Synthetic biology
Ali Tavassoli*
School of Chemistry, University of Southampton, Southampton, UK SO17 1BJ. E-mail: ali1@soton.ac.uk
First published on 10th November 2009
Abstract
There is currently much excitement surrounding the rapidly growing discipline of synthetic biology, which utilizes the design and construction principles of engineering to develop, evolve and standardize biological components and systems. This systematic approach to improving and increasing the programmability and robustness of biological components is expected to lead to the facile assembly of artificial biological components and integrated systems that enable innovative approaches to solving a wide range of societal challenges. Here we discuss the current state of the art and outline the next wave of synthetic biology: integrating individual components into systems.
 Ali Tavassoli | Ali is a lecturer in chemical biology at The University of Southampton. He is the recipient of the European Association for Chemical and Molecular Sciences' silver medal for European Young Chemist, and a Career Establishment Award from Cancer Research UK. He has a multidisciplinary background in synthetic chemistry and molecular biology. His lab's research interests are at the interface of chemistry and biology; developing high throughput tools, screening methodologies and uncovering compounds that tackle diseases through inhibition of specific protein-protein interactions. They are also active in the new field of synthetic biology. For more information see the Tavassoli group website. |
What is synthetic biology?
The polish scientist Waclaw Szybalski envisaged the possibilities offered by the field of molecular biology upon its maturation in 1974: “Up to now we are working on the descriptive phase of molecular biology. … But the real challenge will start when we enter the synthetic biology phase of research in our field. We will then devise new control elements and add these new modules to the existing genomes or build up wholly new genomes. This would be a field with the unlimited expansion potential…”.1 The increasingly facile and rapidly decreasing cost of DNA synthesis and sequencing has made the above a reality today. This, combined with an increased understanding of biological systems (especially bacterial), has allowed genetic-level modification of living organisms. Advances in recombinant DNA technologies have led to the host organism being increasingly viewed as a chassis, with its components utilised for the replication or overproduction of non-native genes, proteins or small molecules. Biological material, especially DNA and proteins, can therefore be considered components that may be readily assembled within the bacterial chassis, and utilised to produce a system whose functionality can be designed and predicted. A good analogy is the computer industry, and the ability to assemble a device from individual components (processor, hard drive, monitor etc…) that interface with each other and function together as a single unit. Synthetic biologists wish to design and construct (de novo), or evolve standardised biological components that functionally interface with each other with similar ease, and can be rationally combined to produce a predetermined outcome.The synthetic biology community is composed of biologists, chemists and engineers. The diverse background of its proponents has lead to two main branches emerging;2 commonly referred to as top-down and bottom-up synthetic biology. Top-down synthetic biologists aim to reprogram cellular behaviour through introduction of non-native genes and reconfiguration of the gene expression networks. The overall intent is to simplify the creation of complex regulatory networks by utilizing engineering tools and mathematical models developed in systems biology, bioengineering and biotechnology. Such networks can then be constructed and programmed (analogous to a computer) to carry out a designated function. Bottom-up synthetic biology utilizes first principles for the de novo construction of synthetic genomes and non-natural molecules that behave analogously to their counterparts in living systems. This approach utilizes the tools of chemistry and biochemistry, with the creation of artificial life being a long-term goal.
We begin by describing the achievements of the ‘first wave’ of synthetic biology: the construction of genetic units and modules, engineering of biosynthetic pathways and networks and work towards minimal genome organisms and chassis. Rather than give a detailed and comprehensive discussion of all projects, we use a few examples to illustrate the achievements of the field. We finish by detailing our thoughts on the next wave of synthetic biology, which is expected to combine and utilize these basic parts to construct complicated and complex systems.
