The crystal structure of an LLL-configured depsipeptide substrate analogue bound to isopenicillin N synthase
Wei Gea, Ian J. Cliftona, Jeanette E. Stokac, Robert M. Adlingtona, Jack E. Baldwin*a and Peter J. Rutledge*b
aChemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, UK OX1 3TA. E-mail: jack.baldwin@chem.ox.ac.uk; Fax: +44 1865 285002; Tel: +44 1865 275670
bSchool of Chemistry F11, The University of Sydney, NSW 2006, Australia. E-mail: p.rutledge@chem.usyd.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 5020
cSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia
First published on 29th October 2009
Abstract
Isopenicillin N synthase (IPNS) is a non-heme iron(II) oxidase, which catalyses the biosynthesis of isopenicillin N (IPN) from the tripeptide δ-L-α-aminoadipoyl-L-cysteinyl-D-valine (LLD-ACV) in a remarkable oxidative bicyclisation reaction. The natural substrate for IPNS is the LLD-configured tripeptide. LLL-ACV is not turned over by the enzyme, but inhibits turnover of the LLD-tripeptide. The mechanism by which this inhibition takes place is not fully understood. Recent studies have employed a range of LLD-configured depsipeptide substrate analogues in crystallographic studies to probe events preceding β-lactam closure in the IPNS reaction cycle. Herein, we report the first crystal structure of IPNS in complex with an LLL-configured depsipeptide analogue, δ-L-α-aminoadipoyl-L-cysteine (1-(R)-carboxy-2-thiomethyl)ethyl ester (LLL-ACOmC). This report describes the crystal structure of the IPNS:Fe(II):LLL-ACOmC complex to 2.0 Å resolution, and discusses attempts to oxygenate this complex at high pressure in order to probe the mechanism by which LLL-configured substrates inhibit IPNS catalysis.
Introduction
Isopenicillin N synthase (IPNS) is a non-heme iron(II)-dependent oxidase with an essential role in the biosynthesis of all penicillin and cephalosporin antibiotics.1 IPNS mediates conversion of the linear tripeptide δ-L-α-aminoadipoyl-L-cysteinyl-D-valine (LLD-ACV, 1) to bicyclic isopenicillin N (IPN, 2) in a single step (Scheme 1). LLD-ACV is biosynthesised in bacteria and fungi from the constituent amino acids L-α-aminoadipic acid, L-cysteine and L-valine by the enzyme ACV synthetase (ACVS), a nonribosomal peptide synthase (NRPS). In the course of the ACVS-mediated condensation, the stereochemistry of the valine residue is inverted to afford the LLD-tripeptide that is the natural substrate for IPNS.2,3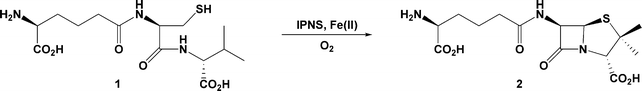 | ||
| Scheme 1 IPNS-mediated conversion of LLD-ACV 1 to IPN 2 using an iron(II) cofactor and molecular oxygen as cosubstrate. | ||
The oxidative bicyclisation reaction mediated by IPNS is unique in nature and of key commercial importance, and has been the subject of extensive research interest over many years.1,4–6 Three principal strategies have been used to investigate IPNS catalysis: spectroscopic studies,5 solution-phase incubation of substrate analogues with the enzyme,4 and X-ray crystallography.6–8 The consensus mechanism for non-heme iron-dependent oxidases involves highly-reactive Fe(IV)-oxo (ferryl) intermediates,9–12 and a range of evidence has been put forward for the involvement of such a species in IPNS catalysis.6,13–16
In 1979, Konomi et al. reported solution-phase incubation studies using the all-L configured tripeptide LLL-ACV 3 (an epimer of the native substrate 1; Fig. 1).17 This compound is not turned over by IPNS, but inhibits turnover of the correctly configured substrate LLD-ACV 1. It was assumed that LLL-ACV is not turned over because it does not fit properly into the active site; however, the exact mechanism of inhibition was not determined. In 2005 we reported the first crystal structure of IPNS with an LLL-configured substrate, the tripeptide analogue δ-L-α-aminoadipoyl-L-cysteinyl-L-3,3,3,3′,3′,3′-hexafluorovaline (LLL-AC6FV, 4; PDB ID for IPNS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe(II):LLL-AC6FV complex: 2bu9).18
:Fe(II):LLL-AC6FV complex: 2bu9).18
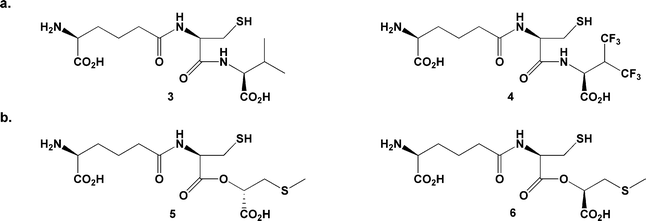 | ||
| Fig. 1 a. ‘All-L’ substrate analogues studied previously LLL-ACV 317 and LLL-AC6FV 4;18b. the epimeric depsipeptides LLD-ACOmC 5 and LLL-ACOmC 6. | ||
The structure of the IPNS:Fe(II):LLL-AC6FV complex revealed that tripeptide 4 does in fact bind in the IPNS active site. It does so with the L-aminoadipoyl and L-cysteinyl residues adopting very similar conformations to those seen for LLD-configured substrates. However, the altered stereochemistry of the third amino acid orients its side-chain away from the iron and ‘up’ into a region of the active site pocket occupied by four water molecules in the IPNS:Fe(II):LLD-ACV complex. The carboxylate group of the L-hexafluorovaline residue is oriented towards iron, and hydrogen-bonded to an iron-bound water molecule and adjacent tyrosine residue (Tyr189). As a result, the iron is ligated by an additional water ligand in the site opposite Asp216 (generally accepted to be the oxygen binding site), held in place by a network of hydrogen bonds. It was proposed that this water ligand, held in place by strong hydrogen bonding, blocks oxygen entry and inhibits the oxidative turnover reaction before it can even begin.18
The current study tests this hypothesis by crystallising IPNS with another LLL-configured substrate analogue and attempting to force oxygen into the crystalline complex at high pressure. We recently reported co-crystallisation of the LLD-depsipeptide δ-L-α-aminoadipoyl-L-cysteine (1-(S)-carboxy-2-thiomethyl)ethyl ester (LLD-ACOmC, 5) with IPNS and oxidative turnover of the crystalline IPNS:Fe(II):LLD-ACOmC complex to an unexpected sulfenate product.19 Herein, we detail the crystal structure of IPNS complexed with the LLL-depsipeptide δ-L-α-aminoadipoyl-L-cysteine (1-(R)-carboxy-2-thiomethyl)ethyl ester (LLL-ACOmC, 6). (Note that compounds 5 and 6 are abbreviated as ACOmC to highlight their structural relationship to the analogues δ-(L-α-aminoadipoyl)-L-cysteinyl-S-methyl-D-cysteine (LLD-ACmC)6 and δ-(L-α-aminoadipoyl)-L-cysteine D-α-hydroxyisovaleryl ester (LLD-ACOV).14)
Results and discussion
As part of ongoing studies using protein crystallography to investigate the mechanism of IPNS catalysis,6,14,15,19–25 the substrate analogue δ-L-α-aminoadipoyl-L-cysteine (1-(R)-carboxy-2-thiomethyl)ethyl ester (LLL-ACOmC, 6) was prepared in five steps from L-α-aminoadipic acid derivative 9, S-p-methoxybenzyl-L-cysteine 10 and tert-butyl acrylate 11 (Scheme 2).16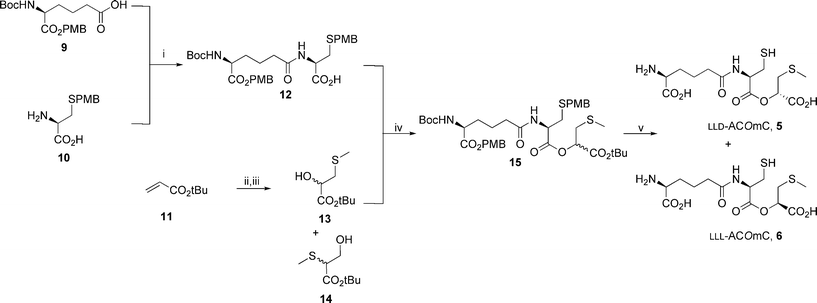 | ||
| Scheme 2 Synthesis of LLL-ACOmC 6. i. 9, iso-butylchloroformate, Et3N, THF, −12 °C, 30 min, then 10, Et3N, H2O, 0 °C–RT, 90 min, 82%; ii. m-CPBA, DCM, reflux, 48 h; iii. NaSCH3, NaHCO3, H2O, 0 °C, 1 h, 80% (over two steps) of a 1:2 mixture of 13:14; iv. PPh3, DEAD, THF, 0 °C, sonication 1 h, then RT 20 h, 63% of a 1:1 mixture of LLL:LLD-15; v. TFA, anisole, reflux, 30 min, then reversed phase HPLC, 43% of LLD-ACOmC 5 and 36% of LLL-ACOmC 6. | ||
LLL-ACOmC 6 was crystallised with IPNS following the previously reported procedure.26 Crystals of IPNS:Fe(II):LLL-ACOmC were observed to form more rapidly and at a wider range of conditions (well and drop buffer concentrations, and concentrations of seed crystals added) than for IPNS:Fe(II):LLD-ACOmC. Large plate-shaped crystals of IPNS:Fe(II):LLL-ACOmC grew in ca. two days compared to ca. four days required for IPNS:Fe(II):LLD-ACOmC.
The crystal structure the anaerobic IPNS:Fe(II):LLL-ACOmC complex was solved to 2.0 Å resolution (Table 1), affording the first structure of IPNS complexed with an LLL-configured depsipeptide (Fig. 2a,b). In this structure, the L-α-aminoadipoyl and L-cysteinyl residues occupy equivalent positions to those observed for LLD-ACV8 and complexes with other LLD-configured substrates.6,14,15,19–24LLL-ACOmC 6 is tethered by a salt bridge to Arg87 through the L-α-aminoadipoyl carboxylate, and to iron by the cysteinyl sulfur. The L-configured third residue occupies a very different region of space from that observed with LLD-ACV 1 or LLD-ACOmC 5, as would be expected. The side-chain of the third residue is positioned away from the active site metal and above Tyr189, into a region occupied by four water molecules in the IPNS:Fe(II):LLD-ACV complex.8 The OmC-carboxylate points towards the iron and is linked by hydrogen bonding to a metal-bound water molecule that is ligated in the site opposite Asp216. The other oxygen atom of this carboxylate is hydrogen bonded to the side-chain phenol of Tyr189.
| a Rmerge = ∑j∑h|Ih,j−〈Ih〉|/∑j∑h〈Ih〉× 100.b Rcryst =∑‖Fobs| − |Fcalc‖/∑|Fobs| × 100.c Rfree = based on 5% of the total reflections.d RMS deviation from ideality for bonds (followed by the value for angles).e Average B factors in order: main chain; side chain; substrate and iron; solvent. | |
|---|---|
| X-Ray source | Rotating Anode, LMB, Oxford, UK |
| Wavelength/Å | 1.5418 |
| PDB acquisition code | 2vbd |
| Resolution/Å | 2.0 |
| Space group | P212121 |
| Unit cell dimensions (a, b, c/Å) | 46.53, 71.31, 100.65 |
| Total number of reflections | 152677 |
| Number of unique reflections | 23160 |
| Completeness (%) | 97.7 |
| Completeness in outer shell (%) | 85.9 |
| Rmerge (%)a | 9.0 |
| Rcryst (%)b | 18.8 |
| Rfree (%)c | 21.7 |
| RMS deviationd | 0.007 (1.0) |
| Average B factorse/Å2 | 13.4, 15.4, 19.1, 19.0 |
| Number of water molecules | 213 |
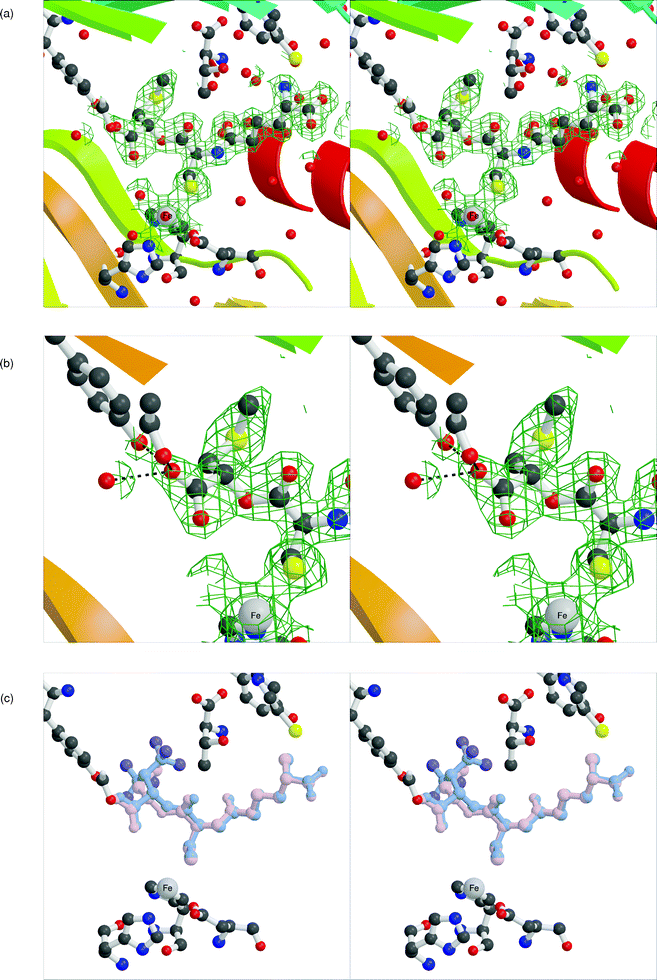 | ||
| Fig. 2 Active site of the anaerobic IPNS:Fe(II):LLL-ACOmC complex to 2.0 Å resolution (PDB ID 2vbd). a. Overview of the whole active site region, showing the full extent of LLL-ACOmC binding to the protein. b. Closer view of the third (L-OmC) residue of the depsipeptide substrate analogue showing hydrogen bonding to Tyr189, Ser281 and an adjacent water molecule (A2213). c. Overlay figure showing superimposition of IPNS-bound LLL-ACOmC with LLL-AC6FV (PDB ID 2bu9). The 2mFo–DFc electron density maps are shown in green and contoured at 1σ. | ||
This arrangement mirrors the binding of LLL-AC6FV 4 to IPNS,18 which ligates to the protein through a similar hydrogen bonding network around the carboxylate of its third residue (Fig. 2c). A similar substrate conformation is also seen in complexes of IPNS with the smaller tripeptides δ-L-α-aminoadipoyl-L-cysteinyl-glycine (LL-ACG, 7) and δ-L-α-aminoadipoyl-L-cysteinyl-D-alanine (LLD-ACA, 8; Fig. 3).23 The less sterically demanding and less hydrophobic side-chains of these analogues allow additional water molecules into the normally hydrophobic region of the active site opposite Asp216, as now observed in the IPNS complex with LLL-ACOmC 6. Furthermore, the glycinyl carboxylate in the IPNS:Fe(II):LL-ACG complex is also linked by hydrogen bonding to an additional water ligand at the iron centre.
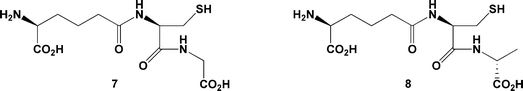 | ||
| Fig. 3 LL-ACG 7 and LLD-ACA 8, less sterically demanding tripeptide substrates, which allow additional water molecules into the active site region when bound to IPNS.23 | ||
It is interesting to note that the methylsulfide side-chain of LLL-ACOmC 6 is accommodated satisfactorily in the region above Tyr189, even without the potential for significant new hydrogen bonding interactions to ameliorate the loss of those usually present in this region. It was observed that several fluorine atoms of LLL-AC6FV were well positioned to interact with Tyr189 and water molecules in this area, and that the resulting hydrogen bonding interactions would mitigate energy penalties incurred by loss of the hydrogen-bonded network of water molecules from this area.18 In contrast, the methylsulfide side-chain of LLL-ACOmC 6 cannot hydrogen bond to Tyr189 or adjacent water molecules in the same way, but this residue is accommodated in this region nonetheless.
Two alternative explanations for the inhibitory effects of LLL-configured substrates were put forward on the basis of the IPNS:Fe(II):LLL-AC6FV crystal structure.18 The most likely explanation is that hydrogen bonding between the hexafluorovalinyl carboxylate, iron-bound water, Tyr189 and other water molecules prevents dioxygen from displacing the metal-ligated water and initiating turnover. Alternatively, it was postulated that oxygen might still bind but then be unable to react because the cysteinyl β-carbon and ‘valinyl’ nitrogen have moved in the LLL-AC6FV complex relative to LLD-ACV 1 (by 0.1 and 0.77 Å respectively).
To test these alternative hypotheses, crystals of IPNS:Fe(II):LLL-ACOmC were exposed to the co-substrate oxygen at high pressure in an attempt to force oxygen binding and substrate turnover. After exposure of the IPNS:Fe(II):LLL-ACOmC complex to high-pressure oxygen for time spans equivalent to those required to bring about turnover of LLD-depsipeptide analogues (20 bar for 1 min; 20 bar for 2 min; 40 bar for 10 min),14–16,19 the crystal structure showed no change relative to the ‘time 0’ electron density map (data not shown). Exposure to higher oxygen pressures for longer times (40 bar for 60 min; 40 bar for 100 min; 40 bar for 300 min) brought changes in the active site electron density that indicate incomplete substrate occupancy—i.e. departure of the substrate (or product(s)). However, these extremely forcing conditions have been shown to bring about a range of non-specific oxidative transformations in previous studies.6,20
Conclusion
Previous research has demonstrated that IPNS tolerates considerable variation in the valinyl section of its substrate LLD-ACV 1, and that many compounds variant at this position are efficiently turned over by the enzyme.4 The IPNS active site appears well able to accommodate—and react with—a large range of D-amino acids in the binding pocket close to the iron centre. However, the crystal structure of the IPNS:Fe(II):LLL-ACOmC complex reported here confirms that for LLL-configured analogues such as LLL-ACOmC 6, the side-chain of the third residue cannot get near the active site iron and sits instead above Tyr189. This leaves the iron unprotected and liable to coordinate with an extra water ligand, which blocks oxygen binding and turnover.4This study demonstrates that although the LLL-ACOmC 6 binds to IPNS and sits comfortably in the active site, the conformation of the compound does not facilitate turnover by IPNS. When LLD-ACV 1 binds to IPNS,8 it displaces the water molecule that is ligated to iron opposite Asp216 in the ‘substrate free’ IPNS:Mn(II) crystal structure.7 So, with LLD-ACV 1 anchored in the active site, the iron is penta-coordinate. In contrast, the equivalent water molecule remains bound to the IPNS:Fe(II):LLL-ACOmC complex and appears to be locked into this site by hydrogen bonding. This leaves the metal hexa-coordinate with a water molecule in the oxygen binding site. The hydrogen bonding network appears sufficiently robust to prevent oxygen from entering the active site, so the turnover reaction is blocked before it can begin. Furthermore, the observed hydrogen bonding around the OmC-carboxylate potentially imparts greater stability to the complex and explains why IPNS crystallises more readily with LLL-ACOmC than with LLD-ACOmC 5. This may also explain why LLL-ACV 3 can compete with LLD-ACV 1 and act as an inhibitor of IPNS catalysis.17
Experimental
Synthesis of LLL-ACOmC 6
LLL-ACOmC 6 was prepared in five steps from the doubly-protected L-α-aminoadipic acid derivative 9, S-p-methoxybenzyl-L-cysteine 10 and tert-butylacrylate 11 (Scheme 2).16 The crude depsipeptide product (a mixture of the LLL- and LLD-diastereomers) was purified by reversed-phase HPLC (10 mM NH4HCO3, Hypersil 5 μ C18 column, 250 × 10 mm internal diameter; λ = 254 nm; 4 mL min−1) to afford pure LLL-ACOmC 6 (Rt = 12.0 min) and LLD-ACOmC 5 (Rt = 13.5 min). Data for 6: δH (200 MHz, D2O) 1.50–1.70 (2H, m, NHCHCH2CH2CH2), 1.70–1.85 (2H, m, NHCHCH2CH2CH2), 2.07 (3H, s, SCH3), 2.31 (2H, t, J 7.0 Hz, NHCHCH2CH2CH2), 2.82–3.02 (4H, m, CH2SH and CH2SCH3), 3.64 (1H, t, J 5.5 Hz, NH2CHL-AA), 4.63–4.72 (1H, m, NHCHL-Cys), 4.94 (1H, dd, J 7.0 Hz, 5.0 Hz, OCHL-Omc); data for 5 is presented in ref. 16.Crystallization and turnover experiments
Crystals of the anaerobic IPNS:Fe(II):LLL-ACOmC complex were grown in a glovebox as detailed previously.26 Suitable crystals were identified using a light microscope, then removed from the glovebox, exchanged into cryoprotectant buffer (a 1:1 mixture of well buffer: saturated Li2SO4 in 40% (v/v) glycerol solution), mounted individually in loops and rapidly cryo-cooled in liquid nitrogen. Crystals for oxygen exposure were transferred to the high-pressure oxygen bomb and exposed to oxygen as reported previously, prior to cryo-cooling.27Data collection and structure determination
Data were collected at 100 K (maintained with an Oxford Cryosystems Cryostream) using Cu-Kα radiation from a Rigaku rotating anode generator and a MAR Research image plate detector at the Laboratory of Molecular Biophysics (LMB), Oxford, UK. Data were processed using MOSFLM28 and the CCP4 suite of programs.29 REFMAC5 was used to refine data,30 the program O to build models.31 The initial phases were generated from the IPNS:Fe(II):ACV structure published previously8 by molecular replacement. Manual rebuilding of side-chains was undertaken where required. Crystallographic coordinates and structure factors have been deposited in the Worldwide Protein Data Bank, accession number 2vbd. The programs MOLSCRIPT,32 BOBSCRIPT33 and Raster3D34 were used to prepare the colour figure (Fig. 2).Abberviations
| AC6FV | (δ-α-aminoadipoyl-cysteinyl-3,3,3,3′3′3′-hexafluorovaline) |
| ACA | (δ-α-aminoadipoyl-cysteinyl-alanine) |
| ACG | (δ-α-aminoadipoyl-cysteinyl-glycine) |
| ACmC | (δ-α-aminoadipoyl-cysteinyl-S-methyl-cysteine); |
| ACOmC | (δ-α-aminoadipoyl-cysteine (1-carboxy-2-thiomethyl)ethyl ester) |
| ACOV | (δ-α-aminoadipoyl-cysteine α-hydroxyisovaleryl ester) |
| ACV | (δ-α-aminoadipoyl-cysteinyl-valine) |
| IPN | (isopenicillin N) |
| IPNS | (isopenicillin N synthase) |
Acknowledgements
We thank Dr Annaleise Howard-Jones, Prof. Chris Schofield, Dr Victor Lee, Dr Zhihong Zhang, Dr Edward Lowe and Dr Jing He for help and discussions.References
- J. E. Baldwin and C. J. Schofield, in The Chemistry of β-Lactams, ed. M. I. Page, Blackie, Glasgow, 1992, pp. 1–78 Search PubMed.
- H. R. V. Arnstein and P. T. Grant, Biochem. J., 1954, 57, 353–359 CAS.
- M. F. Byford, J. E. Baldwin, C.-Y. Shiau and C. J. Schofield, Chem. Rev., 1997, 97, 2631–2649 CrossRef.
- J. E. Baldwin and M. Bradley, Chem. Rev., 1990, 90, 1079–1088 CrossRef CAS.
- R. D. G. Cooper, Bioorg. Med. Chem., 1993, 1, 1–17 CrossRef CAS.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. H. Hensgens, M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721–724 CrossRef CAS.
- P. L. Roach, I. J. Clifton, V. Fulop, K. Harlos, G. J. Barton, J. Hajdu, I. Andersson, C. J. Schofield and J. E. Baldwin, Nature, 1995, 375, 700–704 CrossRef CAS.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hadju and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 21–68 CrossRef CAS.
- C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS.
- P. C. A. Bruijnincx, G. van Koten and R. Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- J. E. Baldwin, in Special Publication No. 52, ed. A. G. Brown, and S. M. Roberts, The Royal Society of Chemistry, London, 1985, pp. 62–85 Search PubMed.
- J. M. Ogle, I. J. Clifton, P. J. Rutledge, J. M. Elkins, N. I. Burzlaff, R. M. Adlington, P. L. Roach and J. E. Baldwin, Chem. Biol., 2001, 8, 1231–1237 CrossRef CAS.
- A. Daruzzaman, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2006, 7, 351–358 CrossRef CAS.
- W. Ge, I. J. Clifton, J. E. Stok, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, J. Am. Chem. Soc., 2008, 130, 10096–10102 CrossRef CAS.
- T. Konomi, S. Herchen, J. E. Baldwin, M. Yoshida, N. A. Hunt and A. L. Demain, Biochem. J., 1979, 184, 427–430 CAS.
- A. R. Howard-Jones, P. J. Rutledge, I. J. Clifton, R. M. Adlington and J. E. Baldwin, Biochem. Biophys. Res. Commun., 2005, 336, 702–708 CrossRef CAS.
- W. Ge, I. J. Clifton, A. R. Howard-Jones, J. E. Stok, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2009, 10, 2025–2031 CrossRef CAS.
- J. M. Elkins, P. J. Rutledge, N. I. Burzlaff, I. J. Clifton, R. M. Adlington, P. L. Roach and J. E. Baldwin, Org. Biomol. Chem., 2003, 1, 1455–1460 RSC.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, C. J. Schofield and P. J. Rutledge, Biochem. J., 2003, 372, 687–693 CrossRef CAS.
- A. R. Grummitt, P. J. Rutledge, I. J. Clifton and J. E. Baldwin, Biochem. J., 2004, 382, 659–666 CrossRef CAS.
- A. J. Long, I. J. Clifton, P. L. Roach, J. E. Baldwin, P. J. Rutledge and C. J. Schofield, Biochemistry, 2005, 44, 6619–6628 CrossRef CAS.
- A. R. Howard-Jones, J. M. Elkins, I. J. Clifton, P. L. Roach, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, Biochemistry, 2007, 46, 4755–4762 CrossRef CAS.
- A. C. Stewart, I. J. Clifton, R. M. Adlington, J. E. Baldwin and P. J. Rutledge, ChemBioChem, 2007, 8, 2003–2007 CrossRef CAS.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, A. J. Long, R. W. Strange, S. S. Hasnain, C. J. Schofield, J. E. Baldwin and J. Hajdu, Eur. J. Biochem., 1996, 242, 736–740 CrossRef CAS.
- P. J. Rutledge, N. I. Burzlaff, J. M. Elkins, M. Pickford, J. E. Baldwin and P. L. Roach, Anal. Biochem., 2002, 308, 265–268 CrossRef CAS.
- A. G. W. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1696–1702 CrossRef.
- Collaborative Computational Project, Number 4, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef.
- T. A. Jones, J. Y. Zou, S. W. Cowan and M. Kjelgaard, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, 47, 110–119 CrossRef.
- P. J. Kraulis, J. Appl. Crystallogr., 1991, 24, 946–950 CrossRef.
- R. M. Esnouf, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 938–940 CrossRef CAS.
- E. A. Merritt and D. J. Bacon, Methods Enzymol., 1997, 277, 505–524 CAS.
| This journal is © The Royal Society of Chemistry 2010 |