Total synthesis of LeA-LacNAc pentasaccharide as a ligand for Clostridium difficile toxin A†
Ping Zhangab, Kenneth Ngb and Chang-Chun Ling*a
aDepartment of Chemistry, Alberta Ingenuity Center for Carbohydrate Science, University of Calgary, Calgary Alberta, T2N 1N4, Canada. E-mail: ccling@ucalgary.ca; Fax: +1 (403) 289-9488; Tel: +1 (403) 220-2768
bDepartment of Biological Science, Alberta Ingenuity Center for Carbohydrate Science, University of Calgary, Calgary Alberta, T2N 1N4, Canada
First published on 13th November 2009
Abstract
The toxins TcdA and TcdB produced by the human pathogen Clostridium difficile gain entrance to host epithelial cells by recognizing cell-surface carbohydrate ligands. Inhibiting the attachment of these toxins to host cells has been proposed to be a viable therapy to treat C. difficile infections. Glycan array screening previously revealed that the LeA-LacNAc pentasaccharide binds strongly to TcdA. Here we report the efficient syntheses of the pentasaccharide and a structurally related tetrasaccharide motif. These compounds will be used to better define the carbohydrate-binding specificity of toxins from C. difficile, which will hopefully lead to the development of improved therapeutics.
Introduction
Clostridium difficile infection is one of the most common hospital-acquired diseases in the industrialized world, affecting hundreds of thousands of people in North America annually.1 The clinical symptoms can range from mild to severe diarrhea, and in extreme cases pseudomembranous colitis.2 The disease is linked to the use of broad-spectrum antibiotics that disrupt the normal microflora found in the colon.3 The resistance of C. difficile to commonly used antibiotics such as clindamycin and fluoroquinolones makes the colon vulnerable to colonization by these opportunistic pathogens when such antibiotics are administered systemically.4 Paradoxically, broad-spectrum antibiotics such as metronidazole and vancomycin remain the most effective and commonly used treatments of C. difficile infections, even though a side-effect of these treatments is the continued disruption of normal microbial populations in the colon. As a result, recurrent infections of C. difficile are unfortunately extremely common and troubling limitation of current treatments. To develop new and more effective treatments, we and others have undertaken molecular studies of virulence factors from C. difficile. Two of the most important virulence factors are the large toxin proteins TcdA and TcdB.5,6 These proteins bind to intestinal epithelial cells and specifically glucosylate a specific Thr residue in the Rho GTPases that are essential to the maintenance of the cellular cytoskeleton.7,8 Because the initial adhesion of toxins to host intestinal epithelial cells is mediated by carbohydrates,9 several novel approaches for treating C. difficile infections have been proposed based on the competitive inhibition of toxin binding to host epithelial cells10–12 Although a specific human receptor has yet to be identified, the trisaccharide αGal(1→3)βGal(1 →4)βGlcNAc was found to bind to toxin A with weak affinity and the crystal structure of the complex was solved.13 To define better the binding specificities of native toxin molecules, TcdA and TcdB, His6-tagged recombinant proteins containing portions of the carboxyl-terminal carbohydrate-binding domains of TcdA and TcdB have been used by our group and others to screen the glycan microarray at the Consortium for Functional Glycomics (CFG).14 Several carbohydrate structures showed strong binding to TcdA, including 1 and 3 (Fig. 1). Compound 3 is a pentasaccharide containing a Lewis A motif linked to the O-3′ position of a lactosamine disaccharide (LeA-LacNAc) while the related tetrasaccharide contains a neolactosamine residue linked to the same lactosamine moiety. Here we report the chemical synthesis of two analogs 2 and 4, which have the same glycan sequences compared to 1 and 3 respectively. A longer 6-aminohexyl linker was chosen to be the aglyconic group due to its higher flexibility when conjugating to scaffolds to generate potentially high affinity inhibitors using the multivalency concept.15,16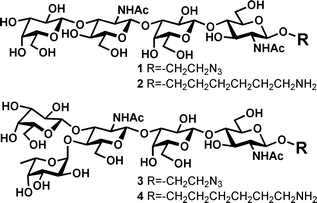 | ||
| Fig. 1 Structures of ligands bounds to toxin A from CFG screen experiments and synthetic targets. | ||
Results and discussion