Components and modules
One of the defining goals of synthetic biology is the application of engineering principles to biological systems to develop and construct networks and pathways that behave in a predictable manner.3 One can envisage synthetic gene networks constructed to emulate digital circuits and devices, potentially allowing programming of cellular behaviour using the same principles as modern computing. One of the earliest examples of this approach is the construction of a genetic toggle switch in E. coli, based on the predictions of a simple mathematical model.4 The switch is composed of two repressors and two constitutive promoters (Fig. 1), with each promoter being inhibited by the repressor that is transcribed by the opposing promoter. The switch is thus flipped between stable states by either inducer (transient chemical or thermal induction, depending on the promoter). The design of the system may seem simplistic from a molecular biology point of view; however, the strength of this approach is the utilization of mathematical models and engineering principles to ensure the bistability of the switch.4 | ||
Fig. 1 Toggle switch design. Repressor A inhibits transcription of repressor B by targeting promoter A. Inducer A binds repressor A, switching on promoter A and transcription of repressor B. Inducer B inhibits repressor B, switching on transcription of repressor A and the reporter gene. Thus the switch can be flipped between stable states using inducer A or inducer B. The behaviour of the toggle switch and the conditions for bistability can be understood using  and and  , where U is the concentration of repressor A, and V is the concentration of repressor B, α1 is the effective rate of synthesis of repressor A and α2 is that for repressor B. β is the cooperativity of repression of promoter B and γ is that for promoter A. , where U is the concentration of repressor A, and V is the concentration of repressor B, α1 is the effective rate of synthesis of repressor A and α2 is that for repressor B. β is the cooperativity of repression of promoter B and γ is that for promoter A. | ||
Another example of a module is a genetic oscillator, of which several examples exist. An early example used three transcriptional repressors in a negative feedback loop,5 with the mathematical model developed to predict transcriptional regulation behaviour being particularly elegant. In this model, the repressor proteins and their corresponding mRNA concentrations are treated as continuous, dynamic variables. Variations in factors such as the dependence of the transcription rate on repressor concentration, the translation rate, and the decay rate of the protein and mRNA, result in one of two solutions: either the system converges towards a steady state, or the system becomes unstable, leading to sustained limit-cycle oscillations. The model indicated that oscillations are favoured in systems with strong promoters coupled to efficient ribosome-binding sites, with comparable protein and mRNA decay rates. The network was therefore constructed to this model, using hybrid promoters (to ensure transcriptional strength and tightness) and a carboxy-terminal tag that reduces the half-life of the repressor proteins, bringing it in line with that for mRNA. The oscillator network periodically induces the production of green fluorescent protein and interestingly, as the periods of oscillation were slower than the cell-division cycle, the oscillator state seems to be transmitted from generation to generation. Other examples6,7 of synthetic oscillators have introduced more robust and tunable oscillators (Fig. 2), and a tunable synthetic oscillator that functions within mammalian cells.8
 | ||
| Fig. 2 A dual-feedback circuit oscillator. Three copies of a hybrid promoter (plac/ara-1—made up of the activation operator site from the araBAD promoter and the repression operator site from lacZYA) drive the expression of araC, lacI and GFP genes, forming negative and positive feedback loops. The promoter is activated by the AraC protein in the presence of arabinose and repressed by the LacI protein in the absence of isopropyl -D-1-thiogalactopyranoside (IPTG). The promoters are activated by the addition of arabinose and IPTG to the growth medium. The increased production of AraC in the presence of arabinose activates the positive feedback loop; the concurrent increase in LacI, however, results in activation of the negative feedback loop. The differential activity of the two feedback loops leads to oscillatory behaviour by the circuit. The oscillatory period is tunable (from around 15 min to 60 min) by varying the IPTG and arabinose levels. | ||
Another key component of digital circuits and computing is a counter, a simple cellular equivalent of which has recently been reported.9 The genetic counter can count up to three induction events, allowing counting of varied, user-defined inputs. The ever-increasing synthetic gene circuit components have enlarged the molecular toolbox available to synthetic biologists and bioengineers, allowing the construction of cellular systems of increasing complexity and sophistication.6,9 Despite these rapid advances in synthetic genomics, assembling such networks with predictable functions remains very challenging and requires considerable retrofitting and fine-tuning of parts for the system to function as intended. The advancement of this field and the realization of its long-term goals are therefore dependant on increasing the predictability and decreasing the amount of fine-tuning required to construct functional networks.10 Another major step forward would be combining these components within the same chassis, to produce a multi-component, multi-functional synthetic network.
