DIBAL-H mediated triple and quadruple debenzylations of perbenzylated cyclodextrins†
Girish K. Rawal, Shikha Rani, Sandra Ward and Chang-Chun Ling*
Alberta Ingenuity Center for Carbohydrate Science, Department of Chemistry, University of Calgary, Calgary Alberta, T2N 1N4, Canada. E-mail: ccling@ucalgary.ca; Fax: +1 (403)289-9488; Tel: +1 (403) 220-2768
First published on 12th November 2009
Abstract
Diisobutylaluminium hydride (DIBAL-H) mediated reductive removal of benzyl groups was investigated for perbenzylated α-, β- and γ-cyclodextrins using DIBAL-H in hexane as the reagent. It was found that under the new conditions, the debenzylation can be better controlled to provide sequentially tri- and tetra-debenzylated products in moderate yields and in a regioselective manner. In the case of α-cyclodextrin, the removal of the third and fourth benzyl groups took a different path involving the secondary rim, compared to β- and γ-cyclodextrins which both gave only 6-O-debenzylated products.
Introduction
Recently, there has been an increasing interest1–4 in developing methodologies for the regioselective substitution of cyclodextrins (CDs). These multi-functionalized CDs have great utility in many areas such as for use as novel host molecules to complex bioactive compounds with high affinities,5 as artificial enzymes6 and as gene delivery vehicles.7 Among all the hydroxyls present in a CD molecule, those attached to the C-6's of the glucopyranosyl units are sterically the most accessible and they are all located at the primary rim of the hydrophobic cavity; the remaining hydroxyl groups are all found at the secondary rim because they are attached to either C-2's or C-3's of the sugar units; they are sterically less accessible. It's relatively straightforward to persubstitute all hydroxyls or regioselectively differentiate one group of hydroxyls from the other.8 However, it is extremely challenging to regioselectively differentiate hydroxyls of the same group due to their identical chemical properties. There have been a few methods reported in literature involving the use of either bulky groups such as trityl9 and its variant10 or geometrically restricted capping reagents to react with close hydroxyl pairs at the primary rim.11–13 Several years ago, Sinaÿ's group introduced a distinct approach14 which involved the use of diisobutylaluminium hydride (DIBAL-H) in toluene as the reagent to remove 1–2 benzyl groups attached to O-6 positions of perbenzylated α-, β- and γ-CDs. The reaction conditions could be tuned to produce either mono- or didebenzylated CDs in moderate to excellent yields. The most prominent feature of the method was the high regioselectivity during the removal of the second benzyl group. For example, for α/β-CDs, the second debenzylation occurs exclusively at the O-6 of the D sugar unit of cyclic oligosaccharides to form 6A,6D-O-didebenzylated α-/β-CD in high yields while in the case of perbenzylated γ-CD, a mixture of 6A,6D/6A,6E-O-didebenzylated products was formed in moderate yield. More recently, we reported our preliminary results15 on the one-pot triple and quadruple debenzylations of perbenzylated α-CD by slightly modified conditions using DIBAL-H in hexane as reagent instead of the original DIBAL-H in toluene (Scheme 1). The simple switch of reagent resulted in much slower debenzylation thus allowing for a better monitoring and control of the progress of the reaction. In addition to the known mono- (2) and didebenzylated α-CD (3), we also observed the formation of two major debenzylated products, namely a triol (4, 32% yield) and a tetraol (5, 10% yield).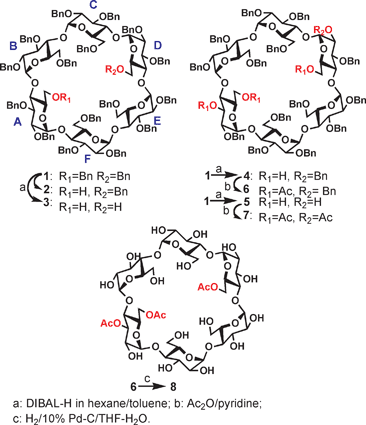 | ||
| Scheme 1 Sequential debenzylations of perbenzylated α-CD. | ||
As shown in the structures, both the third and fourth benzyl removals occurred at the secondary rim of the α-CD cavity and remarkably, the glucopyranosyl unit which had a prior O-6 debenzylation also preferred to have its O-3 benzyl removed. The structures of both 4 and 5 were determined via either a sequence of acetylation/hydrogenation to remove all the benzyl groups (4→6→8) or simply an acetylation (5→7). It is important to note that all acetylated products were stable after the removal of benzyl groups and no acetate migration was observed. For example, in compound 6, we observed that a glucopyranosyl unit had respectively an acetate at O-3 and O-6 positions by a combination of 1D/2D (GCOSY and GTOCSY) NMR experiments and these substitution patterns were further confirmed by a selective 1D TOSCY experiment (see supporting information†); the same acetate-substitution patterns were also observed in the fully debenzylated polyol 8 by NMR experiments. These results suggested that the catalytic hydrogenation step was carried out under extremely mild conditions therefore no acetate migration occurred. In this paper, we report further studies in applying our reported conditions to perbenzylated β- and γ-CDs. A rational to explain the unusual debenzylation path is proposed.
Results and discussion
β-Cyclodextrin
The intriguing debenzylation path of 1 prompted us to investigate the debenzylation of perbenzylated β-CD (9). We treated compound 9 with DIBAL-H in hexane (40 equiv, 0.9 M) under similar heating conditions (50 °C). As shown in Table 1; it was confirmed again that the debenzylation process was sluggish. Even after 12 h, we still isolated 15% starting material, and the previously known 6-O-monodebenzylated β-CD (10) and its 6A,6D-O-didebenzylated analog (11) were isolated in only 45% and 25% yields respectively. However, in the literature, it was reported14a that if DIBAL-H in toluene was used at 0.5 M concentration, compound 11 could be isolated in 87% yield after heating for just 2 h at 50° C. After 24 h, the starting material was completely consumed and we started to observe the formation of some more polar compounds besides the monool 10 and diol 11– both remained as the major components of the reaction mixture until 48 h (entries 2 and 3). The monool 10 finally disappeared after 3 days and the amounts of the two new spots gradually increased and eventually became the majority after 96 h. The less polar spot (Rf 0.5, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 hexane:ethyl acetate) was isolated in 25% and the more polar spot (Rf 0.25, 1:1 ethyl acetate:hexane) in 22% yield (entries 4 and 5).
:3 hexane:ethyl acetate) was isolated in 25% and the more polar spot (Rf 0.25, 1:1 ethyl acetate:hexane) in 22% yield (entries 4 and 5).
The less polar spot was subjected to an acetylation and from 1H NMR, it was found to be a inseparable 1:1 mixture containing two triacetylated β-CD derivatives because 6 acetate peaks were observed (15 and 16); interestingly none of the acetyl groups were found to attach to a secondary hydroxyl group as after a debenzylation, we did not observe any proton resonating above 5.0 ppm (see supporting information†) which was the case for α-CD (compounds 5 and 7). This was further confirmed from the 1H NMR spectra of the fully debenzylated products (not shown). The structures of the two triols were assigned to be 6A,6B,6D-β-O-tridebenzylated β-CD 12 and 6A,6B,6E-β-O-tridebenzylated 13 (Scheme 2) respectively, according to our proposed rational (see later).
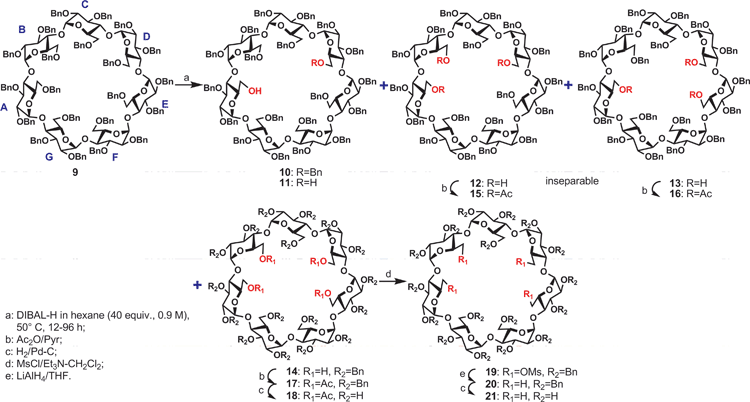 | ||
| Scheme 2 Stepwise debenzylations of perbenzylated β-cyclodextrin. | ||
Similarly, the more polar spot was acetylated and fortunately, the 1H and 13C NMR spectra revealed that it constitutes a single compound 17 with four acetates. Since we did not observe any 1H NMR signals with chemical shifts greater than 5.0 ppm in the spectra of the fully debenzylated 18, we concluded that all the four debenzylations had taken place at the primary rim. In fact, in the 1H spectra of the 14, we also observed 4 sets of signals at 3.02, 2.79, 2.58, 2.48 ppm, each appeared as a doublet of doublets, they indeed corresponded to 4 sets of primary hydroxyl group signals. Thus the triple and quadruple debenzylations of perbenzylated β-CD took a different path than α-CD. The four debenzylation sites in tetraol 14 could not be unambiguously established even with the help of advanced 2D NMR experiments for both 17 and 18 due to significant overlaps in 1H NMR spectra. Thus we performed a series of chemical derivatizations to assist the task. We decided to perform 6-deoxygenation to convert all the 6-O-debenzylated sites to 6-deoxygenated sites. This in principle should generate 4 sets of doublets (H-6s) which should appear at a distinct region (1.0–2.0 ppm), well separated from the rest of the sugar protons (3.2–5.5 ppm). Indeed, the tetraol 14 was smoothly converted to the tetramesylates (19, 84% yield) by treating 14 with methanesulfonyl chloride in anhydrous dichloromethane using triethylamine as a base. The activated sulfonyl ester was then reduced with excess lithium aluminium hydride (LAH) at room temperature (→20); compound 20 was finally fully debenzylated under standard hydrogenation conditions to give the fully debenzylated 21. In the 1H NMR spectra of 21, we observed four sets of doublets at the 1.50–1.80 ppm region (two of them overlapped), correlating to the H-6's of four 6-deoxygenated glucopyranosyl units, which reconfirm our earlier observation that the quadruple debenzylations had occurred only at the primary rim of β-CD.
In order to determine the exact sites of debenzylations, we performed a systematic NMR study of compound 21, recorded in deuterated pyridine. As shown in Fig. 1, the 1H–1H COSY and TOSCY spectra clearly showed 7 anomeric protons at 5.74, 5.60, 5.59, 5.56 (×2), 5.41, 5.40 ppm, which can be divided into four groups according to their chemical shift similarities (Table 2).
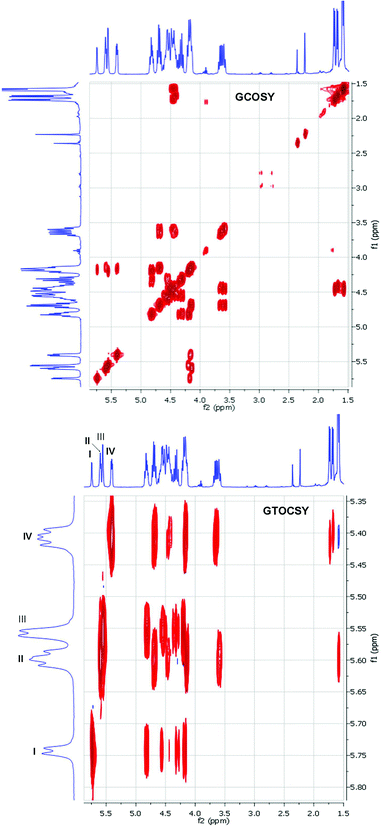 | ||
| Fig. 1 1H–1H GCOSY and GTOCSY spectra of compound 21 in pyridine-d5 recorded at 400 MHz. | ||
| H-1 | H-2 | H-3 | H-4 | H-5 | H-6(a) | H-6b | |
|---|---|---|---|---|---|---|---|
| I | 5.74 | 4.17 | 4.82 | 4.36 | 4.56 | 4.50 | 4.42 |
| II | 5.60 | 4.14 | 4.69 | 3.59 | 4.45 | 1.58 | |
| 5.59 | 4.14 | 4.69 | 3.59 | 4.45 | 1.58 | ||
| III | 5.56 | 4.20 | 4.81 | 4.36 or 4.30 | 3.96 | 4.63–4.38 | |
| 5.56 | 4.20 | 4.81 | 4.30 or 4.36 | 4.96 | 4.63–4.38 | ||
| IV | 5.41 | 4.16 | 4.68 | 3.66 | 4.46 | 1.73 | |
| 5.40 | 4.16 | 4.68 | 3.64 | 4.42 | 1.68 |
The group I consists of one sugar unit which has its anomeric proton resonating at 5.74 ppm; the other three groups consist of two sugar units each with their H-1's at 5.60/5.59 ppm for the group II, at 5.56/5.56 ppm for group III and at 5.41/5.40 ppm for group IV. The GTOCSY experiment revealed that the four sugar units of groups II and IV had 6-deoxygenations because the H-1 proton of each of them correlated with a CH3 at the most upfield region. All 6-deoxygenated sugar units had a more upfield-shifted H-4; the H-4's of group II were observed slightly upfield than those of group IV. All the H-4's of group I and III were found to be more downfield at a very narrow 4.27–4.39 ppm region with the H-4 of group I resonating slightly upfield than H-4's of group III.
The 2D 1H–1H ROESY experiment of compound 21 is shown in Fig. 2. By analyzing the across glycosidic ROE correlations between neighboring H-1 and H-4 pairs, we were able to unambiguously establish the connectivities of the cyclic oligosaccharide. For example, the H-1 of group I unit (non-deoxygenated) has a strong correlation with one of the H-4 of the sugar unit of group III (non-deoxygenated), while both H-1's of group III units correlate with H-4's of group IV (deoxygenated) units; and similarly, the H-1's of the group IV units correlate with the H-4's of group II units (deoxygenated). Additionally, when analyzing the anomeric H-1's of group II units, it was found that one of them correlates with H-4 of the group I monosaccharide, and another one correlates with the H-4 of one of the group III unit. These information suggested that the anomeric O-1 of monosaccharide of group I is connected to the C-4 of one of the unit in group III; both anomeric O-1's of group III are connected to C-4's of group IV and their anomeric O-1's are in turn connected to C-4's of group II; for the anomeric O-1's of group II, one of them is connected to C-4 of group I and another one is connected to C-4 of one of group III unit. The above information allowed us to conclude that the structure of compound 21 is the 6A,6B,6D,6E-tetradeoxygenated β-CD, as shown in Fig. 2. Consequently, the original compound 14 had 6A,6B,6D,6E-O-tetradebenzylations.
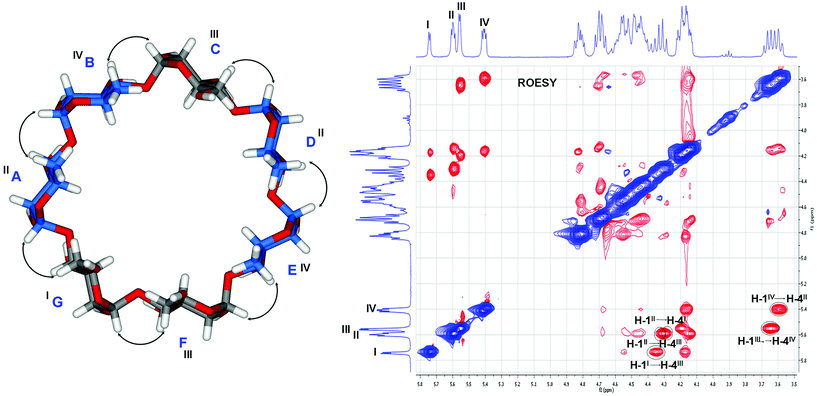 | ||
| Fig. 2 Analysis of 2D 1H–1H ROESY spectrum of compound 21 in pyridine-d5 (Sugar units with carbons in blue have a 6-deoxy functionality). | ||
We noticed that a partially protected analog related to tetraol 14 was previously reported in the literature2b which was synthesized in eight steps with an overall yield of ∼8%. The present method has clear advantage as it gives similarly tetrasubstituted β-CD in 22% yield in one step.
γ-Cyclodextrin
To further explore the potential of the current method, we attempted to study the one-pot triple and quadruple debenzylations of perbenzylated γ-CD (22). Due to the presence of an additional glucopyranosyl unit and three more benzyl groups than β-CD, it was expected that the removal of benzyl groups should be much more complex. Indeed, when we applied similar debenzylation conditions as before, we did not obtain encouraging results. After the treatment of 22 with DIBAL-H in hexane for one day as before, we observed two major spots – which were determined to be the known 6-O-mono and the inseparable 6A,6D/6A,6E-di-debenzylated γ-CDs. By prolonging the reaction time, we observed several more polar spots which were all found to contain a mixture of different triols from the acetylated products (not shown). The 1D 1H and 2D GCOSY and GHSQC NMR spectra of all fully acetylated products revealed an absence of signals above 5.0 ppm (except benzyl protons), indicating that these mixtures were all 6-O-debenzylated.The reaction mixture became even more complex when left reacting further (4 days); according to TLC, numerous small components with similar intensities were detected with no visually major spots produced. However, by following our previous knowledge about the polarity of the tetraols, we were able to isolate one product 23 (Rf 0.35, 9:1 dichloromethane:acetone) in 8% yield after repeated column chromatographies. Compound 23 was characterized as the 6A,6B,6E,6F-O-tetradebenzylated γ-CD as both 23 and its subsequently acetylated form 24 had a C2 symmetry (Scheme 3). Clearly, the triple and quadruple debenzylation of perbenzylated γ-CD has little synthetic advantage due to the extreme low yields and difficulty to isolate products in pure form.
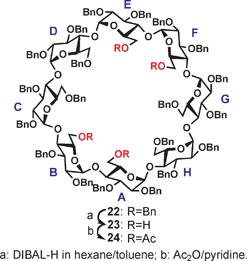 | ||
| Scheme 3 DIBAL-H mediated quadruple debenzylation of perbenzylated γ-CD. | ||
Rational for the observed regioselectivities
Our results were consistent with the literature reports14a that the DIBAL-H mediated removal of first and second benzyls in all perbenzylated CDs remained at the primary rim. However, our results also revealed that there are significant differences at the triple and quadruple debenzylation stages where in the case of perbenzylated α-CD (1), the DIBAL-H preferred attacking a benzyl group attached to a sterically more hindered secondary hydroxyl, specifically the one attached to the O-3 position(s) of the same glucopyranosyl unit which had a prior loss of O-6 benzyl; while in the case of perbenzylated β- (9) and γ-CDs (22), the reducing reagent kept attacking the more accessible benzyl groups at the primary rim. Considering that compound 1 has the smallest cavity size among the three substrates, we think that our results can be rationalized by the steric crowding existing at the primary rim.As depicted in Fig. 3, in the case of perbenzylated α-CD, because of its smaller cavity and conical shape which bring the benzyl groups at the primary rim closer through space, after the second debenzylation (structure I), one of the OAl(i-Bu)2 groups was pushed out to avoid steric crowding and orients itself towards the outside of the cavity (structure II); consequently, the aluminium of the OAl(i-Bu)2 group becomes spaciously close to the O-3 of the same sugar unit which could dynamically coordinate to the O-3 possibly after a ring flip which would bring O-3 and O-6 even closer (structure III). The coordination triggers the polarization of the corresponding benzylic C–O bond; a subsequent attack by hydride on the benzylic methylene carbon from another DIBAL-H molecule (present in excess) causes a regioselective removal of the benzyl at the O-3 position of the same unit to produce triol 4. The other OAl(i-Bu)2 group can also bind to the O-3 of the same ring but to a lesser extent, because of the already decreased crowding, which results in the formation of tetraol 6 in lower yield.
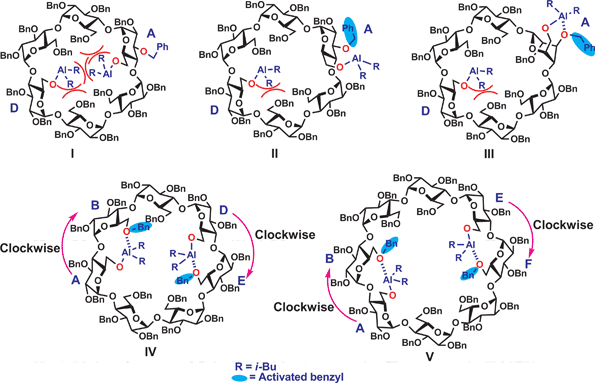 | ||
| Fig. 3 Proposed rationale for the observed regioselectivities in triple and quadruple debenzylations of CDs. | ||
Since the cavity sizes of CD derivatives 9 and 22 were larger, the steric crowding is much less prominent in these cases; thus after the second debenzylation, there was less steric crowding therefore no need for the OAl(i-Bu)2 to reorientate toward the outside of the cavity because of the van der Waals attraction between isobutyl and benzyl groups. This is why we observed debenzylations only at the primary rim in both cases.
The preferred formation of 14 and 23 could be explained by involving the respective clockwise coordination by the aluminium of the O6-Al(i-Bu)2 to the neighboring O6 (Fig. 3). In the case of β-CD, after the second removal of 6-O-benzyl at the D-unit, the aluminium attached at O-6's of A and D units coordinate to the O-6's of B and E units (Structure IV), resulting in the formation of 6A,6B,6D,6E-O-tetradebenzylated β-CD 14; at the stage of the third benzyl removal, a mixture of compounds: 6A,6B,6D-O-tridebenzylated (12) and 6A,6B,6E-O-tridebenzylated β-CD 13 was formed. In the case of γ-CD, the isolated product 6A,6B,6E,6F-O-tetradebenzylated γ-CD 23 originated from one of the didebenzylated intermediates–the 6A,6E-O-didebenzylated-γ-CD, after a clockwise activation of the neighboring 6-O-benzyl groups (Structure V). In the case of α-CD, because of its much reduced ring size at the primary face compared to β- and γ-CDs, if a clockwise coordination by the aluminium of the O6-Al(i-Bu)2 to the neighboring O6 occurred after the removal of second benzyl, the van der Waals repulsion between the benzyl groups and i-Bu groups might be too significant; therefore, debenzylation path via structure V became favored. Sollogoub et al. were the first to investigate the clockwise debenzylation mechanism2b,16 using some bridge-capped and 6-deoxygenated CD derivatives; however, their mechanism did not invoke the clockwise aluminium coordination to the neighboring O6, but using the diminished steric crowding argument. We think that in our cases, the clockwise aluminium coordination is more plausible which explains the observed regioselectivity. A molecular model is constructed (Fig. 4) to show the probable intermediate. It is noteworthy that previously, we also observed that in the 1H NMR spectra of a 6A,6B,6D,6E-tetratritylated α-CD compound, the trityl groups shield the anomeric region of the neighboring glucopyranosyl group in a clockwise fashion.9c
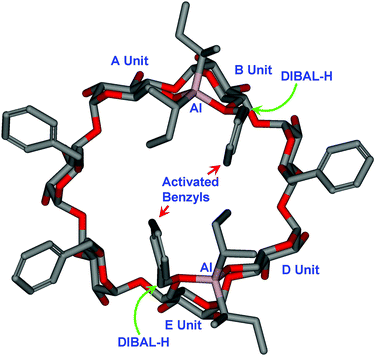 | ||
| Fig. 4 The molecular model of a possible β-CD intermediate that shows the clockwise coordination by aluminium from the O6–Al(i-Bu)2 group in unit A and B, to the neighboring benzylated O6 (unit B and E). The coordination activates the related benzyl group, which facilitates the attack by another DIBAL-H from the exterior of the cavity. (For clarity, all H atoms and benzyl groups attached to the secondary face were removed.) | ||
In conclusion, we have developed a one-pot synthesis of some multi-substituted CD derivatives, previously unable or difficult to obtain, using DIBAL-H mediated debenzylation. Although the yields for individual CD derivatives were low, the method allowed access to triply and quadruply functionalized α- and β-CD with ease; the short reaction sequence makes the syntheses of these novel CD derivatives feasible and we demonstrated that some compounds could be obtained in gram quantities. The unique topology of these CD derivatives should make them ideal candidates for use as scaffolds in the preparation of diverse supramolecular assemblies.
Experimental section
General methods
Optical rotations were determined in a 5 cm cell at 25 ± 2 °C. [a]D values are given in units of 10−1 deg cm2 g−1. Analytical TLC was performed on Silica Gel 60-F254 (Merck, Darmstadt) with detection by quenching of fluorescence and/or by charring with 5% sulfuric acid in water or with a ceric ammonium molybdate dip. All commercial reagents were used as supplied unless otherwise stated. Column chromatography was performed on Silica Gel 60 (Silicycle, Ontario). Organic solutions from extractions were dried with anhydrous Na2SO4 prior to concentration under vacuum at < 40 °C (bath). 1H NMR spectra were recorded at 300, and 400 MHz on Bruker spectrometers. The first order proton chemical shifts δH and δC are reported in δ (ppm) and referenced to either residual CHCl3 (δH 7.24, δC 77.0, CDCl3) or residual CD2HOD (δH 3.30, δC 49.5, CD3OD). 1H and 13C NMR spectra were assigned with the assistance of GCOSY, GHSQC. Matrix-assisted laser desorption/ionization and time-of-flight mass spectra (MALDI-TOF MS) were recorded on a Bruker Daltonics AUTOFLEX III spectrometer using 2,5-dihydroxybenzoic acid (DHP) as matrix by the analytical services of the Department of Chemistry, University of Calgary.2A,2B,2C,2D,2E,2F,3A,3C,3D,3E,3F,6A,6C,6D,6F-Hexadeca-O-benzyl-α-cyclodextrin (4) and 2A,2B,2C,2D,2E,2F,3A,3C,3D,3F,6A,6C,6D,6F-hexadeca-O-benzyl-α-cyclodextrin (5)
To a solution of perbenzylated α-CD (1, 6.0 g, 2.32 mmol) in anhydrous toluene (36.6 mL), was added a solution of DIBAL-H in hexane (66 mL, 1.4 M solution, 92.4 mmol) with stirring under argon atmosphere at room temperature. The reaction mixture was heated to 50 °C for 4 days and then quenched by pouring the reaction mixture carefully into ice-cold water (40 mL) with vigorous stirring. The reaction mixture was subsequently diluted with more EtOAc (100 mL) and filtered off by passing through a thin bed of celite. This celite was then washed thoroughly with more hot EtOAc (3 × 30 mL). More H2O (100 mL) was added to the filtrate and the organic layer was separated. After washing with brine (100 mL) and drying over anhydrous Na2SO4, the organic phase was evaporated. The crude mixture was purified by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 15–40%) as the eluent to afford sequentially diol 3 (278 mg, 5% yield), triol 4 (1.72 g, 32% yield) and tetraol 5 (517 mg, 10% yield) as colorless solids.:AcOEt 7:3); [α]25D +32.6° (c 0.9, CHCl3); 1H and 13C NMR data are identical to the literature.1b:AcOEt 6:4); [α]25D +42.2° (c 0.9, CHCl3); δH (400 MHz, CDCl3) 7.46–7.07 (m, 75H, Aromatic), 5.62 (d, 1H, J = 4.0 Hz, H-1), 5.52 (d, 1H, J = 11.0 Hz, PhCH), 5.32 (d, 1H, J = 10.6 Hz, PhCH), 5.21 (d, 1H, J = 10.8 Hz, PhCH), 5.15 (d, 1H, J = 11.2 Hz, PhCH), 5.09 (d, 1H, J = 3.8 Hz, H-1), 5.01–4.67 (m, 13H, 4 × H-1, 9 × PhCH), 4.59–4.36 (m, 17H, 17 × PhCH), 4.29–4.19 (m, 2H), 4.16–3.53 (m, 30H), 3.48–3.36 (m, 4H), 3.28 (dd, 1H, J = 3.5, 9.9 Hz, H-2), 3.10 (brs, 1H, OH), 2.76 (brs, 1H, OH); δC (100 MHz, CDCl3) 139.66, 139.63, 139.27, 139.19, 139.16, 138.66, 138.41, 138.19, 138.09, 138.01, 137.93, 137.85, 137.56, 128.50, 128.46, 128.34, 128.30, 128.28, 128.21, 128.15, 128.10, 128.08, 128.06, 127.96, 127.93, 127.89, 127.82, 127.73, 127.68, 127.63, 127.61, 127.58, 127.50, 127.45, 127.33, 127.30, 127.25, 127.03, 126.90, 126.85, 126.54, 100.59, 100.33, 99.16, 98.43, 97.88, 97.80 (6 × C-1), 82.54, 82.17, 81.60, 81.52, 81.23, 81.14, 80.72, 80.68, 80.56, 79.81, 79.64, 78.67, 78.08, 77.65, 76.43, 76.34, 75.93, 75.81, 75.75, 74.38, 74.05, 73.38, 73.34, 73.34, 73.29, 72.80, 72.71, 72.46, 72.33 (CBn, C-2, C-3, C-4), 72.17, 72.12, 71.83, 71.61, 71.47, 71.29 (6 × C-5), 70.03, 69.92, 69.63, 69.33, 61.90, 61.85 (6 × C-6); m/z (MS MALDI-TOF) calcd for [C141H150O30 + Na]+ 2346.0, found 2346.4.:4); [α]25D +50.8° (c 1.0, CHCl3); δH (400 MHz, CDCl3) 7.51–7.06 (m, 35H, aromatic), 5.44 (d, 1H, J = 11.2 Hz, PhCH), 5.24 (d, 1H, J = 11.2 Hz, PhCH), 4.94–4.84 (m, 3H, 2 × PhCH + H-1), 4.83–4.70 (m, 4H, 2 × PhCH, H-1), 4.61 (d, 1H, J = 13.0 Hz, PhCH), 4.53 (m, 2H, PhCH), 4.46–4.38 (m, 3H, PhCH), 4.35 (d, 1H, J = 13.0 Hz, PhCH), 4.29 (dd, 1H, J = 9.33, 9.70 Hz, H-3), 4.23 (d, 1H, J = 13.0 Hz, PhCH), 4.17 (dd, 1H, J = 9.16, 8.56 Hz, H-3), 4.04–3.94 (m, 2H, H-3, H-6), 3.90–3.72 (m, 6H, 3 × H-5, 2 × H-4, 1 × H-6), 3.71–3.56 (m, 5H, 4 × H-6, 1 × H-2), 3.45–3.35 (m, 2H, H-4, H-2), 3.29 (dd, 1H, J = 3.4, 10.0 Hz, H-2); δC (100 MHz, CDCl3) 139.71, 139.46, 138.57, 138.46, 138.08, 138.06, 137.24, 128.78, 128.56, 128.33, 128.31, 128.22, 128.19, 128.14, 128.10, 127.93, 127.87, 127.82, 127.70, 127.61, 127.57, 127.46, 127.26, 127.15, 127.01, 126.83, 101.69 (C-1), 100.75 (C-1), 99.92 (C-1), 83.46 (C-4), 82.41 (C-4), 82.10 (C-4), 81.11 (C-3), 80.42 (C-3), 79.10 (C-2), 78.56 (C-2), 77.12 (C-2), 76.29 (PhCH), 75.73 (PhCH), 74.45 (PhCH), 73.44 (PhCH), 73.34 (PhCH), 72.92 (C-3), 72.45 (PhCH), 72.28 (PhCH), 72.13 (C-5), 71.93 (C-5), 71.04 (C-5), 69.73 (C-6), 69.25 (C-6), 61.78 (C-6); m/z (MS MALDI-TOF) calcd for [C134H145O30 + H]+ 2234.0, found 2234.3.3A,6A,6D-Tri-O-acetyl-2A,2B,2C,2D,2E,2F,3B,3C,3D,3E,3F,6B, 6C,6E,6F-hexadeca-O-benzyl-α-cyclodextrin (6)
To the solution of triol 4 (232 mg, 0.1 mmol) in dry dichloromethane (2 mL) was added Ac2O (0.5 mL), anhydrous pyridine (0.5 mL) and a catalytic amount of 4-N,N-dimethylaminopyridine, and the reaction mixture was stirred for 6 h at room temperature. The reaction was monitored by TLC, and when the reaction was complete, the mixture was concentrated under reduced pressure to afford syrup which was purified by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 10–25%) as eluent to give compound 6 (225 mg, 92% yield) as a white solid. Rf 0.65 (hexane:AcOEt 7:3); [α]25D +23.7° (c 1.7, CHCl3); δH (400 MHz, CDCl3) 7.45–7.10 (m, 75H), 5.74 (dd, 1H, J = 10.0, 9.6 Hz, H-3), 5.35 (d, 1H, J = 11.3 Hz, PhCH), 5.32 (d, 1H, J = 11.0 Hz, PhCH), 5.26 (d, 1H, J = 11.0 Hz, PhCH), 5.22 (d, 1H, J = 11.2 Hz, PhCH), 5.11 (d, 1H, J = 10.9 Hz, PhCH), 5.10 (d, 1H, J = 3.5 Hz, H-1), 5.02–4.84 (m, 9H, 5 × PhCH, 4 × H-1), 4.73 (d, 1H, J = 13.0 Hz, PhCH), 4.63–4.55 (m, 2H, PhCH, H-1), 4.55–4.20 (m, 25H, 18 × PhCH, 4 × H-6, 3 × H-3), 4.18–3.95 (m, 8H, 4 × H-5, H-6, 3 × H-3), 3.87 (m, 7H, 2 × H-6, 2 × H-5, 3 × H-4), 3.78–3.63 (m, 4H, 3 × H-6, H-4), 3.62–3.53 (m, 3H, 2 × H-6, H-4), 3.49–3.36 (m, 6H, 6 × H-2), 1.97 (s, 3H, COCH3), 1.92 (s, 3H, COCH3), 1.92 (s, 3H, COCH3); δC (100 MHz, CDCl3) 170.72, 170.35, 170.16 (3 × C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 140.07, 140.04, 139.77, 139.61, 138.78, 138.69, 138.56, 138.51, 138.36, 138.31, 138.23, 138.22, 138.18, 138.13, 128.43, 128.33, 128.30, 128.29, 128.18, 128.13, 128.06, 128.05, 128.01, 127.96, 127.91, 127.82, 127.75, 127.72, 127.70, 127.65, 127.56, 127.53, 127.47, 127.43, 127.38, 127.18, 127.13, 127.07, 126.83, 126.78, 126.62, 126.53, 101.36, 101.08, 100.67, 100.24, 100.21, 99.89 (6 × C-1), 82.07, 81.94, 81.74, 81.62, 80.50, 80.37, 80.32, 80.13, 80.08, 79.36, 78.99, 78.81, 78.73, (5 × C-2, 5 × C-3, 6 × C-4), 76.41, 76.06, 75.96, 75.76, 75.02, 74.21, 73.34, 73.25, 72.95, 72.39, 72.22, 72.18, 72.14 (CBn, 1 × C-5, 1 × C-2), 71.78, 71.59, 71.53, 71.38, 69.96, 69.82 (5 × C-5, 1 × C-3), 69.16 (2 × C-6), 68.96 (C-6), 68.86 (C-6), 63.74 (C-6), 62.94 (C-6), 21.32, 20.94, 20.89 (3 × CH3); m/z (MS MALDI-TOF) calcd for [C147H156O33 + Na]+ 2472.0, found 2472.1.
O), 140.07, 140.04, 139.77, 139.61, 138.78, 138.69, 138.56, 138.51, 138.36, 138.31, 138.23, 138.22, 138.18, 138.13, 128.43, 128.33, 128.30, 128.29, 128.18, 128.13, 128.06, 128.05, 128.01, 127.96, 127.91, 127.82, 127.75, 127.72, 127.70, 127.65, 127.56, 127.53, 127.47, 127.43, 127.38, 127.18, 127.13, 127.07, 126.83, 126.78, 126.62, 126.53, 101.36, 101.08, 100.67, 100.24, 100.21, 99.89 (6 × C-1), 82.07, 81.94, 81.74, 81.62, 80.50, 80.37, 80.32, 80.13, 80.08, 79.36, 78.99, 78.81, 78.73, (5 × C-2, 5 × C-3, 6 × C-4), 76.41, 76.06, 75.96, 75.76, 75.02, 74.21, 73.34, 73.25, 72.95, 72.39, 72.22, 72.18, 72.14 (CBn, 1 × C-5, 1 × C-2), 71.78, 71.59, 71.53, 71.38, 69.96, 69.82 (5 × C-5, 1 × C-3), 69.16 (2 × C-6), 68.96 (C-6), 68.86 (C-6), 63.74 (C-6), 62.94 (C-6), 21.32, 20.94, 20.89 (3 × CH3); m/z (MS MALDI-TOF) calcd for [C147H156O33 + Na]+ 2472.0, found 2472.1.3A,3D,6A,6D-Tetra-O-acetyl-2A,2B,2C,2D,2E,2F,3B,3C,3D,3E, 3F,6B,6C,6E,6F-hexadeca-O-benzyl-α-cyclodextrin (7)
To a solution of tetraol 5 (112 mg, 0.05 mmol) in dry dichloromethane (1.0 mL) was added Ac2O (0.3 mL), anhydrous pyridine (0.3 mL) and a catalytic amount of 4-N,N-dimethylaminopyridine, and the reaction mixture was stirred for 6 h at room temperature. The reaction was monitored and worked up as 6. Compound 7 was obtained by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 10–25%) as eluent to give compound 7 as colorless solid (108 mg, yield 90%). Rf 0.65 (hexane:AcOEt 7:3); [α]25D +39.3° (c 0.6, CHCl3); δH (400 MHz, CDCl3) 7.39–7.11 (m, 35H, aromatic), 5.80 (t, 1H, J = 9.9 Hz, H-3), 5.28 (d, 1H, J = 11.9 Hz, PhCH), 5.17 (d, 1H, J = 11.5 Hz, PhCH), 4.96 (d, 1H, J = 3.1 Hz, H-1), 4.94 (d, 1H, J = 11.3 Hz, PhCH), 4.90 (d, 1H, J = 3.1 Hz, H-1), 4.85 (d, 1H, J = 11.7 Hz, PhCH), 4.67 (d, 1H, J = 13.0 Hz, PhCH), 4.58 (d, 1H, J = 13.2 Hz, PhCH), 4.57 (d, 1H, J = 3.1 Hz, H-1), 4.55–4.46 (m, 2H, PhCH, H-6), 4.46–4.38 (m, 3H, 3 × PhCH), 4.34 (d, 1H, J = 11.7 Hz, PhCH), 4.30–4.18 (m, 3H, 2 × PhCH, H-6), 4.17–4.02 (m, 4H, 2 × H-3, H-5, PhCH), 3.98–3.74 (m, 6H, 2 × H-4, 2 × H-5, 2 × H-6), 3.66 (br d, 1H, J = 9.7 Hz, H-6), 3.60–3.51 (m, 2H, H-4, H-6), 3.39 (dd, 1H, J = 3.1, 9.8 Hz, H-2), 3.36 (dd, 1H, J = 2.9, 10.2 Hz, H-2), 3.32 (dd, 1H, J = 3.1, 10.4 Hz, H-2), 1.92 (s, 3H, COCH3), 1.88 (s, 3H, COCH3); δC (100 MHz, CDCl3) 170.19, 169.97 (2 × CO), 140.17, 140.12, 138.83, 138.57, 138.31, 138.29, 138.17, 128.38 - 126.47, 101.74 (C-1), 101.04 (C-1), 100.45 (C-1), 82.47 (C-4), 80.43 (C-4), 80.37 (C-2), 80.05 (C-3), 79.80 (C-3), 78.77 (C-2), 76.50 (C-2), 75.91 (PhCH), 75.49 (PhCH), 74.00 (PhCH), 73.29 (PhCH), 73.20 (PhCH), 72.19 (C-5), 72.16 (PhCH), 71.81 (PhCH), 71.44 (C-5), 71.26 (C-3), 69.94 (C-5), 69.17 (C-6), 68.71 (C-6), 63.11 (C-6), 21.16 (CH3), 20.89 (CH3); m/z (MS MALDI-TOF) calcd for [C142H152O34 + Na]+ calcd 2424.0, found 2424.0.3A,6A,6D-Tri-O-acetyl-α-cyclodextrin (8)
The perbenzylated derivative 6 (122 mg, 0.05 mmol) was dissolved in a mixture of THF–H2O (3:1, 5 mL) and was stirred with 10% Pd–C (50 mg) under a hydrogen atmosphere overnight. The insoluble Pd–C was removed by filtration through celite and evaporation of the solvent under reduced pressure gave pure compound 8 (52 mg, quantitative yield). [α]25D +134.7° (c 1.4, CH3OH); δH (400 MHz, CD3OD) 5.38 (dd, 1H, J = 9.2, 9.7 Hz, H-3), 5.01 (d, 1H, J = 3.5 Hz, H-1), 4.98–4.91 (m, 4H, 4 × H-1), 4.79 (d, 1H, J = 3.3 Hz, H-1), 4.66 (br d, 1H, J = 10.8 Hz, H-6), 4.58 (br d, 1H, J = 10.8 Hz, 1H, H-6), 4.36 (dd, 1H, J = 6.0, 12.1 Hz, H-6), 4.22 (dd, 1H, J = 6.6, 11.9 Hz, H-6), 4.16–4.09 (m, 1H, H-5), 4.09–4.02 (m, 1H, H-5), 4.02–3.78 (m, 17H), 3.67–3.38 (m, 12H), 2.11 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.09 (s, 3H, COCH3); δC (100 MHz, CD3OD) 172.36, 171.44, 171.42 (3 × CO), 102.49, 102.46, 102.43, 102.11, 102.02, 101.68 (6 × C-1), 82.45, 82.11, 82.08, 82.01, 81.28, 79.98 (6 × C-2), 73.90, 73.81, 73.77, 73.74, 72.62, 72.50, 72.44, 72.33, 72.25, 72.23, 72.14, 71.49, 70.97, 70.24, 69.67 (C-3's, C-4's, C-5's), 63.83 (C-6_OAc), 63.67 (C-6_OAc), 60.51, 60.46, 60.39, 60.17 (4 × C-6_OH), 20.40 (CH3), 19.40 (2 × CH3); m/z (MS MALDI-TOF) calcd for [C42H66O33 + Na]+ 1121.3, found 1121.3; calcd for [C42H66O33 + K]+ 1137.3, found 1137.2.2A,2B,2C,2D,2E,2F,2G,3A,3B,3C,3D,3E,3F,3G,6A,6B,6E-Hexadeca-O-benzyl-β-cyclodextrin (14)
To a solution of perbenzylated β-cyclodextrin 9 (5.0 g, 1.653 mmol) in anhydrous toluene (26.5 mL), was added a solution of DIBAL-H in hexane (47 mL, 1.4 M solution, 66.1 mmol) with stirring under an atmosphere of argon at room temperature. The reaction mixture was heated to 50 °C for 4 days and then quenched by slowly pouring the reaction mixture into ice-cold water (35 mL) with vigorous stirring. The reaction mixture was diluted with EtOAc (80 mL) and filtered through celite. This celite was then washed thoroughly with hot EtOAc (3 × 25 mL). More water (100 mL) was added to the filtrate and the organic layer was separated and washed with brine (100 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 10–50%) as eluent to afford sequentially the diol 11 (376 mg, 8% yield) and two major fractions. The less polar fraction (Rf 0.45, dichloromethane:acetone 85:15), possibly 12 and 13, as confirmed from the acetylated products (see below) contained an inseparable mixture of two triols (1.138 g, 25% yield); the more polar fraction is the tetraol 14 (969 mg, 22% yield).:AcOEt 7:3); [α]25D +34.8° (c 1.2, CHCl3); 1H and 13C NMR data are identical to the literature.1b:hexane 1:1); Rf 0.30 (dichloromethane:acetone 85:15); [α]25D +25.4° (c 0.3, CHCl3); δH (400 MHz, CDCl3) 7.37–7.01 (m, 85H, Ar-H), 5.42 (d, 1H, J = 3.7 Hz, H-1), 5.34 (d, 1H, J = 3.8 Hz, H-1), 5.17 (m, 4H, H-1, 3 × PhCH), 5.03 (d, 1H, J = 3.5 Hz, H-2), 4.98–4.87 (m, 3H, H-1, 2 × PhCH), 4.83–4.34 (m, 31H, PhCH), 4.08–3.59 (m, 35H, H-3, 7 × H-4, 7 × H-5, 7 × H-6, 7 × H-6′), 3.51 (m, 4H, 4 × H-2), 3.42 (dd, 1H, J = 9.7, 3.5 Hz, H-2), 3.39–3.32 (m, 2H, 2 × H-2), 3.03 (dd, 1H, J = 5.3, 5.3 Hz, OH-6), 2.79 (dd, 1H, J = 5.6, 5.6 Hz, OH-6), 2.68 (dd, 1H, J = 6.2, 6.2 Hz, OH-6), 2.48 (dd, 1H, J = 6.5, 6.5 Hz, OH-6); δC (100 MHz, CDCl3) 139.39, 139.25, 139.20, 139.08, 138.93, 138.81, 138.58, 138.49, 138.36, 138.22, 138.17, 