Since both compounds 2 and 4 have a LacNAc disaccharide fragment at the reducing end, a retrosynthetic analysis (Fig. 2) revealed that if a 2 + 2 or 3 + 2 disconnection could be respectively performed for tetrasaccharide 2 and pentasaccharide 4, a disaccharide acceptor 5 could become a common intermediate for the synthesis of both saccharides. Although there are three hydroxyl groups presented in 5, we estimated that the OH-3 of the galactose unit should be the most reactive, because the OH-2 of the same unit was known to be less reactive;17 the OH-3 of the glucosamine unit is sterically shielded by a phthalimido group at C-2 and galactosyl residue at O-4 thus less accessible for glycosylations. A neolactosamine donor is required for the synthesis of tetrasaccharide 2. We decided to use thioglycosides as potential donors because of their versatility and remarkable stability towards many reaction conditions during functional group transformations. Donor 6 with the O-6 and O-4 of the GlcNAc residue orthogonally protected with a benzyl and a p-methoxybenzyl group respectively was the initial choice, as a selective removal of the p-methoxybenzyl group could generate a key alcohol 9, required for the synthesis of a Lewis A trisaccharide donor 8 which is essential for the synthesis of pentasaccharide 4. Alternatively, donor 7 with the O-6 and O-4 of the GlcNAc residue protected with a benzylidene could be used, as a regioselective opening of the benzylidene could also generate donor 9. All disaccharides 5–7 could be prepared using galactosyl donors 10,181117 or 12 and N-phthalimido protected thioglycosides 13 or 14 which can be used as either thioglycoside donor or acceptor. Fucosyl donor 15 was chosen to react with alcohol 9 to form the Lewis A19 trisaccharide donor 8. The phthalic group was selected to be the protecting group of the 2-amino group of glucosamine20 because of its excellent participating property that leads to the exclusive formation of β-glycosides; although the 2,2,2-trichloroethoxycarbonyl (Troc)19e,21 could also be used as a protecting group to give β-glycosides, a clear advantage of using phthalic group for the protection of amine is that all intermediates bearing the phthalimido group can be easily detected by charring with acid in thin-layer chromatography analysis; additionally, the phthalimido group exhibits higher stability in basic conditions compared to Troc group.22 | ||
| Fig. 2 Retrosynthetic analysis for both tetrasaccharide 2 and pentasaccharide 4. | ||
Scheme 1 showed the synthesis of N-phthalimido protected glucosamine thioglycosides 13 and 14. The known compound 1623 was converted to 17 in large scale and high yield by the treatment of 16 with boron trifluoride etherate and p-chlorophenylthiol which is a preferred thiol because it is less malodorous compared to the more coventional ethanethiol or thiophenol. We found that if an in situ acetylation step was carried out after the reaction, we could convert the remaining thiol to the corresponding thioacetate thus essentially eliminating the unpleasent smell. In agreement with the literature,24 the β-OAc of 16 was found to react much faster (4 h) than the corresponding α-OAc (up to 72 h). No chromatography was needed to isolate the thioglycoside 17 as a simple recrystallization from a mixture of ethyl acetate–hexane afforded the pure thioglycoside 17 in 76% yield, illustrating the advantage of using p-chlorophenylthiol as the aglycone. The subsequent removal of acetates was carried out using a milder Zémplen transesterification condition22 with a guanidine/guanidinium buffer as the deacetylation reagent in anhydrous methanol; compound 18 crashed out from the reaction media in pure form and in almost quantitative yield (98%). The more conventional condition (sodium methoxide in methanol) was found to be less suitable as it resulted in a partial opening of the phthalimido group. To synthesize compound 14, a transacetalation was carried out on triol 18 with benzaldehyde dimethyl acetal under the catalysis of camphorsulfonic acid to give the desired alcohol 14 in excellent yield (93%). To regioselectively protect the primary alcohol of 18, the stanylidene acetal-mediated benzylation was found to be efficient; thus a solution of 18 with 1 equivalent of dibutyltin oxide in toluene was heated to reflux to form intermediate stanylidene acetal which allowed the installation of a benzyl group at O-6 in a regioselective manner to give compound 19 in 67% yield. To synthesize alcohol 13, another dibutyltin oxide-mediated alkylation was carried out with p-methoxybenzyl (MBn) chloride; compound 13 was obtained in moderate yield (33%) together with the regioisomer 20 which has the MBn group installed at the O-3 position (30% yield).