Engineering biosynthetic networks
Microbial hosts have long been utilised as chassis for the production of non-native proteins via singular transgenes. An example of traditional genetic engineering is the introduction of the gene that codes for human insulin into E. coli for the production of transgenic protein.11 The integration and engineering of multiple pathways into a bacterial host would enable a similar approach to be taken for the production of complex natural products. Coupling multiple enzymes to create a metabolic pathway allows circumnavigation of much of the problems associated with chemical synthesis of complex natural products, chemical intermediates or even small molecules; enzymes catalyse in a single step what may require multiple steps and purifications by synthetic chemistry. Given the increasingly important role played by natural products (such as Taxol and Vancomycin) in healthcare, there is tremendous potential in developing new biosynthetic pathways for the rapid production of complex compounds from simple, inexpensive starting materials. The synthetic biology approach to engineering such metabolic pathways is to focus on the design and construction of core components, with predictable, tunable function, that can be assembled into larger integrated systems. This is particularly challenging, given the nonlinear nature of biological components and the unpredictability of the interactions of new components with biological systems.A successful implementation of this approach is the production of artemisinic acid, a precursor of the antimalarial drug artemisinin (Fig. 2) in engineered E. coli12 or S. cerevisiae.12,13 Artemisinin is a sesquiterpene lactone endoperoxide, isolated from the sweet wormwood plant (Artemisinin annua), which is highly effective against multi-drug resistant strains of the malaria parasite Plasmodium falciparum. The relative structural complexity of artemisinin, makes its total synthesis challenging, and its production by traditional methods costly.14 The conversion of artemisinic acid to artemisinin in contrast, is efficient and cost effective,15 therefore the microbial synthesis of artemisinic acid should reduce the production costs of artemisinin to make it affordable to those in the developing world.
The first step towards artemisinic acid is the introduction of the amorphadiene synthase gene from A. annua into a suitable host (E. coli12 or S. cerevisiae13). This results in the production of amorphadiene from farnesyl pyrophosphate (FPP), but the increase in demand on FPP results in low yields. Improving the amount of amorphadiene produced required modifying the biosynthetic pathways in the host to increase FPP levels. This was achieved by up-regulating the several genes responsible for FPP biosynthesis, and down-regulating an enzyme converting FPP to sterols. With amorphadiene production optimised, its conversion to artemisinic acid was achieved in one step using a novel cytochrome p450 and its redox partner from A. annua, which catalyses the three-step oxidation reaction within the host strain (Fig. 3). This resulted in a strain of E. coli producing artemisinic acid de novo at levels exceeding 300 mg L−1,16 and transgenic yeast producing artemisinic acid at 115 mg L−1.13 Artemisinic acid is readily isolated from fermentation tanks and converted to artemisinin. This process has proved so efficient that it is being developed for the large-scale production of artemisinin and is expected to dramatically reduce the cost of an adult course of the drug (currently around $2.20).
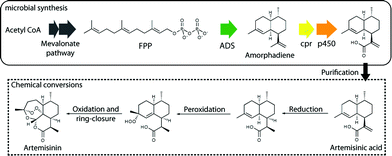 | ||
| Fig. 3 Semi-synthetic production of Artemisinin. The gene coding for amorphadiene synthase (ADS) is introduced into the organism, and converts FPP to amorphadiene. Amprphadiene is converted to artemisinic acid by a novel p450 oxidase and its redox partner (cpr) also introduced into the organism. Artemisinic acid is purified from the fermentation tanks and converted to artemisinin using established chemical steps. | ||
Another area where synthetic biology has the potential to make a significant contribution is the microbial production of biofuels. This approach has the potential to have a dramatic impact, given the increasing energy costs and the need for renewable, environmentally friendly fuels. Metabolic engineering of E. coli and S. cerevisiae for the production of n-butanol17,18 are recent examples of this promising approach. Although still in its infancy, there is much potential for well-characterized biological components that can be combined with a suitable chassis to develop “microbial factories” capable of biosynthesizing complex chemicals and compounds, in higher yields and shorter times than traditional methods.
