Dendron-anchored organocatalysts: the asymmetric reduction of imines with trichlorosilane, catalysed by an amino acid-derived formamide appended to a dendron†
Marek Figlusa, Stuart T. Caldwella, Dawid Walasa, Gulen Yesilbagb, Graeme Cooke*a, Pavel Kočovský*a, Andrei V. Malkov*ac and Amitav Sanyal*b
aWestCHEM, Department of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: graemec@chem.gla.ac.uk; pavelk@chem.gla.ac.uk
bDepartment of Chemistry, Bogazici University, Bebek, Istanbul 34342, Turkey. E-mail: amitav.sanyal@boun.edu.tr
cDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: A.Malkov@lboro.ac.uk
First published on 27th October 2009
Abstract
Asymmetric reduction of ketimines 1a–f with trichlorosilane can be catalysed by the Lewis-basic N-methylvaline-derived formamide anchored to a soluble dendron (11c) with good enantioselectivity (≤94% ee) and low catalyst loading (typically 5 mol%) at room temperature in toluene. This protocol represents an improvement and simplification of the isolation procedure and recovery of the catalyst.
Introduction
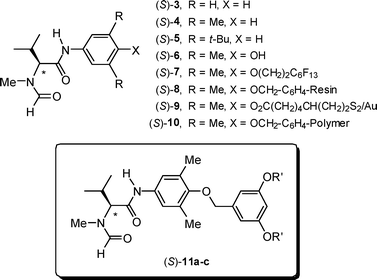
There is an impressive portfolio of protocols for the enantioselective transition metal-catalysed reduction of imines 1,1 which include hydrogenation,2 transfer hydrogenation3 and hydrosilylation,4etc.5 On the other hand, the organocatalytic realm is currently confined to the reduction with Hantzsch dihydropyridine catalysed by chiral Brønsted acids,6 and hydrosilylation with Cl3SiH catalysed by chiral Lewis bases (Scheme 1).7–9 In the last few years Malkov, Kočovský and co-workers have developed a library of Lewis-basic formamides derived from natural amino acids, and those originating from N-methyl valine, such as 3–5 (Fig. 1), proved to be particularly efficient (≤97% ee).7 In order to improve the practicality of the isolation procedure, these catalysts were then modified by appending a fluorous ponytail (7),7c a solid resin (8),7e a gold nanoparticle (9)7i and a soluble polymer (10).7j Herein, we report on an alternative approach, namely anchoring the catalyst to a dendron.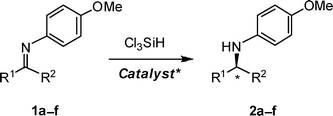
Dendron- and dendrimer-anchored catalysts provide an attractive architecture for performing a range of catalytic reactions.10 In particular, the unique structural characteristics of these systems offer advantages over traditional polymeric supports in terms of providing: (i) a well-defined monodisperse structure; (ii) the ability to tune the accessibility and microenvironment of the catalyst by either attaching it to the terminal functionality, or by isolating it within the dendrimer core; and (iii) the ability to augment the rate and/or the selectivity of the reaction via the so-called “dendrimer effect”.11 Unsurprisingly, these features have more recently inspired the development of dendrimer/dendron-based catalysts for enantioselective transformations.12
Results and discussion
Synthesis
Dendrons with an appended catalytic moiety were synthesized according to Scheme 2. 3,5-Dibenzyloxy benzyl alcohol 12a13 and the phenolic derivative (S)-(−)67e underwent the Mitsunobu reaction with di-(4-chlorobenzyl)-azodicarboxylate (DCAD) and triphenylphosphine (rt, 67 h) to afford the first-generation dendron 11a (17%). Similarly, the reaction of benzyl alcohol 12b13 with phenol (S)-(−)67e (rt, 48 h) furnished the second-generation dendron 11b (23%), and the benzyl alcohol 12c13 (using diethyl azodicarboxylate DEAD) produced the third-generation dendron 11c (26%; rt, 24 h).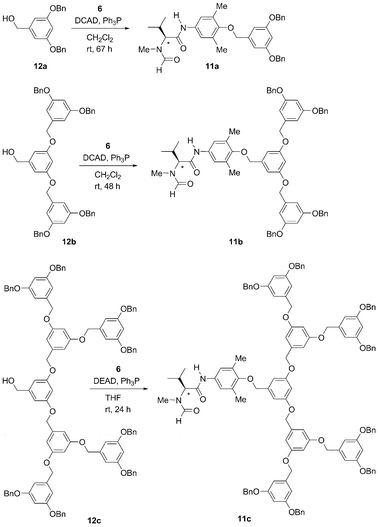 | ||
| Scheme 2 Synthesis of dendron-anchored catalysts 11a–c. | ||
Catalyst screening
The reduction of a substantial portfolio of imines, catalysed by 3–5 and 7–10, was investigated by us earlier.7 Since relatively little variation was observed as a function of the imine structure, the present study was confined to a selected set of representative examples of aromatic imines 1a–f (Scheme 1 and Table 1). The reduction of imine 1a proceeded uneventfully in the presence of each of the three dendron-supported catalysts 11a–c (Table 1, entries 1–3) and, when complete, as indicated by TLC, the mixture was added dropwise to vigorously stirred methanol, which was expected to precipitate the catalyst. However, in the case of the first-generation catalyst (11a), this operation was unsuccessful as no precipitation was observed, leaving a homogeneous solution. Aqueous workup of the latter solution, followed by evaporation and classical chromatography of the residue, afforded amine 2a (89% ee; entry 1). With the second-generation catalyst (11b), the addition of the organic phase to methanol resulted in the formation of a biphasic system with white droplets of the catalyst (at the bottom) surrounded by a “milky” solution, which was separated and centrifuged. This process removed ca. 60% of the catalyst. The supernatant was worked up and evaporated, and the product 2a was purified by chromatography. The precipitation procedure applied to the reaction catalysed by the third-generation catalyst (11c) was more successful: ca. 90% of the catalyst was recovered from the mixture by centrifugation so that the product (2a) thus obtained (after the aqueous workup) was contaminated with only ≤1% of the catalyst (as revealed by HPLC) and was purified by chromatography.| Catalyst (mol%) | 1a–f | R1 | R2 | Yield (%)b | 2 ee (%)c | |
|---|---|---|---|---|---|---|
| a The reaction was carried out at a 0.2 mmol scale with 2.0 equiv. of Cl3SiH in toluene. Trichlorosilane was added at 0 °C, and the mixture was allowed to warm up to room temperature and stirred for 16 h.b Yields of isolated products.c The absolute configuration of the resulting amines was found to be (S)-2 by comparison of their optical rotation and their HPLC behaviour with those of the authentic samples (ref. 7).d Trichlorosilane was added at room temperature instead of 0 °C as 1c was not soluble in toluene at 0 °C.e The reaction was carried out in CH2Cl2.f Ref. 14. | ||||||
| 1 | 11a (10) | 1a | Ph | Me | 88 | 89 |
| 2 | 11b (10) | 1a | Ph | Me | 90 | 87 |
| 3 | 11c (10) | 1a | Ph | Me | 89 | 89 |
| 4 | 11c (5) | 1b | 4-CF3Ph | Me | 81 | 91 |
| 5 | 11c (5)d | 1c | 2-Naphth | Me | 68 | 85 |
| 6 | 11c (5)e | 1c | 2-Naphth | Me | 23 | 70 |
| 7 | 11c (5) | 1d | 4-MeOPh | Me | 78 | 79 |
| 8 | 11c (5) | 1e | Ph | CH2Cl | 69 | 94 |
| 9 | 11c (5) | 1ff | Ph | CH2CO2Et | 89 | 82 |
These experiments demonstrated that only the third-generation catalyst 11c offered the advantages expected for a dendron. Therefore, this catalyst was employed to establish the scope of this method using our standard set of model imines 1b–f (Table 1, entries 4–9). The yields and enantioselectivities proved to be similar to those obtained with homogeneous, non-supported catalysts, reaching a maximum of 94% ee for imine 1e (entry 8). For comparison, Sigamide 5,7c,g one of the most successful catalysts for this transformation known to date,7,8 produced amine 2e in 96% ee.7d Little variation was observed as a function of the substitution pattern. As with Sigamide,7c,d,f toluene was identified as the solvent-of-choice for the dendron-supported catalysts, whereas the less environmentally friendly dichloromethane proved to be inferior (compare entries 5 and 6).
When the regenerated catalyst 11c (10 mol%) was reused for the reduction of imine 1a, the corresponding amine 2a was obtained in the same yield as with the fresh catalysts (90%) but the enantioselectivity dropped to 81% ee (compare with 89% ee, Table 1, entry 3).
Conclusions
In conclusion, three generations of dendron-supported N-methylvaline derivatives 11a–c were prepared as organocatalysts for the enantioselective reduction of imines 1a–f with trichlorosilane, a convenient, non-expensive reducing agent. Application of the third-generation catalyst 11c resulted in a substantial simplification of the isolation procedure, as most of the catalyst (≥90%) can be removed by precipitation and centrifugation.15 Toluene was again identified as the reaction medium-of-choice and the enantioselectivities (≤94% ee) mirrored those attained with Sigamide (5)7 and its congeners.Experimental
Imines 1a–f and amines 2a–f are known compounds, previously prepared in this laboratory;7 synthesis of the phenolic derivative 6 was reported by us recently.7c,e Dendrons 12a–c were prepared using the previously reported methodology.13Formamide (S)-(−)-11a
Di-(4-chlorobenzyl)-azodicarboxylate (DCAD) (436 mg, 1.19 mmol, 1.2 equiv.) was added to a solution of phenol (S)-67c,e (305 mg, 1.09 mmol, 1.1 equiv.), alcohol 12a13 (319 mg, 0.99 mmol) and triphenylphosphine (313 mg, 1.19 mmol, 1.2 equiv.) in dry CH2Cl2 (10 ml). The mixture was stirred at room temperature for 67 h, after which time the resulting precipitate was filtered off and washed with CH2Cl2 (10 mL). The combined organic fractions were concentrated under vacuum and purified by chromatography on a column of silica gel (70 g) with a petroleum ether–ethyl acetate mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford pure formamide (S)-(−)-11a (96 mg, 17%) as a viscous colourless oil: Rf 0.38/0.25 (petroleum ether–ethyl acetate, 1:1); [α]D−58.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, a mixture of amide rotamers in ca. 4:1 ratio; only the data for the major rotamer are given) δ 0.93 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 6.5 Hz, 3H), 2.26 (s, 6H), 2.41–2.54 (m, 1H), 3.02 (s, 3H), 4.47 (d, J = 11.2 Hz, 1H), 4.59 (s, 2H), 5.06 (s, 4H), 6.61 (t, J = 2.2 Hz, 1H), 6.74 (d, J = 2.2 Hz, 2H), 7.24 (s, 2H), 7.31–7.45 (m, 10H), 8.15 (s, 1H), 8.38 (s, 1H); 13C NMR δ 16.44 (CH3), 18.55 (CH3), 19.45 (CH3), 25.37 (CH), 31.53 (CH3), 62.86 (CH), 70.03 (CH2), 73.90 (CH2), 101.48 (CH), 106.55 (CH), 122.37 (CH), 127.46 (CH), 127.93 (CH), 128.51 (CH), 131.58 (C), 133.34 (C), 136.73 (C), 139.83 (C), 152.28 (C), 160.01 (C), 163.87 (CHO), 167.11 (CO); IR (film) ν 3317, 2964, 2929, 1656, 1596 cm−1; MS (FAB) m/z (%) 581 ([M + H]+, 10), 303 (20), 142 (100), 92 (65); HRMS 580.3017 [C36H41O5N2 requires (M + H)+].
:1) to afford pure formamide (S)-(−)-11a (96 mg, 17%) as a viscous colourless oil: Rf 0.38/0.25 (petroleum ether–ethyl acetate, 1:1); [α]D−58.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, a mixture of amide rotamers in ca. 4:1 ratio; only the data for the major rotamer are given) δ 0.93 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 6.5 Hz, 3H), 2.26 (s, 6H), 2.41–2.54 (m, 1H), 3.02 (s, 3H), 4.47 (d, J = 11.2 Hz, 1H), 4.59 (s, 2H), 5.06 (s, 4H), 6.61 (t, J = 2.2 Hz, 1H), 6.74 (d, J = 2.2 Hz, 2H), 7.24 (s, 2H), 7.31–7.45 (m, 10H), 8.15 (s, 1H), 8.38 (s, 1H); 13C NMR δ 16.44 (CH3), 18.55 (CH3), 19.45 (CH3), 25.37 (CH), 31.53 (CH3), 62.86 (CH), 70.03 (CH2), 73.90 (CH2), 101.48 (CH), 106.55 (CH), 122.37 (CH), 127.46 (CH), 127.93 (CH), 128.51 (CH), 131.58 (C), 133.34 (C), 136.73 (C), 139.83 (C), 152.28 (C), 160.01 (C), 163.87 (CHO), 167.11 (CO); IR (film) ν 3317, 2964, 2929, 1656, 1596 cm−1; MS (FAB) m/z (%) 581 ([M + H]+, 10), 303 (20), 142 (100), 92 (65); HRMS 580.3017 [C36H41O5N2 requires (M + H)+].Formamide (S)-(−)-11b
Di-(4-chlorobenzyl)-azodicarboxylate (DCAD) (60 mg, 0.15 mmol) was added to a solution of phenol (S)-67c,e (43 mg, 0.15 mmol), alcohol 12b13 (108 mg, 0.14 mmol) and triphenylphosphine (41 mg, 0.15 mmol) in dry CH2Cl2 (3 ml). The mixture was stirred at room temperature for 48 h, after which time the resulting precipitate was filtered off and washed with CH2Cl2 (10 mL). The combined organic fractions were concentrated under vacuum and purified by chromatography on a column of silica gel (30 g) with a petroleum ether–ethyl acetate mixture (1:1) to afford pure formamide (S)-(−)-11b (33 mg, 23%) as a viscous colourless oil: Rf 0.40/0.23 (petroleum ether–ethyl acetate, 1:1); [α]D−47.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, a mixture of amide rotamers in ca. 4:1 ratio; only the data for the major rotamer are given) δ 0.93 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 6.5 Hz, 3H), 2.28 (s, 6H), 2.44–2.53 (m, 1H), 3.01 (s, 3H), 4.42 (d, J = 11.2 Hz, 1H), 4.70 (s, 2H), 5.00 (s, 4H), 5.04 (s, 8H), 6.59 (s, 3H), 6.71 (s, 4H), 6.73 (s, 2H) 7.23 (s, 2H), 7.33–7.44 (m, 20H), 8.15 (s, 1H), 8.18 (s, 1H); 13C NMR δ 16.47 (CH3), 18.51 (CH3), 19.50 (CH3), 25.24 (CH), 31.53 (CH3), 62.99 (CH), 69.95 (CH2), 70.04 (CH2), 73.90 (CH2), 101.50 (CH), 106.32 (CH), 106.54 (CH), 120.36 (CH), 127.52 (CH), 127.96 (CH), 128.55 (CH), 131.63 (C), 133.32 (C), 136.69 (C), 139.19 (C), 139.87 (C), 152.31 (C), 159.93 (C), 160.10 (C), 163.93 (CHO), 167.01 (CO); IR (film) ν 3315, 3032, 2928, 1656, 1595 cm−1; m/z (FAB) 1006 [(M + H)+, 2%], 531 (95), 303 (30), 219 (100); HRMS 1005.4672 [C64H65O9N2 requires (M + H)+ 1005.4690].Formamide (S)-(−)-11c
Diethyl azodicarboxylate (DEAD) (0.13 mL, 0.81 mmol) was added to a solution of phenol (S)-67c,e (151 mg, 0.54 mmol), alcohol 12c13 (1.04 g, 0.65 mmol) and triphenylphosphine (212 mg, 0.81 mmol) in dry THF (10 mL). The mixture was stirred at room temperature for 24 h, after which time the solution was concentrated under vacuum. The residue was purified by chromatography on a column of silica gel (130 g) with a petroleum ether–ethyl acetate mixture (2:3) to afford a crude product, which was further purified by washing with MeOH (2 × 40 mL) and then ether (2 × 40 mL). Drying of the residue afforded pure formamide (S)-(−)-11c (263 mg, 26%) as a solid foam: Rf 0.49/0.37 (petroleum ether–ethyl acetate, 2:3); [α]D 24.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3, a mixture of amide rotamers in ca. 4:1 ratio; only the data for the major rotamer are given) δ 0.91 (d, J = 6.6 Hz, 3H), 1.04 (d, J = 6.4 Hz, 3H), 2.25 (s, 6H), 2.41–2.50 (m, 1H), 2.96 (s, 3H), 4.32 (d, J = 11.2 Hz, 1H), 4.67 (s, 2H), 4.96 (s, 8H), 4.97 (s, 4H), 5.01 (s, 16H), 6.53 (t, J = 2.2 Hz, 2H), 6.56 (t, J = 2.2 Hz, 4H), 6.59 (t, J = 2.2 Hz, 1H), 6.66 (d, J = 2.2 Hz, 12H), 6.72 (d, J = 2.2 Hz, 2H), 7.18 (s, 2H), 7.27–7.41 (m, 40H), 7.82 (s, 1H), 8.12 (s, 1H); 13C NMR δ 16.47 (CH3), 18.49 (CH3), 19.49 (CH3), 25.18 (CH), 31.49 (CH3), 63.00 (CH), 69.90 (CH2), 70.00 (CH2), 73.87 (CH2), 101.37 (CH), 101.50 (CH), 106.30 (CH), 106.39 (CH), 106.54 (CH), 120.34 (CH), 127.50 (CH), 127.93 (CH), 128.51 (CH), 131.62 (C), 133.28 (C), 136.68 (C), 139.13 (C), 139.91 (C), 152.30 (C), 159.99 (C), 160.10 (C), 163.92 (CHO), 166.96 (CO); IR (film) ν 3312, 3032, 2930, 2873, 1658, 1594 cm−1; MS (FAB) m/z 1855 [(M + H)+, 8%), 846 (10), 606 (13), 423 (45), 303 (100)]; HRMS 1853.8033 [C120H113O17N2 requires (M + H)+ 1853.8039].General procedure for the asymmetric reduction of 1a catalysed by 11a,b
Trichlorosilane (50 μL) was added to a solution of imine 1a (0.22 mmol) and catalyst 11a or 11b (10 mol%) in toluene (1.5 mL) at 0 °C, and the mixture was stirred at room temperature overnight. Chloroform (30 mL) was then added and the solution was washed with aqueous saturated NaHCO3 (10 mL). The aqueous phase was extracted with chloroform (30 mL) and the combined organic solutions were dried over MgSO4. Chloroform was partially evaporated, silica gel (1 g) was added to the residue, and the rest of the solvent was evaporated to dryness. The latter silica gel-adsorbed mixture was loaded onto a column of dry silica gel (15 g) and eluted with a mixture of petroleum ether and ethyl acetate (24:1 or 9:1) to afford pure amine 2a. For yields and enantioselectivity, see Table 1.General Procedure for the Asymmetric Reduction of 1a–f Catalysed by 11c
Trichlorosilane (50 μL) was added to a solution of imine 1 (0.22 mmol) and catalyst 11c (0.022 mmol) in toluene (1.5 mL) at 0 °C and the mixture was stirred at room temperature overnight. The mixture was then poured into rapidly stirred methanol (50 mL), the resulting cloudy mixture was centrifuged, and the clear supernatant was evaporated. The residue was treated with chloroform (30 mL) and the resulting solution was washed with aqueous saturated NaHCO3 (10 mL). The aqueous phase was extracted with chloroform (30 mL), and the combined organic solutions were dried over MgSO4 and evaporated. The crude product was purified by chromatography on a column of silica gel (10 g) using a mixture of petroleum ether and ethyl acetate (15:1) to give the pure amine 2 (89%). For yields and enantioselectivity, see Table 1. The amines thus obtained were identical to the authentic samples prepared earlier by us.7
Acknowledgements
This work was supported by the EPSRC (grant Nos. GR/S87294/01 and EP/E018211/1), the Royal Society of Edinburgh, and AstraZeneca. We thank the University of Glasgow for a graduate fellowship to M.F. A.S thanks TUBITAK (105S353) and TUBA-GEBIP for financial support.Notes and references
- For a general overview of the reduction of imines, see the following: (a) J. D. Morrison, Asymmetric Synthesis, Academic, New York, 1983, vol. 2 Search PubMed; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley & Sons, New York, 1994 Search PubMed; (c) I. Ojima, Catalytic Asymmetric Synthesis, J. Wiley, and Sons, New York, 2nd edn, 2000 Search PubMed; (d) B. R. James, Catal. Today, 1997, 37, 209 CrossRef CAS; (e) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS; (f) B. T. Cho, Tetrahedron, 2006, 62, 7621 CrossRef CAS; (g) E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999, vol. I–III Search PubMed.
- For recent reports on catalytic hydrogenation (with Ti, Ir, Rh and Ru), see ref. 1b–d and the following: (a) D. Xiao and X. Zhang, Angew. Chem., Int. Ed., 2001, 40, 3425 CrossRef CAS; (b) X. B. Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. F. Boogers and J. G. de Vries, Org. Lett., 2003, 5, 1503 CrossRef CAS; (c) C. J. Cobley and J. P. Henschke, Adv. Synth. Catal., 2003, 345, 195 CrossRef CAS; (d) J. Okuda, S. Verch, R. Stürmer and T. S. Spaniol, J. Organomet. Chem., 2000, 605, 55 CrossRef CAS; (e) E. Guiu, B. Muñoz, S. Castillón and C. Claver, Adv. Synth. Catal., 2003, 345, 169 CrossRef CAS; (f) C. J. Cobley, E. Foucher, J.-P. Lecouve, I. C. Lennon, J. A. Ramsden and G. Thominot, Tetrahedron: Asymmetry, 2003, 14, 3431 CrossRef CAS; (g) Y. Chi, Y. G. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120 CrossRef CAS; (h) A. A. Boezio, J. Pytkowicz, A. Côté and A. B. Charette, J. Am. Chem. Soc., 2003, 125, 14260 CrossRef CAS; (i) A. Trifonova, J. S. Diesen, C. J. Chapman and P. G. Andersson, Org. Lett., 2004, 6, 3825 CrossRef CAS; (j) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS; (k) A. Trifonova, J. S. Diesen and P. G. Andersson, Chem.–Eur. J., 2006, 12, 2318 CrossRef CAS . For Rh-catalysed hydrogenation of enamides, see the following: X.-P. Hu and Z. Zheng, Org. Lett., 2004, 6, 3585 Search PubMed.
- (a) J. S. M. Samec and J. E. Bäckvall, Chem.–Eur. J., 2002, 8, 2955 CrossRef CAS; (b) R. Kadyrov and T. H. Riermeier, Angew. Chem., Int. Ed., 2003, 42, 5472 CrossRef CAS; (c) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS; (d) J. B. Åberg, J. S. M. Samec and J.-E. Bäckvall, Chem. Commun., 2006, 2771 RSC. For an overview, see: (e) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC. For the kinetic resolution using transfer hydrogenation combined with an enzymatic reaction, see: (f) J. Paetzold and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620 CrossRef CAS; (g) J. S. M. Samec, A. H. Éll, J. B. Åberg, T. Privalov, L. Eriksson and J.-E. Bäckvall, J. Am. Chem. Soc., 2006, 128, 14293 CrossRef CAS; (h) T. Privalov, J. S. M. Samec and J.-E. Bäckvall, Organometallics, 2007, 26, 2840 CrossRef CAS; (i) C. E. Hoben, L. Kanupp and J.-E. Bäckvall, Tetrahedron Lett., 2008, 49, 977 CrossRef CAS.
- (a) M. T. Reding and S. L. Buchwald, J. Org. Chem., 1998, 63, 6344 CrossRef CAS; (b) X. Verdaguer, U. E. W. Lange and S. L. Buchwald, Angew. Chem., Int. Ed., 1998, 37, 1103 CrossRef CAS; (c) M. C. Hansen and S. L. Buchwald, Org. Lett., 2000, 2, 713 CrossRef CAS; (d) E. Vedejs, P. Trapencieris and E. Suna, J. Org. Chem., 1999, 64, 6724 CrossRef CAS; (e) H. Nishikori, R. Yoshihara and A. Hosomi, Synlett, 2003, 561 CAS; (f) B. H. Lipshutz, K. Noson and W. Chrisman, J. Am. Chem. Soc., 2001, 123, 12917 CrossRef CAS; (g) B. H. Lipshutz and H. Shimizu, Angew. Chem., Int. Ed., 2004, 43, 2228 CrossRef CAS.
- For enzymatic approaches, including reduction, see, e.g.: (a) M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoidian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 41, 3177 CrossRef CAS; (b) R. Carr, M. Alexeeva, M. J. Dawson, V. Gotor-Fernández, C. E. Huphrey and N. J. Turner, ChemBioChem, 2005, 6, 637 CrossRef CAS; (c) C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224 CrossRef CAS. For a brief overview, see: (d) M. Alexeeva, R. Carr and N. J. Turner, Org. Biomol. Chem., 2003, 1, 4133 RSC.
- (a) S. Singh and U. K. Batra, Indian J. Chem, Sect. B, 1989, 28, 1; (b) M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781 CrossRef CAS; (c) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef; (d) M. Rueping, A. P. Antonchik and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 6751 CrossRef CAS; (e) S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS; (f) S. Hoffmann, M. Nicoletti and B. List, J. Am. Chem. Soc., 2006, 128, 13074 CrossRef CAS; (g) J. Zhou and B. List, J. Am. Chem. Soc., 2007, 129, 7498 CrossRef CAS; (h) R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84 CrossRef CAS; (i) S. G. Ouellet, A. M. Walji and D. W. C. MacMillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS.
- (a) A. V. Malkov, A. Mariani, K. N. MacDougall and P. Kočovský, Org. Lett., 2004, 6, 2253 CrossRef CAS; (b) A. V. Malkov, S. Stončius, K. N. MacDougall, A. Mariani, G. D. McGeoch and P. Kočovský, Tetrahedron, 2006, 62, 264 CrossRef CAS; (c) A. V. Malkov, M. Figlus, S. Stončius and P. Kočovský, J. Org. Chem., 2007, 72, 1315 CrossRef; (d) A. V. Malkov, S. Stončius and P. Kočovský, Angew. Chem., Int. Ed., 2007, 46, 3722 CrossRef CAS; (e) A. V. Malkov, M. Figlus and P. Kočovský, J. Org. Chem., 2008, 73, 3985 CrossRef CAS; (f) A. V. Malkov, S. Stončius, K. Vranková, M. Arndt and P. Kočovský, Chem.–Eur. J., 2008, 14, 8082 CrossRef CAS; (g) A. V. Malkov, K. Vranková, S. Stončius and P. Kočovský, J. Org. Chem., 2009, 74, 5839 CrossRef CAS; (h) A. V. Malkov, K. Vranková, R. Sigerson, S. Stončius and P. Kočovský, Tetrahedron, 2009, 65, 9481 CrossRef CAS; (i) A. V. Malkov, M. Figlus, G. Cooke, S. T. Caldwell, G. Rabani, M. R. Prestly and P. Kočovský, Org. Biomol. Chem., 2009, 7, 1878 RSC; (j) A. V. Malkov, M. Figlus, M. R. Prestly, G. Rabani, G. Cooke and P. Kočovský, Chem.–Eur. J., 2009, 15, 9651 CrossRef CAS.
- For other Lewis-basic organocatalysts promoting imine reduction, see: (a) F. Iwasaki, O. Onomura, K. Mishima, T. Kanematsu, T. Maki and Y. Matsumura, Tetrahedron Lett., 2001, 42, 2525 CrossRef CAS; (b) O. Onomura, Y. Kouchi, F. Iwasaki and Y. Matsumura, Tetrahedron Lett., 2006, 47, 3751 CrossRef CAS; (c) Z. Wang, X. Ye, S. Wei, P. Wu, A. Zhang and J. Sun, Org. Lett., 2006, 8, 999 CrossRef CAS; Z. Wang, M. Cheng, P. Wu, S. Wei and J. Sun, Org. Lett., 2006, 8, 3045 CrossRef CAS; (d) Z. Wang, S. Wei, C. Wang and J. Sun, Tetrahedron: Asymmetry, 2007, 18, 705 CrossRef CAS; (e) D. Pei, Z. Wang, S. Wei, Y. Zhang and J. Sun, Org. Lett., 2006, 8, 5913 CrossRef CAS; (f) L. Zhou, Z. Wang, S. We and J. Sun, Chem. Commun., 2007, 2977 RSC; (g) C. Wang, X. Wu, L. Zhou and J. Sun, Chem.–Eur. J., 2008, 14, 8789 CrossRef CAS; (h) P. Wu, Z. Wang, M. Cheng, L. Zhou and J. Sun, Tetrahedron, 2008, 64, 11304 CrossRef CAS; (i) H. Zheng, J. Deng, W. Lin and X. Zhang, Tetrahedron Lett., 2007, 48, 7934 CrossRef; (j) H.-J. Zheng, W. B. Chen, Z.-J. Wu, J.-G. Deng, W.-Q. Lin, W.-C. Yuan and X.-M. Zhang, Chem.–Eur. J., 2008, 14, 9864 CrossRef CAS; (k) S. Guizzetti, M. Benaglia, F. Cozzi, S. Rossi and G. Celentano, Chirality, 2009, 21, 233 CrossRef CAS; (l) F. M. Gautier, S. Jones and S. J. Martin, Org. Biomol. Chem., 2009, 7, 229 RSC; (m) C. Baudequin, D. Chaturvedi and S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 2623 CrossRef.
- For an analogous reduction of ketones with Cl3SiH, catalysed by metal-free Lewis bases, see: (a) F. Iwasaki, O. Onomura, K. Mishima, T. Maki and Y. Matsumura, Tetrahedron Lett., 1999, 40, 7507 CrossRef CAS; (b) Y. Matsumura, K. Ogura, Y. Kouchi, F. Iwasaki and O. Onomura, Org. Lett., 2006, 8, 3789 CrossRef CAS; (c) A. V. Malkov, A. Stewart, J. P. Liddon, P. Ramírez-López, L. Bendová, D. Haigh and P. Kočovský, Angew. Chem., Int. Ed., 2006, 45, 1432 CrossRef CAS.
- For representative recent reviews focusing upon dendrimer/dendron-based catalysts, see, e.g.: (a) A.-M. Caminade, P. Servin, R. Laurent and J.-P. Majoral, Chem. Soc. Rev., 2008, 37, 56 RSC; (b) R. Andrés, E. de Jesús and J. C. Flores, New J. Chem., 2007, 31, 1161 RSC; (c) J. K. Kassube and L. H. Gade, Top. Organomet. Chem., 2006, 20, 61 CAS; (d) A. Berger, R. J. M. Klein Gebbink and G. van Koten, Top. Organomet. Chem., 2006, 20, 1 CAS; (e) D. Méry and D. Astruc, Coord. Chem. Rev., 2006, 250, 1965 CrossRef CAS; (f) J. L. G. Gutierrez, P. G. Gutierrez and F. J. Cruz, Recent Research Trends in Organometallic Chemistry, 2005, 87 Search PubMed.
- For a review of the dendrimer effect in homogenous catalysis, see: B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125 Search PubMed.
- For representative recent examples of dendrimer/dendron-based enantioselective catalysts, see the following: (a) E. Bellis and G. Kokotos, J. Mol. Catal. A: Chem., 2005, 241, 166 CrossRef CAS; (b) Y.-C. Chen, T.-F. Wu, L. Jiang, J.-G. Deng, H. Liu, J. Zhu and Y.-Z. Jiang, J. Org. Chem., 2005, 70, 1006 CrossRef CAS; (c) Y. Li, X.-Y. Liu and G. Zhao, Tetrahedron: Asymmetry, 2006, 17, 2034 CrossRef CAS; (d) L. Yin, R. Li, F. Wang, H. Wang, Y. Zheng, C. Wang and J. Ma, Tetrahedron: Asymmetry, 2007, 18, 1383 CrossRef CAS; (e) Z.-J. Wang, G.-J. Deng, Y. Li, Y.-M. He, W.-J. Tang and Q.-H. Fan, Org. Lett., 2007, 9, 1243 CrossRef CAS; (f) T. Kehat and M. Portnoy, Chem. Commun., 2007, 2823 RSC; (g) R. Breslow, S. Wei and C. Kenesky, Tetrahedron, 2007, 63, 6317 CrossRef CAS; (h) L. Routaboul, S. Vincendeau, C.-O. Turrin, A.-M. Caminade, J.-P. Majoral, J.-C. Daran and E. Manoury, J. Organomet. Chem., 2007, 692, 1064 CrossRef CAS; (i) Y.-N. Niu, Z.-Y. Yan, G.-Q. Li, H.-L. Wei, G.-L. Gao, L.-Y. Wu and Y.-M. Liang, Tetrahedron: Asymmetry, 2008, 19, 912 CrossRef CAS; (j) Y. Liu and M. Shi, Adv. Synth. Catal., 2008, 350, 122 CrossRef CAS; (k) L.-I. Rodríguez, O. Rossell, M. Seco and G. Muller, Organometallics, 2008, 27, 1328 CrossRef CAS; (l) J. Yu, T. V. RajanBabu and J. R. Parquette, J. Am. Chem. Soc., 2008, 130, 7845 CrossRef CAS; (m) F. Zhang, Y. Li, Z.-W. Li, Y.-M. He, S.-F. Zhu, Q.-H. Fan and Q.-L. Zhou, Chem. Commun., 2008, 6048 RSC.
- C. J. Hawker and J. M Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- Imine 1f exists in its enamine form. However, the enamine–imine equilibration can be facilitated, e.g., by addition of AcOH (≤1 equiv.). The resulting imine concentration is sufficient for an effective reduction, as demonstrated by us for Sigamide 5 as catalyst7f,g.
- At 5 mol% catalyst loading, the product after centrifugation is contaminated by less than 0.5% of the catalyst and can be further purified by chromatography or crystallization.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, and 1H and 13C NMR spectra of the dendron-anchored catalysts 11a–c. See DOI: 10.1039/b916601g |
| This journal is © The Royal Society of Chemistry 2010 |