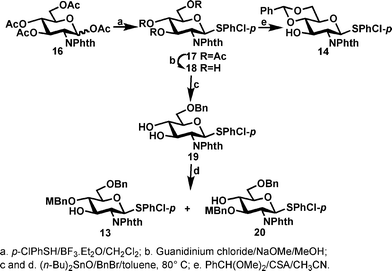 | ||
| Scheme 1 Syntheses of glucosaminyl donors/acceptors. | ||
The synthesis of the common disaccharide 5 (Scheme 2) began with the preparation of acceptor 22 which was obtained by coupling the 6-azidohexan-1-ol 21 with thioglycoside 19 under the activation of N-iodosuccinimide (NIS)/trifluoromethanesulfonic acid (TfOH).
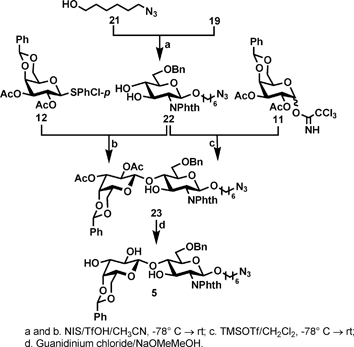 | ||
| Scheme 2 Synthetic route to the common disaccharide acceptor 5. | ||
Despite the presence of two free hydroxyl groups in donor 19, glycosylation proceeded in a remarkably smooth way to afford 22 in 85% yield. Clearly, the primary hydroxyl group of 21 exhibited sufficiently high reactivity to compete with all inter/intramolecular self-condensation side reactions. Coupling of 12 with 22 under the activation of NIS/TfOH condition gave the desired disaccharide 23 in moderate yields (47%). To improve the glycosylation efficiency, we switched the donor to imidate 11 to afford the desired disaccharide 23 in slightly improved yield (60%) under the catalysis of trimethylsilyl trifluoromethanesulfonate (TMSOTf) at −78° C. Finally, a selective deprotection of acetates was carried out using guanidine/guanidium buffer conditions to afford the triol 5 in high yield (84%).
We next investigated the preparation of βGal(1→3)βGlcNPhth disaccharide donor 6 using the designed acceptor 13. We first attempted the glycosylation with the peracetylated galactopyranosyl imidate 10 in anhydrous dichloromethane (Scheme 3) under the catalysis of TMSOTf. By starting the reaction at 0° C and slowly warming the reaction up to room temperature, surprisingly, no desired glycoside 6 was formed even if we used a large excess amount of 10 (up to 3.0 equivalents). The major compound isolated from the reaction mixture was the unreacted acceptor 13 and a small amount of rearranged product 25 from donor. Attempts to change the reaction solvent to others such as anhydrous acetonitrile also failed. When we employed the more reactive imidate donor 11, we were still unable to observe the formation of the desired disaccharide 24. The imidate donor quickly rearranged to form the glycosyl amide 26 and the acceptor 13 was recovered.
 | ||
| Scheme 3 Attempts to prepare disaccharide 6 and 24 from 13. | ||
Previously, it was well-established25 that if a tribenzylated L-fucopyranosyl residue is attached to the O-4 position of a N-phthalimido protected glucosamine acceptor, the OH-3 became inaccessible for glycosylations due to steric hindrance. Here the fact that we used a much smaller and electron-donating MBn group at O-4, and still could not glycosylate the OH-3 position, is surprising. In another experiment (Scheme 4), we reacted the analogous acceptor 19 which had no protecting group at O-4 with a reactive fucosyl imidate donor 28 (1.3 equivalents), we found that both OH-3 and OH-4 were reactive even at low temperature (−78° C); the OH-3 exhibited even higher reactivity than OH-4 as the corresponding disaccharide 29 and 30 were formed and isolated in 52% and 27% yields respectively. We therefore concluded that the low reactivity of OH-3 in 13 was still due to the steric shielding from both the MBn group at O-4 and the 2-phthalimido group; probably both groups form a pocket which traps OH-3 and renders it unreactive towards the oxocarbenium intermediates formed from the glycosyl donors.
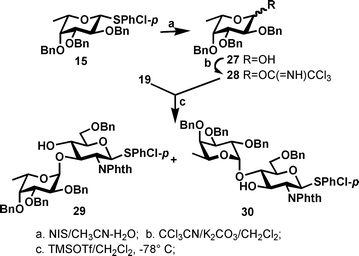 | ||
| Scheme 4 Probing the sterically shielding effect on OH-3 of 2-phthalimido protected acceptor. | ||
Based on the above probing results, the advantage of using 4,6-benzylidenated 14 to synthesize the required disaccharide 9 became evident, because the cyclic benzylidene group provides simultaneous protection to both the O-4 and O-6 positions while making OH-3 available for reactions; most importantly the smaller cyclic acetal relieves steric congestion around OH-3, making it more accessible than OH-3 in 13. Indeed, as shown in Scheme 5, when 14 was subjected to reaction with imidate 10 under the activation of TMSOTf, the desired disaccharide 7 was formed in a smooth manner and isolated in good yield (75%).