Synthetic genomes
This branch of synthetic biology is focused on the chemical synthesis of minimal genome organisms and artificial life in the laboratory. This aim has been greatly advanced by recent progress in DNA synthesis and sequencing technology. Yet, despite the lowered cost of DNA synthesis and sequencing, there are several technical barriers to the synthesis of chromosome-sized stretches of DNA. Current technology allows for the accurate, direct synthesis of a few hundred base pairs; the synthesis of longer fragments requires a combined chemical–biochemical approach, where oligonucleotides (of around 100 bases) are synthesized, combined and ligated to give larger fragments of DNA. This cycle is repeated to yield ever-larger fragments of DNA, and has been used for the construction of viral and bacterial chromosomes.19–21 An early example was the synthesis of the poliovirus cDNA, which upon transcription by RNA polymerase, and translation in a cell free extract resulted in the de novo synthesis of infectious poliovirus.19 The genome of poliovirus (7440 RNA bases) was constructed by assembling purified DNA oligonucleotides (average length of 69 nucleotides) of plus and minus polarity with overlapping complimentary sequences at each end. These were combined into 400–600 base pair segments (the overlapping regions will align, due to the natural base-pairing of DNA) and ligated into a plasmid vector. These vectors were sequenced to ensure the fidelity of the gene fragments and their correct assembly. These DNA fragments were combined (again by overlapping complimentary regions) to give three overlapping DNA fragments of 3026, 1895 and 2682 base pairs each. Combining these three fragments resulted in the full-length cDNA of poliovirus, which was converted to de novo poliovirus RNA by transcription with T7 RNA polymerase.19 A more efficient method was reported shortly after, which was used for the synthesis of the ϕX174 bacteriophage genome (5386 base pairs).21 This methodology has now been used for the synthesis of a bacterial genome (Mycoplasma genitalium) consisting of 582![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 970 base pairs.20,22 The synthetic genome was identical to the wild-type, except for the insertion of an antibiotic marker within a gene associated with adhesion to mammalian cells (GM408), to block pathogenicity. The same group had demonstrated “genome transplantation” in an earlier paper;23 completely replacing the genome of Mycoplasma capricolum with intact genomic DNA from Mycoplasma mycoides. It was therefore hoped that the synthetic genome of M. genitalium could be transplanted in a similar way, thus installing and booting up the synthetic genome within a bacterial chassis. This has so far proved elusive, for reasons that are not clear.
970 base pairs.20,22 The synthetic genome was identical to the wild-type, except for the insertion of an antibiotic marker within a gene associated with adhesion to mammalian cells (GM408), to block pathogenicity. The same group had demonstrated “genome transplantation” in an earlier paper;23 completely replacing the genome of Mycoplasma capricolum with intact genomic DNA from Mycoplasma mycoides. It was therefore hoped that the synthetic genome of M. genitalium could be transplanted in a similar way, thus installing and booting up the synthetic genome within a bacterial chassis. This has so far proved elusive, for reasons that are not clear.Although there has been a significant reduction in the cost of DNA synthesis over the past decade, more robust methods for the synthesis of long oligonucleotides are still needed, and this is an area to which chemists can make significant contributions. New, more efficient DNA coupling reactions would enable the rapid assembly of very large fragments of DNA, and are essential to fully realising the promises of synthetic biology.
Minimal genome chassis
The ability of most host organisms to detect, inactivate and eject foreign DNA through a variety of built in defence mechanisms counteracts the stability of the DNA introduced into the chassis. A bacterial strain with a reduced genome that has its unwanted functions removed, would not only better maintain foreign gene networks; it would also better tolerate the metabolic burden of producing additional introduced enzymes and metabolites. Recently reported genome-reduced strains of E. coli (constructed by rational design) take important steps towards this goal,24 selectively eliminating nonessential genes and those with unwanted functions. This leads to strains with around 15% of their genome eliminated, with comparable growth rates to the wild-type organism, that were able to propagate and maintain recombinant genes and plasmids with higher accuracy. The long-term goal is a minimal genome organism optimised for synthetic biology through dedicated insertion sites and markers. The minimal pathway-footprint of the organism would allow the inserted functions/pathways to proceed more effectively (less resources required for maintaining organism) and potentially less interference of the biological pathways with the inserted ones.A minimal genome organism will not only serve as a potentially ideal chassis, but may also be used to study evolution and the origins of life. The construction of such an organism can be approached from two directions: subtracting genes from an existing organism (as discussed above),24 or the in vitro construction of a living system from components associated with life (DNA, RNA, proteins) and non-natural material.25 The relatively broad definition of what constitutes life26 allows a great deal of creativity and flexibility in this approach. The majority of the research so far has involved encapsulating components within lipid vesicles27 or water–oil emulsions,28 with examples of vesicles constructed that contain cell extracts, enzymes or nucleic acids that are able to carry out basic biological functions, such as protein expression or DNA replication. The fragility of the minimal cells and its dependence on specialized, non-standard feeding material provides a built-in safety feature that should prevent their survival and replication outside the laboratory.
