Low-toxic and safe nanomaterials by surface-chemical design, carbon nanotubes, fullerenes, metallofullerenes, and graphenes
Liang
Yan†
a,
Feng
Zhao†
a,
Shoujian
Li
b,
Zhongbo
Hu
c and
Yuliang
Zhao
*abc
aCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences (CAS), National Center for Nanosciences and Technology of China, Beijing, 100049, China. E-mail: zhaoyuliang@ihep.ac.cn
bThe College of Chemistry and Chemical Engineering, Sichuan University, Chengdu, 610064, China
cThe College of Material Sciences, Graduate University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 14th December 2010
Abstract
The toxicity grade for a bulk material can be approximately determined by three factors (chemical composition, dose, and exposure route). However, for a nanomaterial it depends on more than ten factors. Interestingly, some nano-factors (like huge surface adsorbability, small size, etc.) that endow nanomaterials with new biomedical functions are also potential causes leading to toxicity or damage to the living organism. Is it possible to create safe nanomaterials if such a number of complicated factors need to be regulated? We herein try to find answers to this important question. We first discuss chemical processes that are applicable for nanosurface modifications, in order to improve biocompatibility, regulate ADME, and reduce the toxicity of carbon nanomaterials (carbon nanotubes, fullerenes, metallofullerenes, and graphenes). Then the biological/toxicological effects of surface-modified and unmodified carbon nanomaterials are comparatively discussed from two aspects: the lowered toxic responses or the enhanced biomedical functions. We summarize the eight biggest challenges in creating low-toxicity and safer nanomaterials and some significant topics of future research needs: to find out safer nanofactors; to establish controllable surface modifications and simpler chemistries for low-toxic nanomaterials; to explore the nanotoxicity mechanisms; to justify the validity of current toxicological theories in nanotoxicology; to create standardized nanomaterials for toxicity tests; to build theoretical models for cellular and molecular interactions of nanoparticles; and to establish systematical knowledge frameworks for nanotoxicology.
 Liang Yan | Mr Liang Yan is a graduate student of CAS Key Lab for Biomedical Effects of Nanomaterials & Nanosafety, Chinese Academy of Sciences. He received his BSc in Chemistry at Sichuan University in 2009. He is now studying the chemical functionalization of graphenes as well their biomedical effects. |
 Feng Zhao | Ms Feng Zhao is currently a research fellow at the CAS Key Lab for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences. She received her BSc degree in chemistry from the College of Chemical Engineering, Tsinghua University in 2002, and MSc degree in biology from the College of Biology, Tsinghua University in 2005. Her current research interests mainly include nanotoxicology, and studies on nanoparticles and their interactions with cells or biomacromolecules. |
 Shoujian Li | Prof. Shoujian Li received his PhD in radiochemistry chemistry at Sichuan University. He is the head of the research group “Functional Materials for Radionuclides Separation” of the Chemistry College of Sichuan University. His research interests mainly include the design and synthesis of new functional carbonaceous materials, advanced materials and technologies for spent fuel reprocessing, treatment and disposal of nuclear wastes, including the uses of carbon nanomaterials. |
 Zhongbo Hu | Zhongbo Hu received his PhD from University of Missouri-Columbia in 1994. He is now a Professor and Dean of the College of Materials Science, the Graduate University of Chinese Academy of Sciences. His main research interests are the structures and properties of solid materials. He has authored about 90 peer-reviewed papers. |
 Yuliang Zhao | Yuliang Zhao is a professor and director of the CAS Key Lab for Biomedical Effects of Nanomaterials & Nanosafety, the CAS Institute of High Energy Physics and the National Center for Nanoscience and Technology of China. His research interests mainly include nanochemistry and radiochemistry . He has published about 200 peer-reviewed papers and edited and published 11 books, including “Nanotoxicology”, the world’s first textbook in that field. Professor Zhao is an editorial board member for 8 international SCI journals, including Biomedical Microdevices, Particle & Fibre Toxicology, The Journal of Nanosciences and Nanotechnology and The Journal of Biomedical Nanotechnology. |
Introduction
One of the most important bases of nanotechnology is nanosized and/or nanostructured materials (hereafter referred to as nanomaterials). Some unique physicochemical properties, as compared with the bulk materials of the same chemical composition, have been demonstrated to be capable of changing their biological or toxicological effects in living organisms or the environment. Actually, the study of the favorable biomedical effects and properties of nanomaterials has formed new disciplines: nanomedicine and nanobiotechnology, while the study of the adverse effects of nanomaterials has formed another new branch of nanosciences: nanotoxicology.1In the past decade, both academic communities and the public worldwide have become highly concerned with the adverse effects of nanomaterials. A large number of publications have reported the alterations of toxic responses of nanomaterials in vitro and in vivo as compared with their molecular or larger size forms.1 The nano-factors that mainly dominate the biomedical effects of nanomaterials are summarized in Table 1, some of them have been explored experimentally so far, some have not. Among them, those marked with an asterisk play the most important roles.
| Bulk material or nanomaterials | Nanomaterials |
|---|---|
| • Chemical composition* | • Nanostructure* |
| • Dose (mass concentration)* | • Particles concentration* |
| • Exposure route* | • Particle size/distribution* |
| • Reactivity | • Particle number |
| • Conductivity | • Aggregation/agglomeration* |
| • Morphology (crystalline, amorphous, shape, etc.) | • Surface adsorbability* |
| • Surface areas* | |
| • Physical form (solid, aerosol, suspension, etc.) | • Surface charge* |
| • Self-assembly* | |
| • Purity/Impurities | • Quantum effects |
| • Solubility | •… |
Some of these characteristics of nanomaterials in a biological system can be measured, but some cannot. In some cases, those properties (like huge surface adsorbability, small size, etc.) that endow nanomaterials with new biomedical functions also lead to damage to biological systems. In the current framework of safety evaluation of a bulk material, only 3 factors (dose, chemical composition and exposure route) can approximately determine its safety grade, however, we need more than 10 factors to determine the safety grade of a nanomaterial (Table 1). The diversity of nano-factors makes chemical and physical identification and characterization of nanomaterials in biological systems become much more complex and difficult. On the other hand, nanomaterials are driving the rapid development of biomedicine by making use of their novel properties2 which are mostly rooted in the characteristics listed in Table 1. For example: the quantum effects of quantum dots were employed to image tissues is based on their unique luminescence properties;3 their unique surface and nanostructure endows Gd@C82(OH)224–6 with a high efficacy of inhibition of tumor growth and metastasis, while they are almost nontoxic.7–12 The nanoparticles do not kill cells but possess a high inhibition efficacy of tumor growth, exhibiting great advantages as compared with current clinical anticancer drugs which are highly toxic and play their therapeutic roles by poisoning cancer cells but simultaneously killing normal cells of other organs.
