phosphinate (eqn (3), compound 5)
To a solution of EtOP(O)H2 in CH3CN (1.6 equiv., 32 mmol, 64 mL) and 2-bromostyrene (20 mmol, 3.66 g) were added Pd2dba3 (0.75 mol%, 137 mg) and xantphos (1.6 mol%, 185 mg). After 16 h of reflux, the mixture was concentrated and the resulting oil was diluted in EtOAc (30 mL) and washed with brine (1 × 10 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc–hexanes 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) to afford the desired product as a yellow oil (3.56 g, 64%): 1H NMR (CDCl3, 300 MHz) δ 7.53 (d, J = 8 Hz, 1H, aro CH), 7.20-7.35 (m, 2H, aro CH), 7.14 (d, J = 532 Hz, 1H, H–P), 7.00-7.15 (m, 1H, aro CH), 4.00-4.25 (m, 2H, –CH2–O–), 2.90-3.10 (m, 2H, –CH2–P), 2.00-2.20 (m, 2H, –C–CH2–), 1.37 (t, J = 14, 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 139.6 (d, JPCCC = 16 Hz), 133.2, 130.5, 128.6, 127.9, 124.3, 62.7 (d, JPOC = 7 Hz), 28.9 (d, JPC = 92 Hz), 27.9, 16.5 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 37.4 (d, J = 532 Hz).
:3, v/v) to afford the desired product as a yellow oil (3.56 g, 64%): 1H NMR (CDCl3, 300 MHz) δ 7.53 (d, J = 8 Hz, 1H, aro CH), 7.20-7.35 (m, 2H, aro CH), 7.14 (d, J = 532 Hz, 1H, H–P), 7.00-7.15 (m, 1H, aro CH), 4.00-4.25 (m, 2H, –CH2–O–), 2.90-3.10 (m, 2H, –CH2–P), 2.00-2.20 (m, 2H, –C–CH2–), 1.37 (t, J = 14, 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 139.6 (d, JPCCC = 16 Hz), 133.2, 130.5, 128.6, 127.9, 124.3, 62.7 (d, JPOC = 7 Hz), 28.9 (d, JPC = 92 Hz), 27.9, 16.5 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 37.4 (d, J = 532 Hz).Ethyl (2-bromobenzyl)-H-phosphinate (Scheme 4, compound 4)
To ethyl (1,1-diethoxyethyl)-(2-bromobenzyl)phosphinate (3.03 g, 8 mmol) in 20 mL of 10% ethanol in dichloromethane was added chlorotrimethylsilane (1.5 equiv., 12 mmol, 1.3 mL) under N2 and the clear solution was stirred for 16 h at room temperature.6 The solvent was removed in vacuo and the resulting oil was purified by column chromatography (silica, EtOAc–hexanes 3:7, v/v) to afford the desired product as a yellow oil (2.1 g, 99%): 1H NMR (CDCl3, 300 MHz) δ 7.58 (d, J = 8 Hz, 1H, aro CH), 7.20-7.40 (m, 2H, aro CH), 7.10-7.20 (m, 1H, aro CH), 7.16 (d, J = 544 Hz, 1H, H–P), 4.00-4.25 (m, 2H, –CH2–O–), 3.35-3.55 (m, 2H, C–CH2–P), 1.32 (t, J = 14, 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 133.2 (d, J = 2 Hz), 132.1 (d, J = 6 Hz), 130.7 (d, J = 7 Hz), 129.2 (d, J = 4 Hz), 128.1 (d, J = 4 Hz), 124.8 (d, J = 7 Hz), 63.0 (d, JPOC = 6 Hz), 37.3 (d, JPC = 89 Hz), 16.4 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 34.3 (d, J = 544 Hz); HRMS (EI+) calc. for C9H12BrO2P 262.9837, found 262.9835.General procedure for the cyclization from compound 1 or 2
To the compound 1 or 2 (1 mmol) in toluene (10 mL) was added N-benzylideneaniline (1.2 equiv.) or 1,3,5-tribenzylhexahydro-1,3,5-triazine (0.4 equiv) and the mixture was refluxed for 16 h. The solvent was removed in vacuo, and the resulting oil was diluted in EtOAc (30 mL) and washed with brine (1 × 10 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc–hexanes 3:7, v/v) to afford the desired product.General procedure for the cyclization from compound 3 or 4