The second wave
The first wave of synthetic biology has been multifaceted in its definition, encompassing a variety of areas and disciplines, each with different immediate goals. It has promised much, and as a result has enthused many people for the field. We are close to the beginning of the second phase of synthetic biology in which the basic parts and modules of the first wave will be combined together to create systems-level circuitry. There are significant challenges associated with creating larger functional systems, for example, there is relatively little known about the complexity of biological systems and the response of the system to the introduction of new pathways. The stochastic nature of biochemical reactions also introduces significant genetic noise into biological systems, which seems at odds with attempts to create predictable, robust systems. We are just beginning to unravel and understand the role of noise in cellular processes; it may be that eliminating noise from biological systems is impossible and instead components need to be generated that are resistant to cellular noise and epigenetic variations. Circadian rhythms for example tend to be highly resistant to noise, and as our understanding of these processes improves, so will our ability to emulate their robustness when designing new components.For the synthetic biology community to continue its rapid growth and for the term to become more than just the latest buzzword, it needs to engage and enthuse young scientists to ensure injection of new ideas into the field and evolution of the old ones. The synthetic biology community has developed a fantastic way to achieve these goals through the International Genetically Engineered Machine (iGEM) competition. Since 2004, teams of undergraduates have gathered every autumn at Massachusetts Institute of Technology for the iGEM jamboree, to showcase their synthetic biology summer projects. The students use a standard set of BioBricks (biological components and parts in a standardised vector that contains the same restriction sites to allow easy cloning) supplied to them in late spring as their building blocks. The competition has grown rapidly, from just 5 teams in 2004 to 84 international teams (more than 800 students) in 2008, and 112 teams registered for the 2009 competition. Many of the undergraduates tend to be first or second years, and therefore have a limited scientific background; nonetheless, iGEM has resulted in the conception and construction of several exciting devices, such as bacterial sensors and switches that have led to full-scale laboratory projects (see the iGEM website for specific details of projects). More importantly, iGEM serves as a motivating teaching method and an exceptional way to enthuse young scientists for synthetic biology, ensuring the rapid expansion of the field over the coming years.
There is tremendous potential in the second wave of synthetic biology, and if successful, it will redefine the way we tackle some of the biggest challenges faced by humanity. The ability to engineer life, to rapidly and easily synthesize and construct viral and bacterial genomes and to synthesize artificial living systems presents several unique bioethical and biosafety issues.29 When one considers that the products of the second wave of synthetic biology will be most effective when deployed in living systems and in the general ecosystem, a wide variety of ethical questions and objections may be raised. A detailed discussion of these issues is beyond the scope of this review; however, it is important that scientists are fully engaged and actively involved in all aspects of such discussions, to ensure policy makers and the public are aware of the promise and possibility of synthetic biology, and have a realistic and balanced view of the potential risks.
References
- W. Szybalski, In vivo and in vitro initiation of transcription, Adv. Exp. Med. Biol., 1974, 44, 23–24 CAS.
- S. A. Benner and A. M. Sismour, Synthetic biology, Nat. Rev. Genet., 2005, 6, 533–543 CrossRef CAS.
- E. Andrianantoandro, S. Basu, D. K. Karig and R. Weiss, Synthetic biology: new engineering rules for an emerging discipline, Mol. Syst. Biol., 2006, 2, 0028.
- T. S. Gardner, C. R. Cantor and J. J. Collins, Construction of a genetic toggle switch in Escherichia coli, Nature, 2000, 403, 339–342 CrossRef CAS.
- M. B. Elowitz and S. Leibler, A synthetic oscillatory network of transcriptional regulators, Nature, 2000, 403, 335–338 CrossRef CAS.
- J. Stricker, S. Cookson, M. R. Bennett, W. H. Mather, L. S. Tsimring and J. Hasty, A fast, robust and tunable synthetic gene oscillator, Nature, 2008, 456, 516–519 CrossRef CAS.
- E. Fung, W. W. Wong, J. K. Suen, T. Bulter, S.-g. Lee and J. C. Liao, A synthetic gene-metabolic oscillator, Nature, 2005, 435, 118–122 CrossRef CAS.
- M. Tigges, T. T. Marquez-Lago, J. Stelling and M. Fussenegger, A tunable synthetic mammalian oscillator, Nature, 2009, 457, 309–312 CrossRef CAS.
- A. E. Friedland, T. K. Lu, X. Wang, D. Shi, G. Church and J. J. Collins, Synthetic gene networks that count, Science, 2009, 324, 1199–1202 CrossRef CAS.
- T. Ellis, X. Wang and J. J. Collins, Diversity-based, model-guided construction of synthetic gene networks with predicted functions, Nat. Biotechnol., 2009, 27, 465–471 CrossRef CAS.