Nevertheless, the low solubilities of most carbon nanomaterials result in their difficult manipulation in solvent media, which greatly restricts the experimental explorations of their novel properties and functions, and limits their applications in many fields, including biomedical uses. To this end, surface modification of nanoparticles plays an essential role in changing the physicochemical and surface properties of nanomaterials.13 Chemical modification of nanosurfaces has been developed as the main approach to increase their solubility in water, serum, and various solvents, to enhance their biocompatibility, to reduce toxic responses in biological systems, to create new functions for biomedical or other applications, and to prevent their aggregation because most nanomaterials in solution or aerosol states readily aggregate to form larger-sized particles, etc.
Nanomaterials can be roughly categorized as four groups. (1) Carbon nanomaterials: They consist of carbon atoms but possess different nanostructures, like single-wall carbon nanotubes (SWCNTs), multiwall carbon nanotubes (MWCNTs), fullerenes, metallofullerenes, graphene. (2) Metallic nanomaterials: The nanomaterials of metals, transition metals, their compounds or composites, like quantum dots (QDs), gold nanoparticles, silver nanoparticles, and magnetic nanoparticles, etc. (3) Silicon nanomaterials: They are mainly composed of silicon and its compounds, like silicon or silica nanoparticles. (4) Organic nanomaterials: They are formed via the agglomeration or assembly of organic molecules, like dendrimer, polymers, biomolecules or biomacromolecules, etc.
In this feature article, we focus on processes for the creation of low-toxicity carbon nanomaterials by chemical modifications with surface designs, but do not discuss the other three types of nanomaterials. We will first describe the surface chemistry applicable for nanosurface modification for creating low-toxicity nanomaterials. Then we make comparative discussions on the biological effects of surface-modified and unmodified nanomaterials, to show the reader how the surface modifications change the interaction fashions between biological systems and carbon nanomaterials including carbon nanotubes (CNTs), fullerenes, metallofullerenes, and graphenes.
1. Low-toxicity carbon nanotube materials by surface-chemical design
CNTs,14 rolled-up, seamless cylinders of graphene sheets,15 have always attracted great scientific and applied interest because of their unique physical, mechanical, and chemical properties. CNTs also offer an intriguing platform for different purposes in biomedical fields, such as bioimaging,16,17 delivery of biomacromolecules into cells,18etc. However, the highly hydrophobic surface and non-biodegradable nature of CNTs impose a reduced biocompatibility which not only greatly limits their uses in diverse fields such as biomedical applications but also arouses concerns about their chronic toxicity. Because the non-degradable CNTs were found in the liver of mice 9 months after a single exposure, and could not be degraded biologically, from a toxicological viewpoint their potential chronic toxicities may be highly concerned.19–24 Thus, it is essential to establish relatively simple and applicable ways for preparations of CNT materials that are water-soluble, biocompatible, and optimally biodegradable.(1) Covalent surface modifications. A recent review has well summarized the covalent chemistry for surface modification of CNTs,13 we hence only give a brief discussion herein. The covalent modifications of CNTs are mostly implemented via chemical reactions (like oxidation, halogenation, cycloadditions, and polymer or macromolecule grafting) or electrochemical reactions.
Oxidations are carried out with strong oxidizing agents such as concentrated HNO3,25,26 mixtures of H2SO4 with HNO3, H2O2, or KMnO4, etc. This manipulation can mostly de-cap,27 shorten CNTs,28 or change the shape and even structures of CNTs. Meanwhile, the oxidation also forms various defects on the surface of CNTs, and highly oxygenated CNTs bearing hydroxyl and epoxide groups on the sidewalls, in addition to carbonyl and carboxyl groups located at the edges. The presence of these chemical groups renders CNTs strongly hydrophilic. The weakness of this process is that the products were mostly uncontrollable, and diverse chemical groups coexisted on the surface and the edges of CNTs. A comparatively controllable process is the ozonolysis of the CNT sidewall, then the obtained ozonized CNTs react with several types of reagents to acquire controlled oxygen-functional groups on the CNT surface.29 Moreover, these oxygen-groups are also favorable to further modifications of CNTs via the reactions with amino acids,30octadecylamine,31 and poly(ethylene glycol) (PEG),32etc. These mostly enhance their biocompatibility and were hence widely used in biomedical applications of CNTs.
Halogenation is another useful method to modify CNTs.33,34 Among them, the fluorination reaction is rather significant because the F atom can readily react with many other functional groups, to further design the surface properties of CNTs.35 For example, in the fluorinated CNTs, F can be replaced with alkyl groups using organolithium36 or Grignard reagents37 to obtain the dispersible alkylated CNTs in many organic solvents. Addition of several types of diols38 and diamines39 can also replace the F atom of the fluoro-nanotubes.
The cycloaddition reaction, which primarily occurs on the aromatic sidewalls, was also used to covalently modify CNTs. [2 + 1] cycloadditions to pristine CNTs. The reaction could be carried out with carbene or nitrene generating compounds via the Bingel reaction.40–42 With the 1,3-dipolar cycloaddition reaction CNTs can react with azomethine ylides to form pyrrolideine-fused rings on the tube surface which makes CNTs soluble.43,44 Moreover, the amino-terminated CNTs can be further applied to attach to various biological molecules, e.g.peptides, nucleic acid, amino acids, or drugs,45–50 providing large number of candidates for biomedical selections.