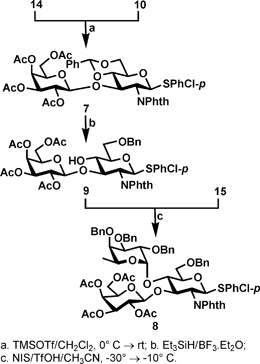 | ||
| Scheme 5 Preparation of neolactosamine and Lewis A donor. | ||
Using the triethylsilane26 as the reducing agent under the catalysis of boron trifluoride etherate, the 4,6-benzylidene group in 7 was regioselectively opened to afford the expected disaccharide alcohol 9 in almost quantitative yield (98%). The free OH-4 in 9 should be much less steric hindered due to the higher rotational freedom of benzyl group attached to the O-6 position thus is readily available for α-fucosylation. We applied the armed-disarmed glycosylation principle by reacting the less reactive thioglycoside 9 (disarmed) with the more reactive thiofucoside 15 (armed) under the activation of NIS/TfOH to afford the desired Lewis A trisaccharide donor 8 in excellent yield (84%)
The success in preparing the requires donors 8 and 9 allowed us to proceed further to synthesize the target tetrasaccharide 2 and pentasaccharide 4. As shown in Scheme 6, under the NIS/TfOH-promoted glycosylation conditions using triol 5 as the acceptor and thioglycoside 9 as the donor, the desired tetrasaccharide 31 was obtained in excellent yield (87%). As expected, despite the presence of three hydroxyl groups, the glycosylation proceeded in a remarkably regioselective manner to give only OH-3′ glycosylated product. The regioselectivity of glycosylation was established by a series of NMR experiments: 1D 1H and 2D 1H-1H COSY NMR revealed a broad doublet (3J 1.9 Hz) at 2.83 ppm, which appeared to correlate with H-2′, suggesting that it's the hydroxyl group (OH-2′) attached to the C-2′ position (the reducing end galactosyl unit). This was further confirmed by a selective 1D TOCSY experiment (see ESI†): we selectively excited the hydroxyl signal at 2.83 ppm, and observed that the magnetization was indeed transferred to the H-1′, H-2′, H-3′ and H-4′ protons (the reducing end galactosyl unit). These results confirmed that the glycosylation occurred at OH-3′. Next, the acetyl and N-phthalimido groups of 31 were concurrently removed in one step by the treatment with hydrazine hydrate in methanol under refluxing conditions. TLC revealed that the deprotection went very clean, only one spot was observed; the intermediate diamine was not isolated but subjected to a regioselective acetylation with acetic anhydride in methanol to provide the diamide which was subsequently debenzylated in 10% acetic acid–methanol under the catalysis of 20% Pd(OH)2 on charcoal to give the targeted tetrasaccharide 2 in 48% overall yield after three steps. Compound 2 was isolated by passing through a C18 Sep-Pak cartridge using a gradient of MeOH–H2O (0% → 10%) as the eluent.
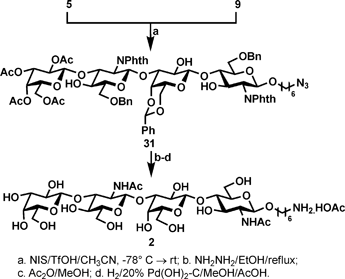 | ||
| Scheme 6 Synthesis of tetrasaccharide 2. | ||
Similarly, when the Lewis A donor 8 was reacted with 5 under similar conditions as for 9, the pentasaccharide 32 was smoothly formed and isolated in very good yield (70%) and excellent regioselectivity (Scheme 7). Following the same deprotecting sequences as for 31, the desired pentasaccharide 4 was obtained in 55% overall yield after three steps.
 | ||
| Scheme 7 Synthesis of pentasaccharide 4. | ||
The structures of both compounds 2 and 4 were confirmed by 1D and 2D NMR experiments, and also by high resolution mass spectrometry (see ESI†).
In conclusion, we have presented a concise route to synthesize a pentasaccharide containing the LeA-LacNAc sequence which has previously been shown to bind to TcdA from C. difficile. By applying the same strategy, a related tetrasaccharide was also prepared. Carefully choosing orthogonally protected donors and acceptors was found to be the key to the success of the syntheses and by effectively applying the armed-disarmed principle, we have significantly reduced the number of required steps thus improved the overall efficiency of the syntheses. These compounds are currently being used for binding and crystallographic studies, which will hopefully better define the molecular basis for carbohydrate binding and ultimately lead to the development of novel therapeutics.