- D. C. Williams, R. M. Van Frank, W. L. Muth and J. P. Burnett, Cytoplasmic inclusion bodies in Escherichia coli producing biosynthetic human insulin proteins, Science, 1982, 215, 687–689 CrossRef CAS.
- J. D. Newman, J. Marshall, M. Chang, F. Nowroozi, E. Paradise, D. Pitera, K. L. Newman and J. D. Keasling, High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli, Biotechnol. Bioeng., 2006, 95, 684–691 CrossRef CAS.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Production of the antimalarial drug precursor artemisinic acid in engineered yeast, Nature, 2006, 440, 940–943 CrossRef CAS.
- G. Schmid and W. Hofheinz, Total Synthesis of Qinghaosu, J. Am. Chem. Soc., 1983, 105, 624–625 CrossRef CAS.
- N. Acton and R. J. Roth, On the Conversion of Dihydroartemisinic Acid into Artemisinin, J. Org. Chem., 1992, 57, 3610–3614 CrossRef CAS.
- M. C. Chang, R. A. Eachus, W. Trieu, D. K. Ro and J. D. Keasling, Engineering Escherichia coli for production of functionalized terpenoids using plant P450s, Nat. Chem. Biol., 2007, 3, 274–277 CrossRef CAS.
- M. Inui, M. Suda, S. Kimura, K. Yasuda, H. Suzuki, H. Toda, S. Yamamoto, S. Okino, N. Suzuki and H. Yukawa, Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli, Appl. Microbiol. Biotechnol., 2008, 77, 1305–1316 CrossRef CAS.
- E. J. Steen, R. Chan, N. Prasad, S. Myers, C. J. Petzold, A. Redding, M. Ouellet and J. D. Keasling, Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol, Microb. Cell Fact., 2008, 7, 36 CrossRef.
- J. Cello, A. V. Paul and E. Wimmer, Chemical synthesis of poliovirus cDNA: generation of infectious virus in the absence of natural template, Science, 2002, 297, 1016–1018 CrossRef CAS.
- D. G. Gibson, G. A. Benders, C. Andrews-Pfannkoch, E. A. Denisova, H. Baden-Tillson, J. Zaveri, T. B. Stockwell, A. Brownley, D. W. Thomas, M. A. Algire, C. Merryman, L. Young, V. N. Noskov, J. I. Glass, J. C. Venter, C. A. Hutchison III and H. O. Smith, Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome, Science, 2008, 319, 1215–1220 CrossRef CAS.
- H. O. Smith, C. A. Hutchison, 3rd, C. Pfannkoch and J. C. Venter, Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15440–15445 CrossRef CAS.
- D. G. Gibson, G. A. Benders, K. C. Axelrod, J. Zaveri, M. A. Algire, M. Moodie, M. G. Montague, J. C. Venter, H. O. Smith and C. A. Hutchison III, One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20404–20409 CrossRef CAS.
- C. Lartigue, J. I. Glass, N. Alperovich, R. Pieper, P. P. Parmar, C. A. Hutchison, III, H. O. Smith and J. C. Venter, Genome transplantation in bacteria: changing one species to another, Science, 2007, 317, 632–638 CrossRef CAS.
- G. Pósfai, G. Plunkett, III, T. Fehér, D. Frisch, G. M. Keil, K. Umenhoffer, V. Kolisnychenko, B. Stahl, S. S. Sharma, M. de Arruda, V. Burland, S. W. Harcum and F. R. Blattner, Emergent properties of reduced-genome Escherichia coli, Science, 2006, 312, 1044–1046 CrossRef CAS.
- A. C. Forster and G. M. Church, Towards synthesis of a minimal cell, Mol. Syst. Biol., 2006, 2, 45.
- D. Deamer, A giant step towards artificial life?, Trends Biotechnol., 2005, 23, 336–338 CrossRef CAS.
- P. L. Luisi, F. Ferri and P. Stano, Approaches to semi-synthetic minimal cells: a review, Naturwissenschaften, 2006, 93, 1–13 CrossRef CAS.
- A. D. Griffiths and D. S. Tawfik, Miniaturising the laboratory in emulsion droplets, Trends Biotechnol., 2006, 24, 395–402 CrossRef CAS.
- M. S. Garfinkel, D. Endy, G. L. Epstein and R. M. Friedman, Synthetic genomics | options for governance, Biosecur. Bioterror., 2007, 5, 359–362 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |