DOI:
10.1039/B916041H
(Perspective)
Org. Biomol. Chem., 2010,
8, 29-38
Synthesis of natural products containing spiroketals via intramolecular hydrogen abstraction
Received 4th August 2009, Accepted 11th September 2009
First published on 7th October 2009
Abstract
Although known for over a quarter of a century, the oxidative radical cyclisation route to spiroketals has found limited use in natural product synthesis in comparison to classical approaches. Its successful application in this field of research forms the subject of this perspective.
 Jonathan Sperry | Jonathan Sperry was born in Bracknell, Berkshire, UK and completed his BSc (Hons) at the University of Exeter, UK in 2002. He was awarded his PhD in 2006 under the supervision of Professor Christopher J. Moody at the same institution working on the synthesis of alkaloid natural products using biomimetic oxidations. Since March 2006 he has been working as a postdoctoral research fellow with Professor Margaret Brimble at the University of Auckland. |
 Yen-Cheng (William) Liu | Yen-Cheng (William) Liu was born in Taipei, Taiwan. He studied medicinal chemistry at The University of Auckland, New Zealand, and obtained a BSc (Hons) degree in 2006 under the supervision of Professor Margaret Brimble working on the synthesis of spiroketals related to the rubromycins. He is currently a fourth year PhD student in the Brimble research group investigating the synthesis of aryl spiroketals by oxidative radical cyclisation involving the intramolecular hydrogen abstraction process. |
 Margaret Brimble | Margaret Brimble was born in Auckland, New Zealand. She completed an MSc in Chemistry at the University of Auckland. She was awarded a UK Commonwealth Scholarship to undertake her PhD studies at Southampton University. In 1986 she was appointed as a lecturer at Massey University, and in 2000 she took up the Chair in Organic and Medicinal Chemistry at the University of Auckland where her research program focuses on the synthesis of spiroacetal-containing natural products, pyranonaphthoquinone antibiotics, alkaloids and peptidomimetics for the treatment of neurodegenerative disorders. |
Introduction
Spiroketals are found in a wide range of natural products that exhibit a broad range of biological properties. Traditionally, the synthesis of spiroketals has been conducted by the acid-catalysed cyclisation of a dihydroxyketone precursor, often producing the thermodynamic products arising from the maximum number of anomeric effects and minimum steric interactions.1a–d Despite the popularity of this method, it is not always amenable to the synthesis of delicate substrates and several natural products are known to contain non-anomeric spiroketals.2a–cThe oxidative radical cyclisation approach provides a valuable alternative to the classical spiroketal synthesis (Scheme 1). It is particularly useful when delicate or acid-labile substrates are involved. If spiroketals of a particular configuration are required, this method often facilitates the synthesis of kinetic products.
 |
| | Scheme 1 Classical and oxidative radical cyclisation approach to spiroketals. | |
The first spiroketal synthesis using a radical cyclisation of an aliphatic alcohol was reported in 1969.3 It was found that a series of diols 1a–c underwent a double intramolecular hydrogen abstraction (IHA) when treated with lead tetraacetate in benzene under reflux, furnishing the spiroketals 2a–c (Scheme 2).
 |
| | Scheme 2 Double IHA of aliphatic diols 1 using lead tetraacetate.3 | |
Using this methodology several years later, Suárez4 carried out the oxidative cyclisation of labdanediol 3 using lead tetraacetate and iodine under light irradiation, demonstrating that the reaction was amenable to complex substrates and proceeds in high yield under photolytic conditions. The reaction provided a 77% yield of spiroketals 4a and 4b in a 3 : 1 ratio (Scheme 3).
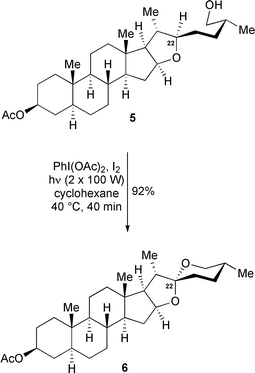
Suárez later investigated the cyclisation of the ε-hydroxy-tetrahydropyran (26-hydroxyfurostan) 5 to spirostan sapogenin 6 and subsequently elucidated the likely mechanism (Scheme 4).5 Initially, the alkoxy radical is formed from a thermal or photochemical homolytic cleavage of the O–I bond in the alkyl hypoiodite 8. The alkyl hypoiodite itself is formed from reaction of alcohol 7 with acetyl hypoiodite which is generated in situ from the reaction between lead tetraacetate and iodine. Initially, both the 1,5 and 1,6-hydrogen shift pathways were thought feasible. However, subsequent use of deuterium-labelled substrates during the cyclisation proved the reaction proceeds exclusively via a 1,6-hydrogen shift5 and the existence of the acetyl hypoiodite was later confirmed by NMR studies.6 Although the formation of five-membered rings generally produces higher yields than their six-membered counterparts,7 the yield for the formation of six-membered rings can be increased if the proton to be abstracted is attached to an oxygen-substituted carbon.8,9 For a successful IHA to occur, the optimum distance between the O-radical and the proton to be abstracted should be around 3 Å.9
 |
| | Scheme 4 Mechanism of the oxidative radical spiroketalisation.5 | |
In 1983, the scope of this reaction was expanded when it was shown that the oxidative radical cyclisation of alcohol 9 could be conducted with mercuric oxide and iodine, affording spiroketals 10a and 10b. Acid-catalysed equilibration of the spiroketal mixture led to the exclusive formation of the thermodynamically favoured spiroketal 10a (Scheme 5).10 This mercury-mediated protocol was subsequently used by Kay in the synthesis of the avian toxin (±)-talaromycin B.11
A major breakthrough came in 1984 when Suárez reported the diastereoselective synthesis of the spiroketal-containing steroid 6 by the photolytic oxidative radical spiroketalisation of alcohol 5 using the non-toxic hypervalent iodine reagent iodobenzene diacetate (PhI(OAc)2) (Scheme 6).12a–b Only one equivalent of PhI(OAc)2 and iodine is required for completion of the reaction, in contrast to the excess of lead tetraacetate and iodine usually required for its variant. Further superiority of this reagent was subsequently demonstrated during the cyclisation of labdanediol 312a,13a–b although in the presence of light, carboxylic acid substrates tend to undergo a Hunsdiecker-type reaction in preference to spiroketal formation.13a,14
In 2000, Markó showed that the diastereoselective synthesis of spiroketals 12 could be achieved by the electrochemical oxidative cyclisation of a series of hydroxy-tetrahydropyrans 11. This analogous procedure proceeds through a similar mechanism, wherein the vital oxidation step is carried out electrochemically (Scheme 7).15
Avermectin A1a
The avermectins are a series of macrocyclic lactone derivatives with potent anthelmintic properties.16 During the total synthesis of avermectin A1a13, the aglycon moiety was constructed using the mercury mediated oxidative radical cyclisation protocol, one of the earliest examples reporting the use of this methodology in a complex molecule synthesis.17a–b Alcohol 14 underwent spiroketalisation upon irradiation in the presence of mercuric oxide and iodine to produce spiroketal 15 as a single diastereomer (Scheme 8). |
| | Scheme 8 Synthesis of spiroketal 15 during the total synthesis of avermectin A1a13 by Danishefsky.17a–b | |
 |
| | Scheme 9 (−)-Calyculin A 16 and synthesis of spiroketals 18a-b by Trost.19 | |
![Salinomycin 19, narasin 20, deoxy-(O-8)-epi-17-salinomycin 21 and CP44,161 22 with a shared [5,6,6]-bis-spiroketal motif.](/image/article/2010/OB/b916041h/b916041h-f1.gif) |
| | Fig. 1 Salinomycin 19, narasin 20, deoxy-(O-8)-epi-17-salinomycin 21 and CP44,161 22 with a shared [5,6,6]-bis-spiroketal motif. | |
Building on previous model work in our own laboratory,24a–b the stereoselective synthesis of the bis-spiroketal fragment present in deoxy-(O-8)-epi-17-salinomycin 21 was successfully achieved.25 Photolytic oxidative radical cyclisation of either spiroketal iodohydrin 23a or 23b in the presence of iodobenzene diacetate and iodine produced a 1.7 : 1 ratio of bis-spiroketals 24a and 24b. Bis-spiroketal 24a possesses the correct absolute stereochemistry present in deoxy-(O-8)-epi-17-salinomycin 21 and thus was considered a suitable intermediate for the synthesis of aldehyde 25 to which the pyran moiety could be appended (Scheme 10). However, despite considerable efforts, displacement of iodide in bis-spiroketal 24a resulted in concomitant silyl group cleavage at C9, curtailing efforts towards the synthesis of aldehyde 25.26 In order to circumvent this problem, an alternative protecting group strategy was sought.
 |
| | Scheme 10 Synthesis of bis-spiroketals 24a and 24b25 and failed conversion to aldehyde 25 by Brimble.26 | |
Having established a suitable route that employed an acetate at C2 during the synthesis of a model bis-spiroketal,26 an investigation into the synthesis of the bis-spiroketal moiety of CP44,161 22 was conducted.27a–b Thus, the 1 : 1 mixture of diastereomeric hydroxyspiroketals 26a-b and 27a-b were subjected to a photolytic oxidative cyclisation in the presence of iodobenzene diacetate and iodine delivering their respective bis-spiroketals 28 and 29. In each case, cis bis-spiroketals 28a and 29a were produced as the major diastereomer. Trans bis-spiroketals 28b and 29b were not observed as they underwent rapid epimerisation at C7 to form cis bis-spiroketals 28c and 29c respectively. Bis-spiroketal 29c has the correct stereochemistry at C2 and the same absolute stereochemistry found in both salinomycin 19 and antibiotic CP44,161 22 (Scheme 11).
 |
| | Scheme 11 Synthesis of bis-spiroketals 28a/c and 29a/c by Brimble.27a–b | |
Ciguatoxin CTX3C
Ciguatoxin CTX3C 30 is the principle polycyclic ether neurotoxin responsible for ciguatera fish poisoning (Fig. 2).28 |
| | Fig. 2 Ciguatoxin CTX3C 30. | |
Building on from previous model work,29 the spiroketal containing the IJKLM portion 32 was synthesised from alcohol 31 using a photolytic oxidative cyclisation in the presence iodobenzene diacetate and iodine. The 1 : 1 mixture of spiroketals produced underwent acid-catalysed equilibration delivering spiroketal 32 exclusively which possesses the correct configuration present in ciguatoxin CTX3C 30 (Scheme 12).30
 |
| | Scheme 12 Synthesis of pentacyclic IJKLM fragment 32 of ciguatoxin CTX3C by Fujiwara.30 | |
Spirolides B and D
Our own laboratory reported the synthesis of the bis-spiroketal fragments related to the marine biotoxins,31 spirolides B 33 and D 34 using iterative oxidative radical cyclisations. Extending a successful synthesis of a racemic model unsaturated 5,5,6-bis-spiroketal,32 the oxidative radical cyclisation was extended to a stereoselective synthesis. Thus, irradiation of either pair of 1 : 1 inseparable diastereomeric alcohols 35a–b or 35c–d in the presence of iodobenzene diacetate and iodine delivered 1 : 1 mixtures of diastereomeric spiroketals 36a-b and 36c-d respectively. Silyl group cleavage from all four diastereomeric spiroketals 36a–d (1 : 1 : 1 : 1) gave the four spiroketal alcohols 37a-d (1 : 1 : 1 : 1) which underwent a second oxidative cyclisation. A 1 : 1 : 1 : 1 mixture of diastereomeric bis-spiroketals 38a–d was obtained which underwent acid-catalysed equilibration to produce bis-spiroketal 38b (18%) which was separable from an inseparable mixture of bis-spiroketals 38a and 38c (3.3 : 1, 68%). The bis-spiroketal 38a possesses the same absolute stereochemistry present in spirolides B and D. Although bis-spiroketals 38a and 38c were further elaborated to bis-spiroketals 39a and 39c respectively, introduction of a synthetic handle at C22 proved troublesome (Scheme 13).33a–b |
| | Scheme 13 Structures of spirolides B 33 and D 34, synthesis of bis-spiroketals 39a/c by Brimble.33a–bcis/trans denotes the arrangement of the both terminal ring oxygen atoms across the central ring; syn/anti denotes the arrangement of the central ring oxygen and the substituents attached to the outer THF ring. | |
Extending this work further,34a–b the synthesis of the fully functionalized bis-spiroketal 40 was achieved. Oxidative cyclisation of a 1 : 1 mixture of diastereomeric alcohols 41 using iodobenzene diacetate and iodine furnished a 1 : 1 mixture of two diastereomeric spiroketals 42 which underwent silyl ether deprotection to give alcohols 43. A second oxidative radical cyclisation of alcohols 43 afforded bis-spiroketals 44a–d as a 1 : 1 : 1 : 1 mixture of diastereomers. The 1 : 1 : 1 : 1 mixture of diastereomeric spiroketals 44a–d was treated with m-CPBA, effecting concomitant epoxidation and equilibration remarkably resulting in one diastereomer 45. Further elaboration gave bis-spiroketal 40, a diastereomer of the bis-spiroketal fragment present in spirolides B and D (Scheme 14).
 |
| | Scheme 14 Synthesis of bis-spiroketal 40 by Brimble.34a–34b | |
Bistramide C
Bistramide C 46 is a bioactive cyclic polyether which exhibits potent in vitro inhibition against numerous tumour cell lines.35 Wipf and co-workers carried out a total synthesis of a stereoisomer of bistramide C using an oxidative cyclisation and subsequently assigned the configuration of the natural product.36 This assignment was later confirmed through total synthesis that also involved a photolytic oxidative radical spiroketalisation.37 Thus, alcohol 47 underwent the key photolytic oxidative cyclisation in the presence of iodobenzene diacetate and iodine followed by reductive removal of the pivaloate to afford a 3.8 : 1 mixture of spiroketal 48 and iodospiroketal 49. Iodospiroketal 49 was treated with azobisisobutyronitrile and tributyltin hydride to produce spiroketal 48 which was used as an intermediate in the total synthesis of (+)-bistramide C 46 (Scheme 15).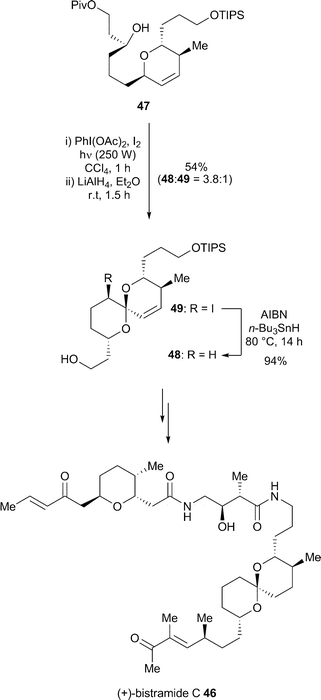 |
| | Scheme 15 Synthesis of (+)-bistramide C 46 by Wipf.37 | |
(+)-Spirolaxine methyl ether
Dallavalle38 reported the total synthesis of the anti-Helicobacter pylori agent (+)-spirolaxine methyl ether 5039 in which spiroketalisation was achieved using an oxidative cyclisation. Upon irradiation in the presence of mercuric oxide and iodine, alcohol 51 underwent oxidative radical cyclisation affording spiroketal 52 as a single diastereomer, a key intermediate in the total synthesis of (+)-spirolaxine methyl ether 50 (Scheme 16). |
| | Scheme 16 Synthesis of (+)-spirolaxine methyl ether 50 by Dallavalle.38 | |
Ritterazines and cephalostatins
The ritterazines40 and cephalostatins41 comprise a family of 45 trisdecacyclic bis-steroidal pyrazines that display potent cytotoxicity against human tumors, ranking among the most potent anticancer agents ever tested by the NCI.42 Selected members of this family that contain spiroketal moieties are shown in Fig. 3. |
| | Fig. 3 Selected ritterazines and cephalostatin 1. | |
Based on extensive model work,43 Fuchs reported the total synthesis of ritterazine M 53 using a photolytic oxidative radical cyclisation of alcohol 58 with iodobenzene diacetate and iodine giving [5,6]-spiroketals 59a-b in a 5.5 : 1 ratio and excellent yield.44 Spiroketal 59a was subsequently used for the total synthesis and structure reassignment of ritterazine M 53 (Scheme 17).