138.08, 138.03, 137.78, 128.37, 128.33, 128.31, 128.25, 128.22, 128.15, 128.11, 128.03, 127.98, 127.90, 127.82, 127.74, 127.69, 127.58, 127.53, 127.26, 127.21, 127.04, 127.00, 126.89, 98.71, 98.61, 98.24, 98.21, 97.98, 97.82, 97.68 (7 × C-1) 81.12, 80.85, 80.78, 80.69, 80.61, 80.47, 80.39, 80.23, 79.34, 79.26, 79.20, 78.78, 78.66, 78.19, 77.83, 76.65, 76.34, 76.21, 76.09, 75.63, 75.59, 75.40, 74.97, 74.78, 74.37, 73.46, 73.36, 73.07, 72.79, 72.75, 72.64, 72.61, 72.39, 72.00, 71.84, 71.77, 71.73 (PhCH, C-3's, C-4's, C-5's, C-6's), 69.41, 69.35, 69.29, 62.15, 62.10, 61.96, 61.82 (7 × C-2); m/z (MS MALDI-TOF) calcd for [C161H172O35 + Na]+ 2688.2, found 2688.0.:1 mixture (200 mg) was acetylated for 6 h as 4 to afford an inseparable mixture (presumably 15 and 16) by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 10–30%) (167 mg, yield 80%). Rf 0.45 (EtOAc:hexane 4:6); characteristic NMR data: δH (400 MHz, CDCl3) 2.01 (s, 3H, COCH3), 1.99 (s, 3H, COCH3), 1.99 (s, 3H, COCH3), 1.98 (s, 3H, COCH3), 1.95 (s, 3H, COCH3), 1.94 (s, 3H, COCH3); δC (100 MHz, CDCl3) 170.39, 170.37, 170.34, 170.31, 170.25 (2 × C) (CO), 20.95, 20.89, 20.86, 20.80; m/z (MS MALDI-TOF) calcd for [C174H184O38 + Na]+ 2904.2, found 2903.8.6A,6B,6D,6E-Tetra-O-acetyl-2A,2B,2C,2D,2E,2F,2G,3A,3C,3D, 3E,3F,3G-6C,6F,6G-heptadeca-O-benzyl-β-cyclodextrin (17)
The pure β-CD tetraol 14 (200 mg, 0.075 mmol) was acetylated with Ac2O (0.5 mL) in a mixture of anhydrous dichloromethane (2 mL) and anhydrous pyridine (0.5 mL) in the presence of a catalytic amount of 4-N,N-dimethylaminopyridine as before. The tetraacetate 17 (182 mg, yield 86%) was obtained as a white solid after column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 15–35%) as eluent. Rf 0.40 (EtOAc:hexane 4:6); [α]25D +20.4° (c 0.4, CHCl3); δH (400 MHz, CDCl3) 7.36–7.01 (m, 85H, Ar-H), 5.37–5.23 (m, 4H, 2 × H-1, 2 × PhCH), 5.21 (d, 1H, J = 10.6 Hz, PhCH), 5.07 (d, 1H, J = 3.7 Hz, H-1), 5.05 (d, 1H, J = 3.5 Hz, H-1), 5.04–4.93 (m, 5H, 3 × H-1, 2 × PhCH), 4.84–4.73 (m, 9H, 9 × PhCH), 4.69–4.33 (m, 26H, 20 × PhCH, 6 × H-6), 4.31–4.25 (m, 2H, 2 × H-6), 4.23–3.97 (m, 17H, 3 × H-4, 3 × H-6, 4 × H-5, 7 × H-3), 3.92 (br d, 1H, J = 8.4 Hz, H-5), 3.88–3.68 (m, 6H, 4 × H-4, 2 × H-5), 3.66–3.58 (m, 3H, 3 × H-6), 3.57–3.45 (m, 5H, 5 × H-2), 3.45–3.39 (m, 2H, 2 × H-2), 2.01 (s, 3H, COCH3), 1.99 (s, 3H, COCH3), 1.99 (s, 3H, COCH3), 1.94 (s, 3H, COCH3); δC (100 MHz, CDCl3) 170.39 (2 × CO), 170.37 (CO), 170.36 (CO), 139.43, 139.28, 139.26, 139.22, 139.17, 138.99, 138.95, 138.63, 138.55, 138.49, 138.34, 138.28, 138.26, 138.17, 138.12, 128.32, 128.28, 128.25, 128.22, 128.17, 128.16, 128.13, 128.11, 128.06, 128.02, 127.98, 127.94, 127.92, 127.87, 127.82, 127.79, 127.73, 127.64, 127.61, 127.51, 127.49, 127.42, 127.15, 127.09, 126.83, 99.19 (C-1), 98.93 (C-1), 98.81 (C-1), 98.63 (C-1), 98.37 (C-1), 98.30 (C-1), 98.23 (C-1), 80.95, 80.87, 80.58, 80.48, 79.80, 79.70, 79.22, 79.03, 78.60, 76.29 (PhCH), 76.16 (PhCH), 75.67 (PhCH), 74.90 (PhCH), 73.43 (PhCH), 73.36 (PhCH), 73.24 (PhCH), 73.20 (PhCH), 72.85 (PhCH), 72.82 (PhCH), 72.75 (PhCH), 72.43 (PhCH), 72.22 (PhCH), 71.79 (C-5), 71.72 (C-5), 71.57 (C-5), 69.83 (C-5), 69.59 (C-5), 69.48 (C-5), 69.32 (C-5), 68.84 (C-6), 68.75 (C-6), 68.58 (C-6), 63.68 (C-6_OAc), 63.59 (C-6_OAc), 63.48 (C-6_OAc), 63.42 (C-6_OAc), 20.91 (CH3), 20.86 (CH3), 20.83 (CH3), 20.72 (CH3); m/z (MS MALDI-TOF) calcd for [C169H180O39 + Na]+ 2856.2, found 2856.0.6A,6B,6D,6E-Tetra-O-acetyl-β-cyclodextrin (18)
Compound 18 was obtained in a similar fashion as 8 (yield: quantitative). δH (400 MHz, CD3OD) 5.04–4.92 (m, 7H, 7 × H-1), 4.67–4.40 (m, 4H, 4 × H-6a_OAc), 4.30–4.08 (m, 4H, 4 × H-6b_OAc), 4.05–3.69 (m, 20H, 7 × H-5, 7 × H-3, 3 × H-6a_OH, 3 × H-6b), 3.63–3.37 (m, 14H, 7 × H-2, 7 × H-4), 2.10 (s, 6H, 2 × Ac), 2.07 (s, 6H, 2 × Ac); δC (100 MHz, CD3OD) 171.45 (CO), 171.39 (CO), 171.29 (CO), 171.26 (CO), 103.01, 103.00, 102.72, 102.71, 102.40, 102.21, 102.04 (7 × C-1), 82.76, 82.73, 82.36, 82.35, 81.89, 81.27, 81.24, 73.46, 73.40, 73.38, 73.32, 73.26, 73.17, 72.89, 72.77, 72.63, 72.38, 72.29, 69.65, 69.60, 69.55, 63.53 (× 3, C-6_OAc), 63.44 (C-6_OAc), 60.45 (C-6_OH), 60.07 (C-6_OH), 59.95 (C-6_OH), 19.40 (× 2, Ac), 19.36 (× 2, Ac); m/z (MS MALDI-TOF) calcd for [C50H78O39 + Na]+ 1325.4, found 1325.1.6A,6B,6D,6E-Tetra-O-methanesulfonyl-2A,2B,2C,2D,2E,2F, 2G,3A,3C,3D,3E,3F,3G,6C,6F,6G-heptadeca-O-benzyl-β-cyclodextrin (19)
To the clear solution of tetraol 14 (156 mg, 0.0585 mmol) in anhydrous dichloromethane (4 ml) and triethylamine (0.0652 ml, 0.469 mmol, 8 eq), was added methanesulfonyl chloride (0.024 ml, 0.305 mmol, 5.2 eq) at 0 °C, and the reaction was continued at 15 °C for 4 h. The reaction was quenched with a few drops of cold water. More dichloromethane (20 mL) was added, and the organic solution was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 20–45%) as the eluent to afford pure tetramesylate 19 as a colorless solid (146 mg, yield 84%). Rf 0.30 (hexane:AcOEt 6:4); δH (400 MHz, CDCl3) 7.37–6.92 (m, 85H, 17× Ph), 5.45 (d, 1H, J = 3.8 Hz, H-1), 5.31 (d, 1H, J = 10.5 Hz, CHPh), 5.25 (d, 1H, J = 11.3 Hz, CHPh), 5.22 (d, 1H, J = 4.7 Hz, H-1), 5.20–5.16 (m, 2H, H-1, CHPh), 5.02 (d, 1H, J = 10.9 Hz, CHPh), 4.99 (d, 1H, J = 3.7 Hz, H-1), 4.98 (d, 1H, J = 3.6 Hz, H-1), 4.93 (d, 1H, J = 3.4 Hz, H-1), 4.91 (d, 1H, J = 3.4 Hz, H-1), 4.92–4.87 (m, 1H, CHPh), 4.81–4.27 (m, 29H, CHPh), 4.21–3.54 (m, 35H, 7 × H-3, 7 × H-4, 7 × H-5, 7 × H-6, 7 × H-6′), 3.52 (d, 1H, J = 3.7 Hz, H-2), 3.49 (dd, 1H, J = 5.5, 3.8 Hz, H-2), 3.47–3.44 (m, 2H, 2 × H-2), 3.44–3.41 (m, 1H, H-2), 3.38 (dd, 1H, J = 3.1, 2.5 Hz, H-2), 3.36 (dd, 1H, J = 3.2, 2.4 Hz, H-2), 2.93 (s, 3H, SO2CH3), 2.90 (s, 3H, SO2CH3), 2.87 (s, 6H, 2 × SO2CH3); δC (100 MHz, CDCl3) 139.36, 139.20, 139.16, 139.15, 139.04, 138.81, 138.78, 138.59, 138.42, 138.38, 138.26, 138.23, 138.19, 138.01, 137.90, 137.77, 128.38, 128.35, 128.33, 128.24, 128.18, 128.15, 128.12, 128.05, 128.03, 127.98, 127.92, 127.86, 127.77, 127.73, 127.67, 127.58, 127.54, 127.48, 127.43, 127.33, 127.17, 127.16, 127.13, 127.09, 127.06, 126.94, 126.86, 126.66, 98.94, 98.84, 98.76, 98.60, 98.28, 98.21, 97.94 (7 × C-1), 80.91, 80.74, 80.67, 80.60, 80.47, 80.14, 79.74, 79.11, 78.97, 78.20, 77.88, 76.66, 76.33, 76.31, 76.08, 75.66, 75.30, 74.66, 73.48, 73.43, 73.36, 73.26, 72.89, 72.73, 72.43, 72.02, 71.76, 71.46, 69.59, 69.28, 69.16, 69.06, 68.98, 37.25, 37.13, 37.07, 37.03 (4 × SO2CH3); m/z (HRMS MALDI-TOF) calcd for [C165H180O43S4 + Na]+ 3000.06734, found 3000.06823.6A,6B,6D,6E-Tetradeoxy-2A,2B,2C,2D,2E,2F,2G,3A,3C,3D,3E, 3F,3G,6C,6F,6G-heptadeca-O-benzyl-β-cyclodextrin (20)
To the clear solution of the tetramesylate 19 (140 mg, 0.0486 mmol) in anhydrous tetrahydrofuran (4 ml), was added lithium aluminium hydride (14.8 mg, 0.389 mmol, 8 eq), and the reaction was continued at room temperature for 15 h. The excess amount of hydride was destroyed by AcOEt (0.5 mL) and the mixture was evaporated to dryness, and the residue was re-dissolved in dichloromethane (15 mL) and the solution was washed with water (5 mL) and brine (5 mL), dried over anhydrous Na2SO4 and evaporated. The crude material was purified by column chromatography on silica gel using a mixture of AcOEt in hexane (gradient 10–20%) as the eluent to afford pure tetradeoxygenated 20 (98.5 mg, yield 78%). Rf 0.4 (hexane:AcOEt 7:3); δH (400 MHz, CDCl3) 7.23–7.03 (m, 85H, 17 × Ph), 5.27–5.22 (m, 2H, CHPh), 5.21 (d, 1H, J = 3.7 Hz, H-1), 5.18 (d, 1H, J = 3.8 Hz, H-1), 5.13 (d, 1H, J = 10.9 Hz, CHPh), 5.08 (d, 1H, J = 11.1 Hz, CHPh), 5.04 (d, 2H, J = 3.7 Hz, 2 × H-1), 4.99 (d, 1H, J = 3.5 Hz, H-1), 4.94–4.88 (m, 3H, H-1, 2 × CHPh), 4.85 (d, 1H, J = 3.4 Hz, H-1), 4.83–4.36 (m, 28H, CHPh), 4.16–3.59 (m, 27H, 7 × H-3, 7 × H-4, 7 × H-5, 3 × H-6, 3 × H-6′), 3.56–2.72 (m, 7H, 7 × H-2), 1.33 (d, 1H, J = 6.2 Hz, CH3), 1.28 (d, 1H, J = 5.8 Hz, CH3), 1.27 (d, 1H, J = 6.0 Hz, CH3), 1.25 (d, 1H, J = 6.4 Hz, CH3); m/z (HRMS MALDI-TOF) calcd for [C161H172O31 + Na]+ 2624.1780, found 2624.1775.6A,6B,6D,6E-Tetradeoxy-β-cyclodextrin (21)
Compound 20 (90 mg, 0.0346 mmol) in a mixture of tetrahydrofuran–H2O (3:1, 4 mL), was hydrogenated in the presence of 10% Pd–C (35 mg) as 8 to give pure compound 21 (34 mg, quantitative yield). [α]25D +85.3° (c 0.2, CH3OH); δH (400 MHz, pyridine-d5) 5.74 (d, 1H, J = 3.5 Hz, H-1), 5.60 (d, 1H, J = 3.5 Hz, H-1), 5.58 (d, 1H, J = 3.6 Hz, H-1), 5.55 (d, 2H, J = 3.1 Hz, 2 × H-1), 5.41 (d, 1H, J = 3.8 Hz, H-1), 5.40 (d, 1H, J = 3.7 Hz, H-1), 4.86–4.78 (m, 3H, 3 × H-3), 4.73–4.65 (m, 4H, 4 × H-3), 4.62–4.39 (m, 13H, 7 × H-5, 3 × H-6, 3 × H-6′), 4.35 (dd, 1H, J = 9.2 Hz, H-4), 4.31 (dd, 2H, J = 9.3 Hz, 2 × H-4), 4.23–4.12 (m, 7H, 7 × H-2), 3.66 (dd 1H, J = 8.6 Hz, H-4), 3.64 (dd, J = 8.6 Hz, 1H, H-4), 3.60 (overlap two dd, J = 9.0 Hz, 2H, 2 × H-4), 1.73 (d, 3H, J = 6.2 Hz, -CH3), 1.68 (d, 3H, J = 6.2 Hz, -CH3), 1.58 (d (two overlapping), 6H, J = 6.2 Hz, 2 × -CH3); δC (100 MHz, pyridine-d5) 104.62, 104.60, 104.31, 104.28, 104.01, 103.91, (7 × C-1), 90.25, 90.22, 90.13, 90.05 (4 × C-4), 83.67, 83.52, 83.38 (3 × C-4), 75.12, 75.07, 74.81, 74.79, 74.76, 74.69 (7 × C-2, 7 × C-3), 74.43, 74.40, 74.34 (3 × C-5), 68.58, 68.46, 68.39, 68.36 (4 × C-5), 62.02, 61.92, 61.83 (3 × C-6), 18.72, 18.62, 18.59 (4 × CH3); m/z (MS MALDI-TOF) calcd for [C42H70O31 + Na]+ 1093.4, found 1093.0.2A,2B,2C,2D,2E,2F,2G,2H,3A,3B,3C,3D,3E,3F,3G,3H,6A,6B,6E,6F-Eicosa-O-benzyl-γ-cyclodextrin (23)
To a solution of perbenzylated γ-cyclodextrin 22 (3.0 g, 0.868 mmol) in anhydrous toluene (13.5 mL) was added a solution of DIBAL-H (25 mL, 1.4 M solution in hexane, 34.7 mmol) with stirring under argon atmosphere, and the mixture was heated to 50 °C for 4 days and then quenched by pouring the reaction mixture into ice-cold water (40 mL). The spot with an Rf 0.35 (dichloromethane:acetone, 9:1) was isolated by repeated column chromatographies on silica gel using a mixture of AcOEt in hexane (gradient 15–50%, and then repurification using a mixture of dichloromethane:acetone 9:1) as eluent to afford 23 (215 mg, 8% yield). Rf 0.35 (dichloromethane:acetone 9:1); [α]25D +40.0° (c 0.4, CHCl3); δH (400 MHz, CDCl3) 7.38–7.07 (m, 50H, Ar-H), 5.36 (d, 1H, J = 3.5, H-1), 5.25–5.20 (m, 2H, H-1, PhCH), 5.12–5.03 (m, 1H, PhCH), 4.91 (d, 1H, J = 3.5, H-1), 4.87 (d, 1H, J = 3.5, H-1), 4.84–4.37 (m, 23H), 4.09–3.61 (m, 26H), 3.58–3.51 (m, 2H), 3.49 (dd, 1H, J = 9.1, 3.5, H-2), 3.36 (dd, 1H, J = 9.6, 3.4, H-2), 3.26 (s, 1H, OH), 2.59 (s, 1H, OH); δC (100 MHz, CDCl3) 139.53, 139.14, 138.70, 138.63, 138.34, 138.20, 138.14, 137.90, 128.43, 128.38, 128.32, 128.29, 128.27, 128.22, 128.15, 128.12, 128.09, 128.00, 127.87, 127.79, 127.74, 127.69, 127.63, 127.55, 127.43, 127.18, 127.02, 126.85, 98.44, 98.26, 97.93, 97.53 (C-1), 81.19, 80.87, 80.35, 80.24, 79.53, 78.95, 78.10, 76.15, 76.01, 75.93, 75.27, 74.78, 74.12, 73.46, 73.35, 73.30, 72.85, 72.78, 72.62, 72.45, 72.14, 71.69, 71.35, 68.92, 68.82, 62.07, 61.64 (C-6); m/z (MS MALDI-TOF) calcd for [C188H200O40 + Na]+ 3120.3, found 3120.0.6A,6B,6E,6F-Tetra-O-acetyl-2A,2B,2C,2D,2E,2F,2G,2H,3A,3C, 3D,3E,3F,3G,3H,6C,6D,6G,6H-eicosa-O-benzyl-γ-cyclodextrin (24)
The tetraol 23 (155 mg, 0.05 mmol) was acetylated in a similar fashion as before to afford the desired tetraacetate 24 after column chromatography on silica gel using a mixture of AcOEt–hexane (gradient 10–20%) as eluent (140 mg, 86% yield). Rf 0.40 (EtOAc:hexane 4:6); [α]25D +26.6° (c 0.2, CHCl3); δH (400 MHz, CDCl3) 7.44–7.02 (m, 50H, Ar-H), 5.46 (d, 1H, J = 10.6, PhCH), 5.37 (d, 1H, J = 10.6, PhCH), 5.24 (d, 1H, J = 3.7, H-1), 5.11 (d, 1H, J = 3.5, H-1), 5.08 (d, 1H, J = 3.1, H-1), 5.04 (d, 1H, J = 3.5, H-1), 4.97 (2 × d, 2H, J = 11.0, PhCH), 4.90–4.77 (m, 4H, PhCH), 4.61–4.41 (m, 12H), 4.31–3.91 (m, 14H), 3.85–3.71 (m, 3H), 3.69–3.57 (m, 3H), 3.53–3.42 (m, 4H, 4 × H-2), 1.94 (s, 3H, Ac), 1.92 (s, 3H, Ac); δC (100 MHz, CDCl3) 170.27, 170.25 (CO), 139.58, 139.52, 139.16, 138.92, 138.65, 138.36, 138.33, 138.22, 138.07, 128.33, 128.29, 128.24, 128.14, 128.09, 127.96, 127.95, 127.81, 127.73, 127.61, 127.58, 127.50, 127.47, 127.10, 126.95, 99.23, 98.35, 98.25, 98.16 (C-1), 81.07, 80.99, 80.95, 80.80, 80.66, 80.57, 79.37, 79.04, 78.95, 78.20, 76.47, 76.37, 75.28, 74.98, 73.58, 73.31, 73.17, 72.74, 72.40, 71.77, 71.58, 69.44, 69.24, 68.86, 68.79, 63.35, 20.89 (Ac), 20.78 (Ac); m/z (HRMS MALDI-TOF) calcd for [C196H208O44 + Na]+ 3288.39363, found 3288.39382.