To compound 3 or 4 (5 mmol) in toluene (50 mL) was added the corresponding imine (1.2 equiv.) and the mixture was refluxed for 16 h. Then, caesium carbonate (1.2 equiv.) and Pd(PPh3)4 (2 mol%) were added, and the mixture was refluxed for 24 h. The solvent was removed in vacuo, and the resulting oil was diluted in EtOAc (30 mL) and washed with brine (1 × 10 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc–hexanes 3:7, v/v) to afford the desired product.Formation in situ of the imine from an aldehyde (Table 2, entry 3)
To the compound 3 (5 mmol) in toluene (50 mL) were added benzylamine (1.2 equiv., 0.65 mL) and paraformaldehyde (1.2 equiv., 198 mg), and the mixture was refluxed for 16 h. Then, caesium carbonate (1.2 equiv.) and Pd(PPh3)4 (2 mol%) were added and the mixture was refluxed for 24 h. The solvent was removed in vacuo, and the resulting oil was diluted in EtOAc (30 mL) and washed with brine (1 × 10 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc–hexanes 3:7, v/v) to afford the desired product as a yellow oil (760 mg, 53%).Heck product (Scheme 5, compound 10)
To compound 9 (0.5 mmol, 221 mg) in DMF (2.5 mL) was added N,N-diisopropylethylamine (2 equiv., 1 mmol, 0.2 mL), 1,1′-bis(diphenylphosphino)ferrocene (2.2 mol%, 6.1 mg) and Pd(OAc)2 (2 mol%, 2.2 mg). After 40 h of reflux, the solvent was removed in vacuo, and the resulting oil was diluted in EtOAc (10 mL) and washed with brine (1 × 20 mL). The organic layer was dried and concentrated. The resulting oil was purified by column chromatography (silica, EtOAc 100%) to afford the desired product as a pink oil (54.7 mg, 35%): 1H NMR (CDCl3, 300 MHz) δ 7.20-7.60 (m, 6H, aro CH), 7.13 (t, J = 15, 7 Hz, 1H, aro CH), 6.94 (d, J = 5 Hz, 1H, aro CH), 6.85 (t, J = 14, 7 Hz, 1H, aro CH), 6.67 (d, J = 8 Hz, 1H, –CCH–Ph), 4.00-4.25 (m, 1H, –NH–), 3.90-4.00 (m, 1H, –CH2–O–), 3.45-3.70 (m, 3H, P–CH2–N– and P–CH2–C–), 3.35-3.45 (m, 1H, –CH2–O–), 3.15-3.25 (m, 1H, P–CH2–C–), 1.09 (t, J = 14, 7 Hz, 3H, CH3–); 13C NMR (CDCl3, 75.45 MHz) δ 145.5 (d, J = 2 Hz), 137.5 (d, J = 3 Hz), 133.0 (d, J = 7 Hz), 131.9 (d, J = 11 Hz), 131.56, 131.54, 129.6, 128.9 (d, J = 2 Hz), 128.88, 128.81, 128.5 (d, J = 2 Hz), 127.3, 119.9, 118.3, 60.9 (d, JPOC = 7 Hz), 44.3 (d, JPC = 119 Hz), 31.4 (d, JPC = 78 Hz), 16.6 (d, JPOCC = 6 Hz); 31P NMR (CDCl3, 121.47 MHz) δ 54.2 (s); HRMS (EI+) calc. for C18H20NO2P 313.1232, found 313.1230.
Acknowledgements
We thank the National Institute of General Medical Sciences/NIH (1R01 GM067610) for the financial support of this research.Notes and references
- P,N-heterocycles: (a) L. Maier, Phosphorus Sulfur Relat. Elem., 1981, 11, 149 Search PubMed; (b) D. G. Hewitt and M. W. Teese, Aust. J. Chem., 1984, 37, 205 CAS; (c) Y. G. Trishin, V. I. Namestnikov and V. K. Bel'skii, Russ. J. Gen. Chem., 1999, 69, 1588 CAS; (d) M. M. Campbell, N. I. Carruthers, S. J. Mickel and P. M. Winton, J. Chem. Soc., Chem. Commun., 1984, 200 RSC; (e) A. Yiotakis, S. Vassiliou, J. Jiracek and V. Dive, J. Org. Chem., 1996, 61, 6601 CrossRef CAS; (f) J. Grembecka, A. Mucha, T. Cierpicki and P. Kafarski, J. Med. Chem., 2003, 46, 2641 CrossRef CAS; (g) K. Issleib, H. Winkelmann and H.-P. Abicht, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1974, 4, 191 Search PubMed; (h) K. Issleib, H. Oehme and E. Leißring, Chem. Ber., 1968, 101, 4032 CrossRef CAS; (i) F. Mathey, Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain, Elsevier, Oxford, 2001 (ISBN: 978-0-08-043952-5) Search PubMed.