 |
| | Scheme 17 Total synthesis of ritterazine M 53 by Fuchs.43,44 | |
Due to their scarcity from nature and unknown mechanism of action, several studies towards the synthesis of analogues of the ritterazines and the cephalostatins have been reported, several of which employed the oxidative radical spiroketalisation. Suárez synthesized a series of C22 and C25 stereoisomers of cephalostatin and upon probing the acid stability of the spiroketal products, it was discovered that compounds with the greatest acid-catalysed reactivity possessed the natural stereochemistry.45a,b Also using this methodology, Fuchs reported the synthesis of a series of [5,5]-spiroketal analogues of cephalostatin46 and Shair reported the synthesis of the ‘northern’ halves of ritterazines B (54), F (55), G (56), and H, which ultimately led to the reassignment of the [5,5]-spiroketal moiety of ritterazines B and F.47 These studies have proven the value of this mild spiroketalisation reaction in the remote functionalisation of steroid nuclei.
Conclusions
The oxidative radical cyclisation provides an excellent method for the synthesis of spiroketals and this perspective article highlights the utility of this approach in the field of natural product synthesis. However, the broad importance of this methodology in organic synthesis, particularly in carbohydrate chemistry,48a–c should not be understated. It is envisaged that this method will continue to provide a valuable synthetic tool in complex molecule synthesis.
References
- For general reviews on the synthesis and biological properties of spiroketals, see:
(a) B. Rama Raju and A. K. Saikia, Molecules, 2008, 13, 1942–2038 Search PubMed;
(b) T. K. Mead and B. N. Brewer, Curr. Org. Chem., 2003, 7, 227–256 CrossRef CAS;
(c) M. A. Brimble and D. P. Furkert, Curr. Org. Chem., 2003, 7, 1461–1484 CAS;
(d) M. F. Jacobs and W. Kitching, Curr. Org. Chem, 1998, 2, 395–436 CAS.
- For reviews regarding non-anomeric spiroketals, see:
(a) J. E. Aho, P. M. Pikho and T. K. Rissa, Chem. Rev., 2005, 105, 4406–4440 CrossRef CAS;
(b) S. Favre, P. Vogel and S. Gerber-Lemaire, Molecules, 2008, 13, 2570–2600 Search PubMed;
(c) M. A. Rizzacasa and A. Pollex, Org. Biomol. Chem., 2009, 7, 1053–1059 RSC.
- V. M. Mićović, S. Stojčić, M. Bralović, S. Mladenović, D. Jeremić and M. Stefanović, Tetrahedron, 1969, 25, 985–993 CrossRef CAS.
- A. G. González, C. G. Francisco, R. Freire, R. Hernández, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1976, 17, 2725–2728 CrossRef.
- C. G. Francisco, R. Freire, R. Hernández, M. C. Medina and E. Suárez, Tetrahedron Lett., 1983, 24, 4621–4624 CrossRef CAS.
- J. L. Courtneidge, J. Lusztyk and D. Pagé, Tetrahedron Lett., 1994, 35, 1003–1006 CrossRef CAS.
- G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095–7129 CrossRef CAS.
- C. G. Francisco, R. Freire, A. J. Herrera, I. Peréz-Martín and E. Suárez, Org. Lett., 2002, 4, 1959–1961 CrossRef CAS.
- C. G. Francisco, A. J. Herrera, A. R. Kennedy, D. Melián and E. Suárez, Angew. Chem., Int. Ed., 2002, 41, 856–858 CrossRef CAS.
- I. T. Kay and E. G. Williams, Tetrahedron Lett., 1983, 24, 5915–5918 CrossRef CAS.
- I. T. Kay and D. Bartholomew, Tetrahedron Lett., 1984, 25, 2035–2038 CrossRef CAS.
-
(a) J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1984, 25, 1953–1956 CrossRef;
(b) P. de Armas, J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, J. Chem. Soc., Perkin Trans. 1, 1989, 405–411 RSC.
-
(a) J. de Pascual Teresa, J. G. Urones, A. Montaña and P. Basabe, Tetrahedron Lett., 1985, 26, 5717–5720 CrossRef;
(b) J. G. Urones, P. Basabe, I. S. Marcos, D. D. Martín, M. J. Sexmero, M. H. Peral and H. B. Broughton, Tetrahedron, 1992, 48, 10389–10398 CrossRef CAS.
- J. I. Concepción, C. G. Francisco, R. Freire, R. Hernández, J. A. Salazar and E. Suárez, J. Org. Chem., 1986, 51, 402–404 CrossRef CAS.
- I. E. Markó, Tetrahedron Lett., 2000, 41, 4383–4387 CrossRef CAS.
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Õiwa and S. Õmura, Antimcrob. Agents. Chemother., 1979, 15, 361–367 Search PubMed.
-
(a) S. J. Danishefsky, D. M. Armistead, F. E. Wincott, H. G. Selnick and R. Hungate, J. Am. Chem. Soc., 1987, 109, 8117–8119 CrossRef CAS;
(b) S. J. Danishefsky, D. M. Armistead, F. E. Wincott, H. G. Selnick and R. Hungate, J. Am. Chem. Soc., 1989, 111, 2967–2980 CrossRef CAS.
- S. Matsunaga and N. Fusetani, Tetrahedron Lett., 1991, 32, 5605–5606 CrossRef CAS and references therein.
- B. M. Trost and J. A. Flygare, Tetrahedron Lett., 1994, 35, 4059–4062 CrossRef CAS.
- H. Kinashi, N. Ōtake, H. Yonehara, S. Sato and Y. Saito, Tetrahedron Lett., 1973, 14, 4955–4958 CrossRef.
- J. L. Occolowitz, D. H. Berg, M. Debono and R. L. Hamill, Biol. Mass Spectrom., 1976, 3, 272–277 CAS.
- J. W. Westley, J. F. Blount, R. H. Evans, Jr. and C.-M. Liu, J. Antibiot., 1977, 30, 610–612 CAS.
- J. Tone, R. Shibatawa, M. Maeda, K. Inoue, S. Nishiyama, M. Ishiguro, W. P. Cullen, J. B. Routien, L. R. Chappell, C. E. Moppett, M. T. Jefferson, and W. D. Celmer, 18th Intersociety Conference on Antimicrobial Agents Chemotherapy, Atlanta, Ga, Oct. 2–4, 1978.
-
(a) M. A. Brimble, G. M. Williams, R. Baker and M. James, Tetrahedron Lett., 1990, 31, 3043–3046 CrossRef CAS;
(b) M. A. Brimble, G. M. Williams and R. Baker, J. Chem. Soc., Perkin Trans. 1, 1991, 2221–2227 RSC.
- M. A. Brimble and G. M. Williams, J. Org. Chem., 1992, 57, 5818–5822 CrossRef CAS.
- P. R. Allen, M. A. Brimble and F. A. Fares, J. Chem. Soc., Perkin Trans. 1, 1998, 2403–2411 RSC.
-
(a) P. A. Allen, M. A. Brimble and H. Prabaharan, Synlett, 1999, 295–298 CAS;
(b) P. R. Allen, M. A. Brimble and F. A. Fares, J. Chem. Soc., Perkin Trans. 1, 2001, 379–389 RSC.
- M. Satake, M. Murata and T. Yasumoto, Tetrahedron Lett., 1993, 34, 1975–1978 CrossRef CAS.
- D. Domon, K. Fujiwara, Y. Ohtaniuchi, A. Takezawa, S. Takeda, H. Kawasaki, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 8279–8283 CrossRef CAS.
- D. Domon, K. Fujiwara, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 8285–8288 CrossRef CAS.
- T. Hu, J. M. Curtis, Y. Oshima, M. A. Quilliam, J. A. Walter, W. M. Watson-Wright and J. L. C. Wright, J. Chem. Soc., Chem. Commun., 1995, 2159–2161 RSC.
- V. Caprio, M. A. Brimble and D. P. Furkert, Tetrahedron, 2001, 57, 4023–4034 CrossRef CAS.
-
(a) D. P. Furkert and M. A. Brimble, Org. Lett., 2002, 4, 3655–3658 CrossRef CAS;
(b) M. A. Brimble and D. P. Furkert, Org. Biomol. Chem., 2004, 2, 3573–3583 RSC.
-
(a) K. Meilert and M. A. Brimble, Org. Lett., 2005, 7, 3497–3500 CrossRef CAS;
(b) K. Meilert and M. A. Brimble, Org. Biomol. Chem., 2006, 4, 2184–2192 RSC.
- J.-F. Biard, C. Roussakis, J.-M. Kornprobst, D. Gouiffes-Barbin, J.-F. Verbist, P. Cotelle, M. P. Foster, C. M. Ireland and C. Debitus, J. Nat. Prod., 1994, 57, 1336–1345 CrossRef CAS and references therein.
- P. Wipf, Y. Uto and S. Yoshimura, Chem.–Eur. J., 2002, 8, 1670–1681 CrossRef CAS.
- P. Wipf and T. D. Hopkins, Chem. Commun., 2005, 3421–3423 RSC.
- R. Nannei, S. Dallavalle, M. Lucio, A. Bava and G. Nasini, J. Org. Chem., 2006, 71, 6277–6280 CrossRef CAS.
- A. Arnone, G. Assante, G. Nasini and O. Vajna de Pava, Phytochemistry, 1990, 29, 613–616 CrossRef CAS.
- S. Fukuzawa, S. Matsunaga and N. Fusetani, Tetrahedron, 1995, 51, 6707–6716 CrossRef CAS.
- G. R. Pettit, M. Inoue, Y. Kamano, D. L. Herald, C. Arm, C. Dufresne, N. D. Christie, J. M. Schmidt, D. L. Doubek and T. S. Krupa, J. Am. Chem. Soc., 1988, 110, 2006–2007 CrossRef CAS.
- Reviews regarding the isolation, biological activity and synthesis of the cephalostatins and ritterazines are available
(a) B. R. Moser, J. Nat. Prod., 2008, 71, 487–491 CrossRef CAS;
(b) S. Lee, T. G. LaCour and P. Fuchs, Chem. Rev., 2009, 109, 2275–2314 CrossRef CAS.
- S. Lee, T. G. LaCour, D. Lantrip and P. L. Fuchs, Org. Lett., 2002, 4, 313–316 CrossRef CAS.
- S. Lee and P. L. Fuchs, Org. Lett., 2002, 4, 317–318 CrossRef CAS.
-
(a) C. Betancor, R. Freire, I. Pérez-Martín, T. Prangé and E. Suárez, Org. Lett., 2002, 4, 1295–1297 CrossRef CAS;
(b) C. Betancor, R. Freire, I. Pérez-Martín, T. Prangé and E. Suárez, Tetrahedron, 2005, 61, 2803–2814 CrossRef CAS.
- J. S. Lee and P. L. Fuchs, Org. Lett., 2003, 5, 2247–2250 CrossRef CAS.
- S. T. Phillips and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 6589–6598 CrossRef CAS.
- Recent examples
(a) A. Martín, I. Pérez-Martín, L. M. Quintanal and E. Suárez, Org. Lett., 2007, 9, 1785–1788 CrossRef CAS;
(b) A. Martín, L. M. Quintanal and E. Suárez, Tetrahedron Lett., 2007, 48, 5507–5511 CrossRef CAS;
(c) C. G. Francisco, A. J. Herrera and E. Suárez, J. Org. Chem., 2002, 67, 7439–7445 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.