Acknowledgements
We wish to acknowledge the Alberta Ingenuity, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the University of Calgary for supporting the current project, and Dr. Ping Zhang for technical assistance.Notes and references
- (a) T. Lecourt, A. Herault, A. J. Pearce, M. Sollogoub and P. Sinaÿ, Chem.–Eur. J., 2004, 10, 2960–2971 CrossRef CAS; (b) M. Sollogoub, Eur. J. Org. Chem., 2009, 1295–1303 CrossRef CAS; (c) S. Guieu and M. Sollogoub, Angew. Chem., Int. Ed., 2008, 47, 7060–7063 CrossRef CAS; (d) N. Masurier, O. Lafont, R. Le Provost, D. Lesur, P. Masson, F. Djedaini-Pilard and F. Estour, Chem. Commun., 2009, 589–591 RSC.
- (a) S. Guieu and M. Sollogoub, J. Org. Chem., 2008, 73, 2819–2828 CrossRef CAS; (b) O. Bistri, P. Sinaÿ, J. J. Barbero and M. Sollogoub, Chem.–Eur. J., 2007, 13, 9757–9774 CrossRef CAS.
- P. Balbuena, D. Lesur, M. J. G. Alvarez, F. Mendicuti, C. Ortiz-Mellet and J. M. G. Fernandez, Chem. Commun., 2007, 3270–3272 RSC.
- M. Petrillo, L. Marinescu, C. Rousseau and M. Bols, Org. Lett., 2009, 11, 1983–1985 CrossRef CAS.
- (a) W. H. Tang, I. W. Muderawan, T. T. Ong and S. C. Ng, Tetrahedron: Asymmetry, 2007, 18, 1548–1553 CrossRef CAS; (b) S. Riela, F. D'Anna, P. Lo Meo, M. Gruttadauria, R. Giacalone and R. Noto, Tetrahedron, 2006, 62, 4323–4330 CrossRef CAS; (c) Y. Liu, L. Li, X. Y. Li, H. Y. Zhang, T. Wada and Y. Inoue, J. Org. Chem., 2003, 68, 3646–3657 CrossRef CAS; (d) M. V. Rekharsky and Y. Inoue, J. Am. Chem. Soc., 2002, 124, 813–826 CrossRef CAS; (e) R. F. Gomez-Biagi, R. B. C. Jagt and M. Nitz, Org. Biomol. Chem., 2008, 6, 4622–4626 RSC; (f) O. Bistri, P. Sinaÿ and M. Sollogoub, Tetrahedron Lett., 2006, 47, 4137–4139 CrossRef CAS; (g) A. W. Coleman, C. C. Ling and M. Miocque, Angew. Chem., Int. Ed. Engl., 1992, 31, 1381–1383 CrossRef.
- (a) J. Bjerre, C. Rousseau, L. Marinescu and M. Bols, Appl. Microbiol. Biotechnol., 2008, 81, 1–11 CrossRef CAS; (b) R. Breslow, Acc. Chem. Res., 1995, 28, 146–153 CrossRef CAS; (c) J. Bjerre, E. H. Nielsen and M. Bols, Eur. J. Org. Chem., 2008, 745–752 CrossRef CAS; (d) J. Bjerre, T. H. Fenger, L. G. Marinescu and M. Bols, Eur. J. Org. Chem., 2007, 704–710 CrossRef CAS; (e) F. Ortega-Caballero, J. Bjerre, L. S. Laustsen and M. Bols, J. Org. Chem., 2005, 70, 7217–7226 CrossRef CAS; (f) C. Rousseau, F. Ortega-Caballero, L. U. Nordstrom, B. Christensen, T. E. Petersen and M. Bols, Chem.–Eur. J., 2005, 11, 5094–5101 CrossRef CAS.
- (a) F. Ortega-Caballero, C. Ortiz-Mellet, L. Le Gourrierec, N. Guilloteau, C. Di Giorgio, P. Vierling, J. Defaye and J. M. G. Fernández, Org. Lett., 2008, 10, 5143–5146 CrossRef CAS; (b) A. Díaz-Moscoso, P. Balbuena, M. Gómez-García, C. Ortiz-Mellet, J. M. Benito, L. Le Gourriérec, C. Di Giorgio, P. Vierling, A. Mazzaglia, N. Micali, J. Defaye and J. M. G. Fernández, Chem. Commun., 2008, 2001 RSC; (c) A. Méndez-Ardoy, M. Gómez-García, C. Ortiz-Mellet, N. Sevillano, M. D. Girón, R. Salto, F. Santoyo-González and J. M. G. Fernández, Org. Biomol. Chem., 2009, 7, 2681–268 RSC; (d) S. Menuel, S. Fontanay, I. Clarot, R. E. Duval, L. Diez and A. Marsura, Bioconjugate Chem., 2008, 19, 2357–2362 CrossRef CAS; (e) A. McMahon, E. Gomez, R. Donohue, D. Forde, R. Darcy and C. M. O'Driscoll, J. Drug Delivery Sci. Technol., 2008, 18, 303–307 Search PubMed; (f) N. Mourtzis, M. Paravatou, I. M. Mavridis, M. L. Roberts and K. Yannakopoulou, Chem.–Eur. J., 2008, 14, 4188–4200 CrossRef CAS.
- (a) K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS; (b) A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977–1996 CrossRef CAS.
- (a) J. Boger, D. G. Brenner and J. R. Knowles, J. Am. Chem. Soc., 1979, 101, 7630–7631 CrossRef; (b) C.-C. Ling, A. W. Coleman and M. Miocque, Carbohydr. Res., 1992, 223, 287–291 CrossRef CAS; (c) S. Ward, P. Zhang and C. C. Ling, Carbohydr. Res., 2009, 344, 808–814 CrossRef CAS.
- (a) L. Poorters, D. Armspach and D. Matt, Eur. J. Org. Chem., 2003, 1377–1381 CrossRef CAS; (b) D. Armspach, L. Poorters, D. Matt, B. Benmerad, F. Balegroune and L. Toupet, Org. Biomol. Chem., 2005, 3, 2588–2592 RSC.
- E. Engeldinger, D. Armspach and D. Matt, Chem. Rev., 2003, 103, 4147–4173 CrossRef CAS.
- (a) I. Tabushi, K. Shimokawa, N. Shimizu, H. Shirakata and K. Fujita, J. Am. Chem. Soc., 1976, 98, 7855–7856 CrossRef CAS; (b) I. Tabushi, Y. Kuroda, K. Yokota and L. C. Yuan, J. Am. Chem. Soc., 1981, 103, 711–712 CrossRef CAS; (c) I. Tabushi, L. C. Yuan, K. Shimokawa, K. I. Yokota, T. Mizutani and Y. Kuroda, Tetrahedron Lett., 1981, 22, 2273–2276 CrossRef CAS; (d) I. Tabushi, T. Nabeshima, H. Kitaguchi and K. Yamamura, J. Am. Chem. Soc., 1982, 104, 2017–2019 CrossRef CAS; (e) I. Tabushi, T. Nabeshima, K. Fujita, A. Matsunaga and T. Imoto, J. Org. Chem., 1985, 50, 2638–2643 CrossRef CAS.
- D. Armspach and D. Matt, Carbohydr. Res., 1998, 310, 129–133 CrossRef CAS.
- (a) A. J. Pearce and P. Sinaÿ, Angew. Chem., Int. Ed., 2000, 39, 3610–3612 CrossRef CAS; (b) B. du Roizel, J. P. Baltaze and P. Sinaÿ, Tetrahedron Lett., 2002, 43, 2371–2373 CrossRef.
- G. K. Rawal, S. Rani and C.-C. Ling, Tetrahedron Lett., 2009, 50, 4633–4636 CrossRef CAS.
- O. Bistri, P. Sinaÿ and M. Sollogoub, Tetrahedron Lett., 2005, 46, 7757–7760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR data of synthesized compounds. See DOI: 10.1039/b915450g |
| This journal is © The Royal Society of Chemistry 2010 |