- C. Queffélec, P. Ribière and J.-L. Montchamp, J. Org. Chem., 2008, 73, 8987 CrossRef CAS.
- Reviews: (a) L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef CAS; (b) J.-L. Montchamp, Speciality Chemicals Magazine, 2006, 26, 44 Search PubMed; (c) J.-L. Montchamp, J. Organomet. Chem., 2005, 690, 2388 CrossRef CAS. Hydrophosphinylation: (d) S. Deprèle and J.-L. Montchamp, J. Am. Chem. Soc., 2002, 124, 9386 CrossRef CAS; (e) S. Deprèle and J.-L. Montchamp, Org. Lett., 2004, 6, 3805 CrossRef CAS; (f) P. Ribière, K. Bravo-Altamirano, M. Antczak, J. D. Hawkins and J.-L. Montchamp, J. Org. Chem., 2005, 70, 4064 CrossRef CAS; (g) M. I. Antczak and J.-L. Montchamp, Synthesis, 2006, 3080 CAS; (h) S. Deprèle and J.-L. Montchamp, J. Org. Chem., 2001, 66, 6745 CrossRef CAS; (i) S. Gouault-Bironneau, S. Deprèle, A. Sutor and J.-L. Montchamp, Org. Lett., 2005, 7, 5909 CrossRef CAS; (j) S. Deprèle and J.-L. Montchamp, J. Organomet. Chem., 2002, 643–644, 154 CrossRef CAS. Cross-coupling: (k) J.-L. Montchamp and Y. R. Dumond, J. Am. Chem. Soc., 2001, 123, 510 CrossRef CAS; (l) Z. Huang, K. Bravo-Altamirano and J.-L. Montchamp, C. R. Chimie, 2004, 7, 763 Search PubMed; (m) K. Bravo-Altamirano, Z. Huang and J.-L. Montchamp, Tetrahedron, 2005, 61, 6315 CrossRef CAS; (n) K. Bravo-Altamirano, I. Abrunhosa-Thomas and J.-L. Montchamp, J. Org. Chem., 2008, 73, 2292 CrossRef CAS; (o) K. Bravo-Altamirano and J.-L. Montchamp, Org. Lett., 2006, 8, 4169 CrossRef CAS; (p) L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 4101 CrossRef CAS; (q) L. Coudray, K. Bravo-Altamirano and J.-L. Montchamp, Org. Lett., 2008, 10, 1123 CrossRef CAS. Base-promoted alkylation: (r) I. Abrunhosa-Thomas, C. E. Sellers and J.-L. Montchamp, J. Org. Chem., 2007, 72, 2851 CrossRef CAS; (s) I. Abrunhosa-Thomas, P. Ribière, A. C. Adcock and J.-L. Montchamp, Synthesis, 2006, 325 CAS.
- (a) X. Cheng, R. Goddard, G. Buth and B. List, Angew. Chem., Int. Ed., 2008, 47, 5079 CrossRef; (b) E. D. Matveeva and N. S. Zefirov, Dokl. Chem., 2008, 420, 137 CrossRef CAS; (c) V. V. Belakhov, Y. D. Shenin and B. I. Ionin, Russ. J. Gen. Chem., 2008, 78, 305 CAS; (d) N. S. Zefirov and E. D. Matveeva, ARKIVOC, 2008,(i), 1 CAS; (e) S. Bhagat and A. K. Chakraborti, J. Org. Chem., 2007, 72, 1263 CrossRef CAS; (f) E. D. Matveeva, T. A. Podrugina, M. V. Prisyajnoy and N. S. Zefirov, Russ. Chem. Bull., 2006, 55, 1209 Search PubMed; (g) H.-J. Cristau, A. Herve and D. Virieux, Tetrahedron, 2004, 60, 877 CrossRef CAS; (h) E. D. Matveeva, T. A. Podrugina, E. V. Tishkovskaya, L. G. Tomilova and N. S. Zefirov, Synlett, 2003, 2321 CrossRef CAS; (i) H.-J. Cristau, A. Coulombeau, A. Genevois-Borella, F. Sanchez and J.-L. Pirat, J. Organomet. Chem., 2002, 643–644, 381 CrossRef CAS; (j) R. A. Cherkasov and V. I. Galkin, Russ. Chem. Rev., 1998, 67, 857 RSC.
- Y. R. Dumond, R. L. Baker and J.-L. Montchamp, Org. Lett., 2000, 2, 3341 CrossRef CAS.
- (a) M. J. Gallagher and H. Honegger, Tetrahedron Lett., 1977, 18, 2987 CrossRef; (b) M. J. Gallagher and H. Honegger, Aust. J. Chem., 1980, 33, 287 CAS; (c) J. G. Dingwall, J. Ehrenfreund, R. G. Hall and J. Jack, Phosphorus Sulfur Relat. Elem., 1987, 30, 571 Search PubMed; (d) J. G. Dingwall, J. Ehrenfreund and R. G. Hall, Tetrahedron, 1989, 45, 3787 CrossRef CAS; (e) E. K. Baylis, Tetrahedron Lett., 1995, 36, 9385 CrossRef CAS; (f) E. K. Baylis, Tetrahedron Lett., 1995, 36, 9389 CrossRef CAS; (g) W. Froestl, S. J. Mickel, R. G. Hall, G. von Sprecher, D. Strub, D. P. A. Baumann, F. Brugger, C. Gentsch, J. Jaekel, H.-R. Olpe, G. Rihs, A. Vassout, P. C. Waldmeier and H. Bittiger, J. Med. Chem., 1995, 38, 3297 CrossRef CAS.
- Reviews: (a) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS; (b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS; (c) V. T. Abaev and O. V. Serdyuk, Russ. Chem. Rev., 2008, 77, 177 Search PubMed; (d) M. Kienle, S. R. Dubbaka, K. Brade and P. Knochel, Eur. J. Inorg. Chem., 2007, 4166 CAS; (e) J.-C. Hierso, M. Beaupérin and P. Meunier, Eur. J. Org. Chem., 2007, 3767 CAS; (f) B. Schlummer and U. Scholz, Speciality Chemicals Magazine, 2005, 25, 22 Search PubMed; (g) B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599 CrossRef CAS; (h) B. H. Yang and S. L. Buchwald, J. Organomet. Chem., 1999, 576, 125 CrossRef CAS.
- K. Bravo-Altamirano and J.-L. Montchamp, Org. Synth., 2008, 85, 96 CAS . See also ref. 3o.
- Reviews: (a) M. Oestreich, The Mizoroki-Heck Reaction, Wiley, Oxford, 2009 (ISBN: 0470033940) Search PubMed; (b) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS; (c) J. T. Link, Org. React., 2002, 60, 157 CAS; (d) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS; (e) W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2 CrossRef. For an interesting example using a phosphinate ester, see: (f) F. Hong, J. Xia and Y. Xu, J. Chem. Soc., Perkin Trans. 1, 1994, 1665 RSC.
- Y. Belabassi, S. Gouault-Bironneau, J.-L. Montchamp, 1,1′-Bis(diphenylphosphino)ferrocene, in Encyclopedia of Reagents for Organic Synthesis (eEROS), update article, 2006 Search PubMed.
- (a) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS; (b) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS; (c) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328 CrossRef CAS; (d) V. Kotov, C. C. Scarborough and S. S. Stahl, Inorg. Chem., 2007, 46, 1910 CrossRef CAS; (e) M. M. Rogers, J. E. Wendlandt, I. A. Guzei and S. S. Stahl, Org. Lett., 2006, 8, 2257 CrossRef CAS; (f) J. L. Brice, J. E. Harang, V. I. Timokhin, N. R. Anastasi and S. S. Stahl, J. Am. Chem. Soc., 2005, 127, 2868 CrossRef CAS; (g) S. R. Fix, J. L. Brice and S. S. Stahl, Angew. Chem., Int. Ed., 2002, 41, 164 CrossRef CAS.
- (a) J. P. Wolfe, Eur. J. Org. Chem., 2007, 571 CrossRef CAS; (b) J. S. Nakhla, J. W. Kampf and J. P. Wolfe, J. Am. Chem. Soc., 2006, 128, 2893 CrossRef CAS; (c) M. B. Bertrand, M. L. Leathen and J. P. Wolfe, Org. Lett., 2007, 9, 457 CrossRef CAS; (d) M. B. Bertrand and J. P. Wolfe, Tetrahedron, 2005, 61, 6447 CrossRef CAS; (e) J. P. Wolfe and J. S. Thomas, Curr. Org. Chem., 2005, 9, 625 CrossRef CAS; (f) J. E. Ney and J. P. Wolfe, J. Am. Chem. Soc., 2005, 127, 8644 CrossRef CAS.
- Biological activity of aminophosphinic acids: (a) N. J. Wardle, S. W. A. Bligh and H. R. Hudson, Curr. Org. Chem., 2007, 11, 1635 CrossRef CAS; (b) F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899 CrossRef CAS; (c) M. Collinsová and J. Jirácek, Curr. Med. Chem., 2000, 7, 629 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional procedures and spectral data. See DOI: 10.1039/b917428a |
| This journal is © The Royal Society of Chemistry 2010 |