Experimental section
General methods
Optical rotations were determined in a 5 cm cell at 25 ± 2 °C. [α]D values were given in units of 10−1 deg cm2 g−1. Analytical TLC was performed on Silica Gel 60-F254 (Merck, Darmstadt) with detection by quenching of fluorescence and/or by charring with 5% sulfuric acid in water or with a ceric ammonium molybdate dip. All commercial reagents were used as supplied unless otherwise stated. Column chromatography was performed on Silica Gel 60 (Silicycle, Ontario). Molecular sieves were stored in an oven at 100 °C and flame-dried under vacuum before use. Organic solutions from extractions were dried with anhydrous Na2SO4 prior to concentration under vacuum at < 40 °C (bath). 1H NMR spectra were recorded at 300 and 400 MHz on Bruker spectrometers. The first order proton chemical shifts δH and δC were reported in δ (ppm) and referenced to either residual CHCl3 (δH 7.24, δC 77.0, CDCl3) or residual CD2HOD (δH 3.30, δC 49.5, CD3OD). 1H and 13C NMR spectra were assigned with the assistance of GCOSY, GHSQC; for oligosaccharides with two galactose and two glucosamine residues, residues closer to the non-reducing end were defined with the “I” suffix (ex. Gal-I, GlcN-I) while the ones closer to the reducing end were defined with the “II” suffix (ex: Gal-II, GlcN-II). Microanalyses and electrospray ionization (ESI) mass spectrometry were performed by the analytical services of the Department of Chemistry, University of Calgary. For high resolution mass determination, spectra were obtained by voltage scan over a narrow range at a resolution of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and recorded by the analytical services of the Department of Chemistry, University of Alberta.
000 and recorded by the analytical services of the Department of Chemistry, University of Alberta.Guanidine/guanidinium chloride solution
To a solution of guanidinium chloride (9.6 g, 100 mmol) in a mixture of anhydrous MeOH (900 mL) and CH2Cl2 (100 mL), was added a solution of NaOMe in anhydrous MeOH (1.5 M, 15 mL), and the resulting clear solution was kept at rt and used for subsequent de-O-acetylations.Data for 13 (Found: C, 65.47; H, 5.30; N, 2.02%. C35H32NO7SCl requires C, 65.06; H, 4.99; N, 2.17%); [α]D +13.5 (c 0.7, CHCl3); δH (400 MHz, CDCl3) 7.92–7.78 (m, 2H, Phth), 7.78–7.68 (m, 2H, Phth), 7.45–7.29 (m, 7H, Ar), 7.21–7.12 (m, 4H, Ar), 6.84 (d, 2H, J 8.6 Hz, Ar), 5.54 (d, 1H, J 10.2 Hz, H-1), 4.70–4.56 (m, 4H, Bn), 4.42 (ddd, 1H, J 10.3, 8.7, 4.2 Hz, H-3), 4.20 (dd, 1H, J 10.3, 10.3 Hz, H-2), 3.88–3.79 (m, 2H, H-6a + H-6b), 3.78 (s, 3H, OMe), 3.67 (ddd, 1H, J 9.8, 3.9, 2.1 Hz, H-5), 3.57 (dd, 1H, J 9.5, 8.7 Hz, H-4), 2.25 (d, 1H, J 4.4 Hz, OH-3); δC (100 MHz, CDCl3) 159.50, 138.13, 134.22, 134.20, 131.63, 130.41, 130.11, 129.64, 128.97, 128.45, 127.77, 127.75, 123.81, 114.08 (Ar), 83.06 (C-1), 79.28 (C-5), 78.72 (C-4), 74.37 (Bn or MBn), 73.51 (MBn or Bn), 72.73 (C-3), 68.88 (C-6), 55.57 (C-2), 55.26 (OMe–MBn); m/z (ESI-MS) calcd for [C35H32NO7SCl + Na]+ 668.2, found 668.2.
Data for 20 (Found: C, 65.46; H, 5.25; N, 1.96%. C35H32NO7SCl requires C, 65.06; H, 4.99; N, 2.17%); [α]D +52.0 (c 0.6, CHCl3); δH (400 MHz, CDCl3) 7.86–7.80 (m, 1H, Phth), 7.77–7.64 (m, 3H, Phth), 7.42–7.28 (m, 7H, Ar), 7.15 (d, 1H, J 8.5 Hz, Ar), 6.95 (d, 2H, J 8.6 Hz, Ar), 6.45 (d, 2H, J 8.6 Hz, Ar), 5.51 (d, 1H, J 10.1 Hz, H-1), 4.65 (d, 1H, J 12.1 Hz, Bn or MBn), 4.63 (d, 1H, J 11.8 Hz, MBn or Bn) 4.53 (d, 1H, J 11.8 Hz, MBn or Bn), 4.47 (d, 1H, J 12.1 Hz, Bn or MBn), 4.23 (dd, 1H, J 9.9, 8.1 Hz, H-3), 4.18 (dd, 1H, J 10.1, 10.1 Hz, H-2), 3.87–3.76 (m, 3H, H-4 + H-6a + H-6b), 3.69 (ddd, 1H, J 9.5, 4.8, 4.8 Hz, H-5), 3.63 (s, 3H, OMe–MBn), 2.90 (d, 1H, J 2.7 Hz, OH-4); δC (100 MHz, CDCl3) 158.93, 137.68, 134.24, 134.11, 133.94, 133.91, 131.55, 130.38, 130.11, 129.64, 128.97, 128.54, 127.93, 127.77, 123.47, 123.21, 113.53 (Ar), 83.29 (C-1), 79.31 (C-3), 78.00 (C-5), 74.20 (Bn or MBn), 73.89 (C-4), 73.76 (MBn or Bn), 70.50 (C-6), 54.90 (OMe–MBn), 54.43 (C-2); m/z (ESI-MS) calcd for [C35H32NO7SCl + Na]+ 668.2, found 668.2.
Method A. Thioglycoside 12 (1.028 g, 2.15 mmol) and diol 22 (563 mg, 1.07 mmol) were dissolved in anhydrous CH2Cl2 (6 mL) under Ar, and 4 Å molecular sieves (1.3 g) were added. After stirring at rt for 2 h, the mixture was cooled to −78° C, and NIS (532 mg, 2.25 mmol, 2.1 equiv.) was added. After stirring for another 5 min, TfOH (30 μL) was added dropwise, and the reaction was slowly warming up to rt within 2 h. Et3N (1 mL) was added to quench the reaction. The reaction mixture was worked up as 22 and glycoside 23 (434 mg, 47% yield) was obtained by column chromatography on silica gel using 15% acetone–toluene as eluent.
Method B. A mixture of imidate 1117 (1.659 g, 3.34 mmol) and diol 22 (700 mg, 1.34 mmol) was dissolved in anhydrous CH2Cl2 (6.5 mL) under Ar, and 4 Å molecular sieves (1.9 g) were added. After stirring at rt for 1 h, the mixture was cooled to −78° C, and TMSOTf (18 μL) was added dropwise, and the temperature of the reaction was slowly warmed up to rt. TLC (30% AcOEt–toluene) showed the all starting materials disappeared. The reaction was quenched by the addition of Et3N (0.5 mL) and the mixture was filtered off. After removing the solvent under vacuum, the residue was purified by column chromatography on silica gel using 15% AcOEt–toluene as the eluent to give the desired disaccharide 23 (725.2 mg, 63.2% yield) as a white foam (Found: C, 61.40; H, 5.94; N, 6.16%. C44H50N4O14 requires C, 61.52; H, 5.87; N, 6.52%);. [α]D +9.3 (c 0.6, CHCl3); δH (400 MHz, CDCl3) 7.95–7.78 (m, 2H, Phth), 7.78–7.63 (m, 2H, Phth), 7.53–7.30 (m, 10H, Ph + Bn), 5.44 (s, 1H, PhCH), 5.34 (dd, 1H, J 10.4, 8.0 Hz, H-2–Gal), 5.22 (d, 1H, J 8.5, H-1–GlcN), 4.86 (dd, 1H, J 10.4, 3.6 Hz, H-3–Gal), 4.77 (d, 1H, J 12.0, Bn), 4.53 (d, 1H, J 12.0, Bn), 4.50 (d, 1H, J 8.0, H-1–Gal), 4.44 (ddd, 1H, J 10.9, 8.3, 0.7 Hz, H-3–GlcN), 4.33 (dd, 1H, J 3.6, 0.7 Hz, H-4–Gal), 4.26 (dd, 1H, J 12.7, 1.3 Hz, H-6a–Gal), 4.18 (dd, 1H, J 10.9, 8.5 Hz, H-2–GlcN), 4.02 (d, 1H, J 0.6 Hz, OH-3–GlcN), 3.99 (dd, 1H, J 12.7, 1.6 Hz, H-6b–Gal), 3.84 (ddd, 1H, J 9.8, 6.3, 6.3 Hz, OCHaHb), 3.80–3.69 (m, 3H, H-6a–GlcN + H-6b–GlcN + H-4–GlcN), 3.65 (m, 1H, H-5–GlcN), 3.49 (m, 1H, H-5–Gal), 3.43 (ddd, J 9.7, 7.2, 5.7 Hz, OCHaHb), 3.04 (t, 2H, J 7.0 Hz, CH2N3), 2.07 (s, 3H, Ac), 2.03 (s, 3H, Ac), 1.53–1.37 (m, 2H, OCHaHbCH2), 1.36–1.21 (m, 2H, CH2CH2N3), 1.21–1.04 (m, 4H, 2 × CH2–chain); δC (100 MHz, CDCl3) 170.58 (2 × CO), 168.91 (CO), 138.16, 137.22, 133.95, 131.80, 129.03, 128.47, 128.09, 127.86, 127.81, 126.28, 101.19 (PhCH), 100.97 (C-1–Gal), 98.32 (C-1–GlcN), 81.28 (C-4–GlcN), 74.26 (C-5–GlcN), 73.61 (Bn), 73.01 (C-4–Gal), 71.69 (C-3–Gal), 69.58 (C-3–GlcN), 69.45 (OCHaHb), 68.63 (C-2–Gal), 68.35 (C-6–Gal), 67.98 (C-6–GlcN), 66.59 (C-5–Gal), 56.05 (C-2–GlcN), 51.13 (CH2N3), 29.10 (OCHaHbCH2), 28.58 (CH2CH2N3), 26.20 (CH2–chain), 25.41 (CH2–chain), 20.78 (Ac), 20.74 (Ac); m/z (ESI-HRMS) calcd for [C44H50N4O14 + Na]+ 881.3216, found 881.3213.
Acknowledgements
We wish to acknowledge the Alberta Ingenuity, NSERC and the University of Calgary for supporting the current project, and Prof. D. Bundle and Dr. A. Morales-Izquierdo from the Department of Chemistry, University of Alberta for acquiring the high resolution mass spectra.Notes and references
- (a) C. L. McDonald, G. E. Killgore, A. Thompson, R. C. Owens Jr., S. V. Kazakova, S. P. Sambol, S. Johnson and D. N. Gerding, N. Engl. J. Med., 2005, 353, 2433–2441 CrossRef; (b) H. Kato, T. Yokoyama and Y. Arakawa, J. Med. Microbiol., 2005, 54, 167–171 CrossRef CAS.
- (a) J. G. Bartlett, T. Chang, N. S. Taylor and A. B. Onderdonk, Rev. Infect. Dis., 1979, 1, 370–378 Search PubMed; (b) M. S. Rubin, L. E. Bodenstein and K. C. Kent, Dis. Colon Rectum, 1995, 38, 350–354 Search PubMed.
- J. G. Bartlett, N. Engl. J. Med., 2002, 346, 334–339 CrossRef.
- (a) S. L. Gorbach, N. Engl. J. Med., 1999, 341, 1689–1691 CrossRef; (b) J. G. Bartlett, A. B. Onderdonk, R. L. Cisneros and D. L. Kasper, J. Infect. Dis., 1977, 136, 701–705 CAS.
- D. E. Voth and J. D. Ballard, Clin. Microbiol. Rev., 2005, 18, 247–263 CrossRef CAS.
- N. S. Taylor, G. M. Thorne and J. G. Bartlett, Infect. Immun., 1981, 34, 1036–1043 CAS.
- (a) W. P. Ciesla, Jr. and D. A. Bobak, J. Biol. Chem., 1998, 273, 16021–16026 CrossRef CAS; (b) I. Just, J. Selzer, M. Wilm, C. von Eichel-Streiber, M. Mann and K. Aktories, J. Biol. Chem., 1995, 375, 500–503 CAS; (c) I. Just, M. Wilm, J. Selzer, G. Rex, C. von Eichel-Streiber, M. Mann and K. Aktories, J. Biol. Chem., 1995, 270, 13932–13936 CrossRef CAS.
- For a review, see: T. Jank, T. Giesemann and K. Aktories, Glycobiology, 2007, 17, 15R–22R Search PubMed.
- K. D. Tucker and T. D. Wilkins, Infect. Immun., 1991, 59, 73–78 CAS.
- I. Ofek, D. L. Hasty and N. Sharon, FEMS Immunol. Med. Microbiol., 2003, 38, 181–191 CrossRef CAS.
- T. Dingle, S. Wee, G. L. Mulvey, A. Greco, E. N. Kitova, J. Sun, S. Lin, J. S. Klassen, M. M. Palcic, K. K.-S. Ng and G. D. Armstrong, Glycobiology, 2008, 18, 698–706 CrossRef CAS.
- (a) K. D. Tucker and T. D. Wilkins, Infect. Immun., 1991, 59, 73–78 CAS; (b) S. Teneberg, I. Lönnroth, J. F. Torres López, U. Galili, M. O. Halvarsson, J. Angström and K. A. Karlsson, Glycobiology, 1996, 6, 599–609 CrossRef CAS.
- (a) A. Greco, J. G. S. Ho, S.-J. Lin, M. M. Palcic, M. Rupnik and K. K.-S. Ng, Nat. Struct. Mol. Biol., 2006, 13, 460–461 CrossRef CAS; (b) J. G. S. Ho, A. Greco, M. Rupnik and K. K.-S. Ng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18373–18378 CrossRef CAS.
- http://www.functionalglycomics.org/.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- (a) P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 CrossRef CAS; (b) P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS.
- U. Greilich, P. Zimmermann, K.-H. Jung and R. R. Schmidt, Liebigs Ann. Chem., 1993, 1993, 859–864 CrossRef.
- R. R. Schmidt, J. Michel and M. Roos, Liebigs Ann. Chem., 1984, 1343–1357 CrossRef CAS.
- (a) R. U. Lemieux and H. Driguez, J. Am. Chem. Soc., 1975, 97, 4063–4068 CrossRef CAS; (b) K. L. Matta and S. S. Rana, Carbohydr. Res., 1983, 117, 101–111 CrossRef CAS; (c) J. März and H. Kunz, Synlett, 1992, 589–590 CrossRef; (d) L. Yan and D. Kahne, J. Am. Chem. Soc., 1996, 118, 9239–9248 CrossRef CAS; (e) L. Manzoni, L. Lay and R. R. Schmidt, J. Carbohydr. Chem., 1998, 17, 739–758 CrossRef CAS.
- (a) J. Banoub, P. Boullanger and D. Lafont, Chem. Rev., 1992, 92, 1167–1195 CrossRef CAS; (b) A. F. G. Bongat and A. V. Demchenko, Carbohydr. Res., 2007, 342, 374–406 CrossRef.
- (a) S. Kusumoto, H. Yoshimura, M. Imoto, T. Shimamoto and T. Shiba, Tetrahedron Lett., 1985, 26, 909–912 CrossRef CAS; (b) T. B. Windholz and D. B. R. Johnston, Tetrahedron Lett., 1967, 8, 2555–2557 CrossRef; (c) T. Mukaiyama and K. Matsubara, Chem. Lett., 1992, 1755–1758 CAS; (d) K. Matsubara and T. Mukayama, Chem. Lett., 1993, 2145–2148 CrossRef CAS; (e) M. Schultz and H. Kunz, Tetrahedron Lett., 1992, 33, 5319–5322 CrossRef CAS; (f) U. Ellervik and G. Magnusson, Carbohydr. Res., 1996, 280, 251–260 CrossRef CAS.
- U. Ellervik and G. Magnusson, Tetrahedron Lett., 1997, 38, 1627–1628 CrossRef CAS.
- R. U. Lemieux, T. Takeda, B. Y. Chung, In Synthetic Methods for Carbohydrates, H. S El Khadem, ed., ACS Symposium Series: Washington, DC, 1976; Vol. 39, pp. 90–115 Search PubMed.
- L. Olsson, S. Kelberlau, Z. J. Jia and B. Fraser-Reid, Carbohydr. Res., 1998, 314, 273–276 CrossRef CAS.
- (a) U. Ellervik and G. Magnusson, J. Org. Chem., 1998, 63, 9314–9322 CrossRef CAS; (b) T. Zhu and G.-J. Boons, Carbohydr. Res., 2000, 329, 709–715 CrossRef CAS.
- S. D. Debenham and E. J. Toone, Tetrahedron: Asymmetry, 2000, 11, 385–387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures for compounds 12, 14–15, 17–18, 25–26, 28–30 and 1H and 13C NMR spectra of all synthesized compounds. See DOI: 10.1039/b914193f |
| This journal is © The Royal Society of Chemistry 2010 |