(2) Noncovalent surface modification. Noncovalent processes are other important ways to modify the surface of CNTs. Because of their benzene ring structures, CNTs can noncovalently interact with aromatic polymers or biomolecules through π–π stacking, electrostatic interactions, hydrogen bonding, van der Waal's forces, and so on. Owing to the weak interactions between the CNT surface and the external moiety, the noncovalent functionalizations can change the surface properties but are less able to alter the functional properties of CNTs. Compared with covalent processes, noncovalent modification is implemented via weak interaction forces like surface adsorption onto the sidewalls of CNTs, π–π stacking, electrostatic interaction, hydrogen bonds, van der Waal's forces, etc. This advantage makes them powerful approaches to control biological behavior, such as the toxicity and biocompatibility of CNTs.
Aromatic molecules such as porphyrin derivatives51 and fluorescein (FITC) terminated PEG chains52 were coated onto the CNT surface with π–π interactions.53,54 For example, making use of the π–π interaction between pyrene and the graphitic surface of CNTs, polymers or biomolecules containing the pyrene moiety can be readily adsorbed onto the surface of CNTs, obtaining biocompatible CNTs.55
To design and prepare water-soluble CNTs, many amphiphiles were used to modify CNTs by van der Waal's forces or hydrophobic effects between the hydrophobic domains and the CNTs’ surface.56,57 The obstacle is that some amphiphile-modified CNTs are not stable in the living body.58 It was also found that some amphiphiles might be able to lyse cell membranes and to denature proteins.59 Thus, much attention is required when using amphiphile-modified CNTs in a biological environment. A multiscale simulation was developed to yield the adsorption profile with CNTs, which may be helpful to direct the future research on a selective solubilisation or manipulation of CNTs.60
Polymers, such as PEG, PEGylated phospholipids, etc. have good biocompatibility. They were hence used to regulate the surface properties of CNTs via noncovalent interactions.32,61 In addition, biomolecules like proteins62 can also interact with CNTs through formation of noncovalent conjugates. Making use of the specific recognition properties of the biosystem, CNT–biomolecule conjugates can become high quality materials for biosensor fabrication,63drug delivery,64,65 and cancer therapy,66etc. For example, streptavidin was adsorbed onto the graphitic surface to form the CNT–protein conjugates,67 then they could be internalized by cells when combined with a nanotube–biotin transporter.68Glucose oxidase was also wrapped onto the surface of CNTs, the formed CNT–enzyme conjugate has the potential to be then used as carbon electrode for high sensitive detection of glucose.69 However, the DNA molecule–SWCNT conjugate can be internalized with the presence of nucleases in the serum, indicating that some of the DNA–CNT conjugates may be unstable in the biological environment.70 In addition, an aligned CNT array membrane electrode, which was used as a nanostructured supporting platform for polypyrrole films, had the capability to improve the controlled release of neurotrophin-3 (NT-3), opening a way to release drugs driven by electrochemistry.71
At this point, it is worth noting that the noncovalently modified CNTs can be de-functionalized to restore the properties of pristine CNTs under some conditions.58 This is a of great benefit if we intend to use the intrinsic properties of CNTs for diverse purposes. However, in some cases this unstable feature, easy decomposition of external moieties from the noncovalently modified surfaces of CNTs, is not expected to be suitable for biomedical applications in vivo.2,72,73
Covalent and noncovalent reactions of CNTs with polymers and biomolecules are also important processes because they can enhance the solubility of the CNTs at a low degree of functionalization with a long polymer chains. Moreover, polymer- or biomolecule-conjugated CNTs can be also used for the control of biocompatibility and toxicity of CNTs in drug delivery, biomolecule detection, and bioimaging.
(1) Lowered toxicity responses. Owing to the lack of solubility and dispersion of CNTs in an aqueous solution, unmodified CNTs disperse poorly in water solution but easily float in air. So, in fundamental studies or applications, surfactants are often used to make CNT disperse homogeneously in a medium, which are tenable only on the premise that these surfactants do not influence the toxicological behavior of the CNTs.58 However, it is quite difficult to claim this point because of the ultra-strong adsorbability of CNT surfaces.
We prepared a stable suspension of unmodified CNT samples for cytotoxicity studies by ultrasonication of CNTs in a culture medium without adding surfactants.74 In this method, the CNT samples have to be used immediately. The SWCNTs, MWCNT10 (10–20 nm in diameter), and C60 exhibited quite different cytotoxicities to alveolar macrophages (AM). CNT exposure results in a loss of the phagocytic ability and in ultrastructural injury of AM. The comparative toxicities of three types of carbon nanomaterials to AM exhibited the following sequence order on a mass basis: SWNTs > MWNT10 > quartz > C60 (Fig. 1).74 As all of three types of carbon nanomaterials consist of carbon atoms with the only differences being in their nanostructures, the results suggest that the cytotoxicity of carbon nanomaterials in vitro is highly dependent on their nanostructure.
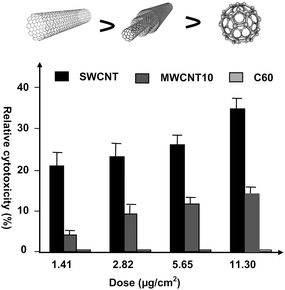 | ||
| Fig. 1 Nanostructure is an important factor for nanotoxicity. The ranking of cytotoxicity of carbon nanomaterials on a mass basis is: SWCNTs > MWCNTs > C60 (ref. 74). | ||
Unmodified CNT particles are easily adsorbed onto the skin of workers in the workplace of production or manipulation of CNTs. Accordingly, the dermal cytotoxicity of CNTs has attracted much attention.75,76 SWCNT exposure to human epidermal keratinocytes results in oxidative stress and cellular toxicity with changes in the ultrastructure and morphology of the cultured skin cells, and unrefined SWCNTs led to accelerated oxidative stress.75 However, in other dermatological trials, CNTs showed no sign of side-effects, such as skin irritation and allergic reactions.77 The reason for this big difference may be attributed to different preparation processes which resulted in different properties of the CNTs. Different preparation processes could produce not only different impurities, but also different structures, different lengths, different sizes, different surface defects, and different unintentional moieties, etc. All these influence the consequence of toxicity tests.
It is very important to note that, as the metallic impurities in CNTs were known to induce significant toxic responses in vitro and in vivo,78,79 is important to identify what toxic responses are from residual metals in CNTs. To this end, a quantitative measurement of the concentration of metal impurities in CNTs is key, although this is most difficult and has remained an open issue for a long time. Recently, a quantitatively analytical method for metal impurities in CNTs has been established by using NAA (neutron activation analysis) technique as a nondestructive standard method of quantification and ICP-MS as a practical analytical method.80 In the absence of a true reference material for CNTs, the NAA method, characterized by nondestructive, high accuracy, high sensitivity, and coincident multielement measurement, can provide the best estimate of the true value of metallic impurities in CNTs. ICP-MS is a desktop instrumental method with which, by using a set of reference data simultaneously determined by both ICP-MS and NAA in the literature, one can obtain the absolute concentration of impurities from the ICP-MS measurement only.
In the workplace of production or manipulation, the easiest exposure route of CNTs is via inhalation of the CNTs that have been unintentionally released in the air. Thus, to estimate the pulmonary toxicity of CNTs is another important topic.20,81–83 The intratracheal administration of soot containing a high content of CNTs to guinea pigs did not induce any measurable inflammation or changed in pulmonary functions after 4 weeks.84 Nevertheless, a long exposure of guinea pigs to carbon nanomaterials induced inflammation; both the respiratory process and the pathological reaction in lung tissues were largely dependent on the time-length of exposure and the characteristics of the CNTs.85 Furthermore, intratracheal injection of CNTs into mice included dose-dependent epithelioid granulomas, and exhibited more toxicity than carbon black and quartz.20 The granulomas observed were not induced by the graphite in CNTs, despite the fact that free graphite can also induce granulomas.86 The biopersistence of unmodified MWCNTs in the lung was a great concern, which had the capacity to stimulate the production of TNF-α in the lung cell and induce both inflammatory and fibrotic reactions.81 Non-dose-dependent multifocal granulomas were observed when the mice were exposed to unmodified CNTs via intratracheal instillation.22
Interestingly, the unmodified SWCNTs can be greatly ingested by macrophage cells, but toxic effects were less observed.87 The cytotoxicity of the unmodified SWCNTs was dose- and time-dependent.19,74,88 The unmodified MWCNTs exhibited clastogenic and an eugenic effect on the epithelial cells,89 and have the capacity to induce asbestos-like, length-dependent, and pathogenic effects in the body cavity of mice.23
The biodistribution of the unmodified SWCNTs via intravenous (i.v.) injection in rabbits was detected through intrinsic near-infrared fluorescence.58 At 24 h after i.v. administration, SWCNTs were found only in the liver at significant concentrations. Due to the limited sensitivity of NIR fluorescence method, later, a more sensitive and quantitative method for measuring the isotope ratios of 13C-labelling SWCNTs was developed to determine the biodistribution of SWCNTs in vivo (Fig. 2A). The unmodified SWCNTs were found to distribute in the entire body, and mainly accumulated in the liver, lungs, and spleen over an extended period of time.90
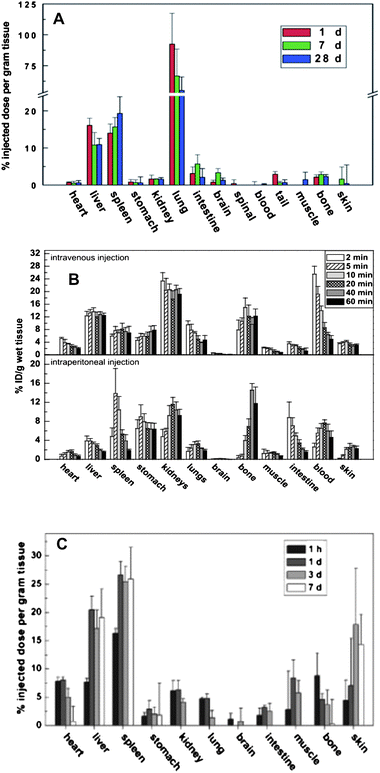 | ||
| Fig. 2 (A) Biodistribution histograms of unmodified SWCNTs in mice at different time points post exposure viai.v. injection. These were quantified by the isotope (13C) labeling technique, which has been demonstrated to be the most sensitive method for determination of biodistributions and metabolism kinetics of carbon nanomaterials in vivo(ref. 90). (B) The biodistribution histograms of OH-modified SWCNT in mice at different time points post exposure viai.v. injection. It was quantified by the isotope (131I) labeling technique (ref. 92). (C) The biodistribution histograms of PEG-modified SWCNT in mice at different time points post exposure viai.v. injection. It was quantified by the isotope (13C) labeling technique (ref. 94). | ||
After surface modification, the hydrophilic CNTs generally became less toxic to human T cells than the unmodified CNTs. However, the toxicity of pristine hydrophobic CNTs was lower than that of oxidized MWCNTs, most likely due to their better dispersion in aqueous solution. At a higher dose (400 μg ml−1), the oxidized MWCNTs became more toxic and induced a reduction in cell viability.21 The dose is always a key factor for the degree of toxicity. We surface-modified CNTs by a covalent hydroxylation reaction and then labeled them with 125I isotope for a sensitive quantification of hydroxylated SWCNTs in mice. We found that the hydroxylated SWCNTs mainly accumulated in the bone, kidney and stomach of mice, and were excreted mainly by the renal route (Fig. 2B).91 The biodistributions of hydroxylated SWCNTs labeled with 131I-isotope show excellent agreement with the results from hydroxylated SWCNTs labeled by the 125I-isotope, proving the reliability of the labeling technique with different isotopes.92 Comparing the results in Fig. 2A and 2B, we can easily find the difference in biodistribution of the unmodified SWCNTs and the modified SWCNTs. The former are mainly accumulated in the liver, lungs, and spleen,90 while the latter changed their biodistribution, accumulation and metabolism kinetics.93
For example, the PEGylated SWCNTs labeled with 13C can distribute throughout most organs within 1 h except brain, intestine and muscle. After 7 days, they mainly accumulated in the liver, spleen and skin (Fig. 2C).94 The PEGylated SWCNTs exhibited a longer blood-circulation time after intravenous injection in mice than the surfactant-suspended pristine SWCNTs in rabbits.58 More importantly, the tumor shows a higher uptake of PEGylated SWNTs, which indicates their potential use for drug delivery and cancer chemotherapy.
Degradability of CNTs directly influences their toxicity and biomedical functions in vivo. Recently, when the SWCNTs were oxidatively treated with a very strong acid H2SO4/H2O2, SWCNTs were partially biodegraded in vitro in the presence of horseradish peroxidase (HRP) and low concentrations of hydrogen peroxide.95 Furthermore, the hypochlorite and reactive radical intermediates of the human neutrophil enzyme myeloperoxidase can catalyse the degradation of SWCNTs.96 Molecular modeling revealed that the interactions between basic amino acids of the enzyme and the carboxyls on the SWCNTs played an important role in positioning the SWCNTs near the catalytic site. Moreover, PEG-functionalized SWCNTs can increase the stability of liposomes when deposited on liposomes to form microcapsules via layer-by-layer deposition, which enhanced the stability of liposomes with the increasing number of deposited layers.97
The biological behavior of MWCNTs has been less studied. However, the surface chemistry for SWCNTs described above is generally applicable to MWCNTs. We can also use surface modification to change the mthod of interaction of MWCNTs with biological systems. For example, taurine-modified MWCNTs exhibited a biodistribution different from unmodified ones, mainly accumulating in the liver and were retained for a long time post-exposure without biodegradation.98 When MWCNTs were modified with 14C-taurine and tween, this difference in surface modification greatly changed the interaction consequences of MWCNTs with blood proteins. The taurine-modified MWCNTs (tau-MWNTs) in mouse serum exhibited a strong protein adsorption onto the MWCNT surfaces, while the tween-modified MWCNTs (tween-MWNTs) prevented protein adsorption on the tube surface (Fig. 3). A change of surface modification greatly altered their biological consequences. This difference greatly dominates MWCNTs biological fate in vivo, including toxicity, metabolism kinetics, and biomedical functions.
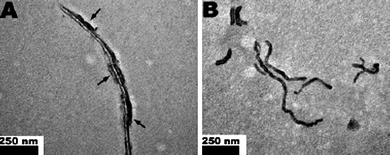 | ||
| Fig. 3 Different surface modifications greatly changed the way MWCNTs interacted with proteins in blood. The TEM photos show the difference in MWCNTs modified by taurine (A: tau-MWNTs) or by tween (B: tween-MWNTs) in mouse serum. The arrows in (A) indicate proteins were heavily absorbed on tau-MWNTs, while (B) those that were not absorbed on tween-MWNTs (ref. 98). This alteration in protein adsorption greatly changes the biological fate of MWCNTs in vivo. | ||
(2) Enhanced biomedical functions. CNTs have been widely studied for their biomedical functions, based on their unique structures, optical and electronic properties. Meanwhile, much effort has been paid to overcome two important obstacles: their toxicity and biocompatibility. Both are closely related to the surface chemistry of CNTs.
Both the band-gap fluorescence in individual semiconducting SWCNTs99 and the band-gap allowing for photoluminescence in the NIR range make CNTs applicable for bioimaging in vivo or in vitro. For example, the antibody-conjugated SWCNTs (emissions of 1000–1600 nm) were studied for selective probing of cell surface receptors and cell imaging.100 Their quantum yields are dependent on the chemical environment,101 bundles,102 sidewall defects,103 and their length.104 Importantly, an appropriate functionalization could induce more intense luminescence through trapping of the excitation energy, such as better dispersion and functionalization of CNTs,105 and the types of surfactants.106,107 The PL-PEG-modified SWCNTs, prepared via solubilizing SWCNTs in sodium cholate and then substituting sodium cholate with a PL-PEG group, had much higher quantum yield than the SWCNTs directly dispersed in PL-PEG. Therefore, the PL-PEG-modified SWCNTs may be developed for near-infrared imaging in mice by detecting the intrinsic near-infrared photoluminescence at a low dose.108
Because of the sharp electronic density of states at the van Hove singularities, SWCNTs exhibits strong resonance Raman scattering. The sharp and strong peaks can be easily distinguished from fluorescence backgrounds, and the Raman signals show no quenching or bleaching under a wide range of imaging conditions.109 It is, hence, rather suitable for bioimaging;17 for example, the RGD-modified SWCNTs were applied for tumors imaged via Raman signal detection.110 With the Multiplexed Multicolor Raman imaging technique, a drastic up-regulation of epidermal growth factor receptor expression on LS174T human colon cancer cells during tumor growth in nude mice was observed.16 Both fluorescent and radioactive-labeled CNTs were used for biomedical imaging in biological tissues/organs.111,112
Both the surface of CNTs and the graphitic network of CNTs provide excellent platforms to attach biomacromolecules and drug molecules to CNTs through covalent or noncovalent interactions. Accordingly, CNTs can be an ideal carrier for drug delivery into cells or tissues. Through surface modification of CNTs with biomacromolecules or drug molecules, the CNT complex obtained can combine both the unique properties of CNTs and the functions of biomacromolecules or drugs, which makes these CNTs complex materials that possess novel functions and advanced applications in biomedical fields.
Their high loading capacity and their ability to penetrate into cells without the aid of any external transport system makes the surface-modified CNTs to be considered a priority drug-carrier and a great opportunity. For example, SWCNT-PTX, prepared viaconjugation of paclitaxel (PXT) to a branched polyethylene glycol (PEG) chain on SWCNTs through a cleavable ester bond, exhibited a much higher inhibition efficacy of tumor growth than its current commercial formulation, Taxol of PXT.113 The SWCNT-PTX prolonged blood circulation and enhanced the tumor uptake of the PXT. Another surface-modified CNT, SWCNT-Pt(IV) prepared via tethering one of the axial ligands of c,c,t-[Pt(NH3)2Cl2(OEt)(O2CCH2CO2H)] to the amine-functionalized SWCNT surface viaamide linkages, was successfully used for the delivery of the anticancer drug cisplatin.64 The cis-[Pt(NH3)2Cl2] was released through intracellular reduction. Moreover, the targeted SWCNT-Pt[IV] was also obtained via attaching the platinum(IV) complex, c,c,t-[Pt(NH3)2Cl2(OEt)(O2CCH2CONH-PEG-FA)], to the amine-functionalized SWCNTs.114Folic acid contained in the platinum(IV) complex offered a way of targeting tumor cells. It significantly enhanced the cell-killing properties of the platinum(IV) complex. Interestingly, CNTs can also act as free-radical scavengers,115 and the 3D CNT monolith further has the function of a metal ion scavenger.116 When CNTs are modified with drug molecules, it may alter their pharmacokinetics. At the same time, the stability and the toxicity of the modified complexes also become key factors for their practical uses in vivo. CNTs coated with surfactant molecules have not usually been stable in the blood serum, and molecules on the surface of CNTs could be displaced by blood proteins.57 A proper modification by the designed surface of CNTs can make them more able to cross the membrane and enter cells without obvious toxicity. Their high aspect ratios and high surface area afford CNTs several unique biological properties in application, such as DNA repair. Moreover, biomolecule-modified CNTs are also a therapeutic platform for genetic diseases, as well a molecular probe for disease detection. For example, SWCNTs have been successfully applied to transport several types of biomolecules, such as peptides,117proteins,118 and plasmid DNA119 into cells.
When streptavidin, a protein clinically used in anticancer therapies, was bound on to the CNTs’ surface and used to form SWCNT–streptavidin conjugates, they could be readily delivered into human promyelocytic leukemia (HL60) cells and human T cellsvia the endocytosis pathway, while the streptavidin by itself could not enter cells.68 Also the noncovalent protein–CNT conjugates118 can be readily transported into various mammalian cellsvia the endocytosis pathway; the internalized conjugates can still retain their biological function.
Phospholipid modified SWCNTs (via the formation of a cleavable disulfide linkage) can carry siRNA, transport and release it into mammalian cells.18,65 These SWCNTs–siRNA conjugates showed a 2-fold advantage over transfection by lipofectamine. most likely due to the huge surface area of SWCNTs for efficient siRNA cargo loading, the high intracellular transporting capacity of SWCNTs, and the high degree of endosome/lysosome escape.65
The excellent electronic properties and the unique shape of CNTs lead them to be a best candidate for fabricating high-sensitivity sensors. For example, CNTs or surface-modified-CNT based biosensors have been fabricated and successfully used to detect proteins, glucose, DNA–DNA hybridization, etc.120,121 On one hand, the semiconductors of CNTs exhibit significant conductance changes due to the change in surface electron density of CNTs after physisorption of different gases122 or biomacromolecules,123 providing a simple way to detect the concentration of the biomacromolecules. On the other hand, CNTs can be modified with those biomacromolecules which have recognition functions such as single-stranded DNA, antibodies, proteins, etc. This provides a way to selectively detect biomacromolecules. In addition, by surface modifications with different functional groups, the modified CNTs can be fabricated into microarrays to detect multiple protein molecules synchronously.124 However, it was found that the surrounding environment can sensitively affect the detection limits and accuracy of the CNT-based biosensors, especially when the detection method is based on the change of conductance.13,125 To gain a stable and size-controllable surface-modified CNT-complex is very important for the development of CNT-based biosensors. In addition, silver-coated carbon nanotubes that have adsorbed Fab mAb can be used to indirectly detect benzoylecgonine, based on label-free surface-enhanced Raman scattering spectroscopy.126
Those drugs that are not stable in blood could be delivered into a given tissue or organ with the aid of surface-designed CNTs. For instance, doxorubicin, a clinical medicine for the treatment of human cancers, was loaded onto branched PEG-modified SWCNTs via supramolecular π–π stacking.66 The obtained CNT-complex exhibited an enhanced therapeutic efficacy and remarkable reduction of toxicity compared with free doxorubicin and DOXIL. If integrating the excellent optical and electronic properties of CNTs with their surface-modification, we may develop more powerful approaches for human disease therapy. For example, when Cy3-labeled single-stranded DNA was transported into a cell by CNTs, the endosomal rupture and release of DNA from the CNTs carrier could be successfully triggered by the optoelectronic excitations of CNTs under NIR radiation, which made good use of the strong optical absorbance in the NIR special spectral window of SWCNTs.106 Then the released DNA can diffuse freely across the nuclear membrane into nucleus, and exhibited no apparent cytotoxicity. Further surface modification with folate, which can recognize and target tumorous cells, could endow PL-PEG-FA-SWCNTs with an additional capacity to selectively destroy cancer cells. The CNT surface modified with neutralite avidin-derivitized mAbs showed the capacity to thermally ablate human Burkitt's lymphoma cellsin vitro with NIR light.127 As there are many varieties of biomolecules or chemical-groups of different functions, by attaching them to the surface of CNTs one could create diverse CNT-based materials to meet the desired objectives.
2. Low-toxic [60]fullerene materials by surface-chemical design
When exploring biological/toxicological effects or the applications of fullerenes, the first barrier one needs to overcome is how to detect them in vivo. Only the detection has been already of special difficulty, to say nothing of its quantitative measurement in vivo. So far, the best way established to solve this point is isotope labeling. In general, the isotope labeling can be classified into two categories. One is to replace the skeleton carbon atoms with C isotopes, such as 13C and 14C,128 which does not alter the physicochemical properties of the fullerenes. The other is to covalently bind radionuclides, such as 125I, onto the surface of fullerenevia the addition reaction.129Radioactive labeling was demonstrated to be the best way for quantitative determination of biodistribution and the metabolism kinetics of fullerene nanomaterials and their derivatives in vivo.
The electronic structure of [60]fullerene is similar to that of the electron-deficient alkenes. Thus, the fullerene surface reacts easily with nucleophiles through a good dienophile of [6,6] bonds. For example, cycloadditions were much employed to prepare a variety of [60]fullerene cycloadducts, which has been summarized in a review discussing the functionalization of C60via cycloaddition.130 More importantly, the cycloadducts of fullerene derivatives can be useful reactants in which some free functional groups can further react with other appendages with special functions, such as drug molecules, amino acids, and biomacromolecules, etc. These types of fullerene complex are particularly suitable for the regulation of the toxic responses of [60]fullerenes by surface design via controlling the type and the chain length of appendages. There are several processes for this purpose, mainly including cyclopropanation, Diels–Alder reaction, and [3 + 2] cycloaddition reaction.
Several cyclopropanations can prepare cyclopropanated [60]fullerenes, among them the most popularly used one is the Bingle reaction which is a nucleophilic addition with very high yields under relatively mild reaction conditions. For example, [60]fullerene surface was modified with diethyl bromomalonate in toluene at room temperature with NaH as auxiliary.131 Moreover, malonate can also react with [60]fullerenes in the existence of iodine132 or with CBr4133 in a base environment. Using the Bingle reaction, a [60]fullerene based dendrimer with an excellent solubility in water has been successfully prepared.134
In the last several years, the concept of multifunctional nanoparticles for biomedical purposes like cancer nanotechnology has been widely investigated.135 It generally needs a defined dimensional nanostructure capable of loading many functional groups. [60]Fullerenes, because of their spherical surface with thirty unsaturated bonds, are excellent candidates for this purpose. For example, bis-functionalized [60]fullerenes can avoid clustering in reasonably dilute solutions in appropriate solutions.136 To gain the particulate forms, which are more likely to have multiple functions, [60]fullerene was bis-functionalized with diethyl bromomalonate to form C60(C(COOH)2)2 which readily aggregated and formed particles of a mean diameter of 170 nm in aqueous solution.137 Based on a similar strategy, polyadducts of fullerene bearing different organic groups can be synthesized. The presence of organic fragments on the surface of fullerenes not only endow polyadducts with more functional properties, but also open a way to further functionalize fullerenes in order to reduce their toxicity or meet a specific demand of practical applications.
The [6,6] double bonds of [60]fullerenes are similar to those of dienophiles. So, in analogy to reactions with dienophiles, the Diels–Alder cycloaddition reaction can be applicable to [60]fullerenes with different dienes. Nevertheless, there are still some differences between them like different reactive conditions. The diene derivatives of fullerenes are good precursors for linkage of some special organic moieties onto the surface of [60]fullerenes, in particular, for the purposes of altering the solubilities, surface properties and improving their biocompatibilities. For example, the Diels–Alder reaction of [60]fullerenes with cyclopentadiene was carried out at room temperature in a toluene solution of [60]fullerenes.138 Another example is the poly(amidoamine) fullerodendrimer which was obtained via Diels–Alder reaction of [60]fullerenes with anthracenyl dendron. It is soluble in polar solvents and stable for several months when stored in the dark at room temperature.139 Another useful process for fullerene surface modification is [3 + 2] cycloaddition of [60]fullerene,140 which introduced a wide variety of substituents and functional groups onto the fullerene surfaces to obtain fullerene derivatives with changed surface properties. The readers may refer to the review in ref. 130 for more details.
When designing chemical processes for surface modification of fullerene, the very important question below needs to be taken into account. There exist 30 unsaturated double bonds on the surface of one fullerene cage: how many additional chemical groups/molecules can be added to the cage without destroying the cage stability and integrity? Theoretically, we can add 60 additional chemical groups onto the surface of one cage. However, when all the double bonds are opened, the cage may no longer retain its cage structure. Therefore, it is important to know the maximum numbers we can add to the cage. We studied the influence of the structural properties on the stability of the fullerene cage in the processes of hydroxylation of [60]fullerene using experimental techniques including laser-induced dissociation associated with a time-of-flight measurement, synchrotron radiation XPS, and FT-IR spectroscopy.141 The stabilities of a family of fullerenols (C60(OH)42, C60(OH)44, C60(OH)36, C60(OH)32, and C60(OH)30) as a function of structural parameters, the hydroxyl number, intensity of the impure group, and the ratio of the carbonyl to hydroxyl groups, were investigated. We found that the molecular stability largely depends on the quantity of impure groups, especially the highly oxygenated carbons in fullerenols, not just on the hydroxyl number. This is different from the previous consideration that the stability of fullerenols only depends on the hydroxyl number. The use of C60(OH)n>36 in practical applications should proceed with caution, since these could lead to unstable open-cage structures. We also found that the formation of impure groups was controllable to gain fullerenols of high stability. The use of M[OH-/C60] > 0.3 for fullerenol synthesis could achieve relatively pure fullerenols of less impure groups on the cage surface. Fullerenols having a structure of R[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–OH] < 0.2 were mostly stable, indicating the importance of the structural balance between the quantity of hydroxyl and impure groups.
O/C–OH] < 0.2 were mostly stable, indicating the importance of the structural balance between the quantity of hydroxyl and impure groups.
(1) Lowered toxicity responses. The toxicity of unmodified [60]fullerenes was studied by many groups. In an earlier study, scientists repeatedly dripped the [60]fullerenes toluene solution onto back skin of a hairless mouse under ultraviolet irradiation. It resulted in the formation of erythema as an acute disease on the skin, but not carcinoma.142 It uncertain whether the toxic response was caused by the organic solvent toluene or the fullerene itself. Similarly, there were no health hazards related to skin irritation and allergic risks after exposure to human skin and the eyes of an albino rabbit to a water suspension of fullerene soot.143 The metabolism behavior of [60]fullerenes was measured viadetection of14C which was used to label [60]fullerenes after intravenous injection into female Sprague–Dawley rats.128 The [60]fullerenes were very rapidly cleared from the circulation within 1 min, and the major organ of accumulation is the liver. Though the unmodified [60]fullerenes were not acutely toxic, they were chronically accumulated in the liver, resulting in difficult clearance from the body. Furthermore, unmodified [60]fullerenes were found in the yolk sac and embryos after exposure of pregnant mice to poly[vinylpyrrolidone] in water containing [60]fullerenes; it produced harmful effects on both conceptuses.144 Note that the toxicities of [60]fullerenes depend on the dose, nanosurface, size, aggregation, bio-species, injection model, and the environment, etc.; here we focus on the nanosurface modification.
Unmodified [60]fullerenes could cross the membrane into cellvia a transmembrane transporter after intraperitoneal injection into Swiss mice.145 Computer simulations indicated that [60]fullerenes could rapidly aggregate in water but disaggregated after entering the membrane interior.146 At higher concentrations, the unmodified [60]fullerenes altered the structural and elastic properties of the lipid bilayer, but caused less mechanical damage to the membrane. However, the simulation results suggest that the unmodified [60]fullerenes entering the bilayer interior could be likely to form micropores which further induced membrane leakage and then cytotoxicity.147,148 Interestingly, the influence of unmodified [60]fullerenes on cell proliferation and differentiation exhibited a cell line dependence. For example, the [60]fullerenes had the capacity to promote the differentiation of Wistar rat limb bud cells, and the differentiation levels of the cells reached a 2–3 fold increase compared to the control.149 However, completely opposite results were observed from embryonic midbrain cells when interacting with [60]fullerenes, which inhibited the differentiation and proliferation of the cells; and this inhibition could be decreased via the addition of antioxidant enzymes such as catalase and SOD.144 Similar inhibition phenomenon was also observed in the human keratinocytes interacting with [60]fullerenes.125
In addition to animal and cellular experiments, another way to evaluate the fullerene toxicity is the study of their interactions with proteins.150 [60]Fullerenes generated singlet oxygen in a model membrane of rat liver microsomes when exposed to UV or visible light. The oxidative damage was induced in terms of lipid peroxidation and damage to proteins, which shows a time- and concentration-dependent manner.
Different surface modifications of C60 result in different biomedical functions. When fullerene was modified with carboxylic acid, the fullerene derivative effectively inhibited glutathione reductase activity, and the inhibition efficacy varied with the number of carboxyls.151 The C3-tris-malonyl-[60]fullerenes and D3-tris-malonyl-[60]fullerenes derivatives possessed a different capacity to inhibit the activity of nitric oxide synthase isoforms.152 While the trisamine [60]fullerene adducts inhibited neuronal nitric oxide synthase in a manner completely reversible by calmodulin.153
The surface modification can improve the performance of fullerenesin vivo or in vitro. When [60]fullerene surfaces were functionalized with organic moieties or biomolecules, the modified [60]fullerenes could be well dispersed in variety of solvents which were friendly towards biosystems. For example, surface modification can transform [60]fullerenes from hydrophobic to hydrophilic, which greatly alters the interaction between the [60]fullerene surface and biomolecules in vivo, perhaps not only reducing their toxic responses but also enhancing their biomedical functions.
(2) Enhanced biomedical functions. The surface modification can greatly enhance or even create new biomedical functions of fullerenes. Lots of experimental results indicated that the interactions of fullerenes with biomolecules can be changed via simple surface modifications. When [60]fullerenes were conjugated with water-soluble amino acid and dipeptide derivatives, they showed the capacity to penetrate lipid bilayer and into the liposomes. The modified fullerenes, localized inside the artificial membrane, could transport the bivalent metal ions into phosphatidylcholine liposomes via transmembrane transportation.154 A [60]fullerene–peptide conjugate, prepared with covalently addition of the 1,2-dihydro-1,2-methanofullerene[60]-61-carboxylic acid to the α-amino group of the peptide T, showed prominent chemotactic potency and weak inhibition of HIV-1 protease.155 Then, several surface-modified [60]fullerenes were designed to inhibit the HIV protease.156 The [60]fullerene–oligonucleotide conjugate was able to combine with three different DNAs, forming a duplex, a triple helix and a triple helix with hairpin structure.157 The anti-HIV functions of fullerenes have been investigated by several groups based on their different surface designs.155,158,159
Among surface-modified fullerenes, fullerenols (hydroxylated fullerene) are the simplest but most important derivatives. They include powerful antioxidants and free radical scavengers both in vitro and in vivo.160–162 For example, we found that after surface modification with ∼22 hydroxyls, the C60(OH)22 is able to protect against NO-induced cell apoptosis and to accelerate the repair of the endothelial cell.137 At the same time, it does not affect the growth of the intact CMECs. Further investigation demonstrated that C60(OH)22 can protect rBCECs or A549 cells against H2O2-induced oxidative damage and stabilize the mitochondrial membrane potential.8C60(OH)20 nanoparticles also exhibited an anti-tumor activity in vivo, and showed specific immunomodulatory effects to T cells and macrophages both in vivo and in vitro.163,164 However, if the surface-modified group is changed to –C(COOH)2, the C60(C(COOH)2)2 showed quite a different capacity to protection against oxidative cellular damage (Fig. 4). The C60(OH)x mainly accumulated in the solid tumor tissues,129 which was attributed to the EPR effect and phagocytosis of mononuclear phagocytes. So far, a better understanding of the antitumor mechanism has not been reached, and more studies need to be conducted. Note that none of these medical functions were found with unmodified C60.
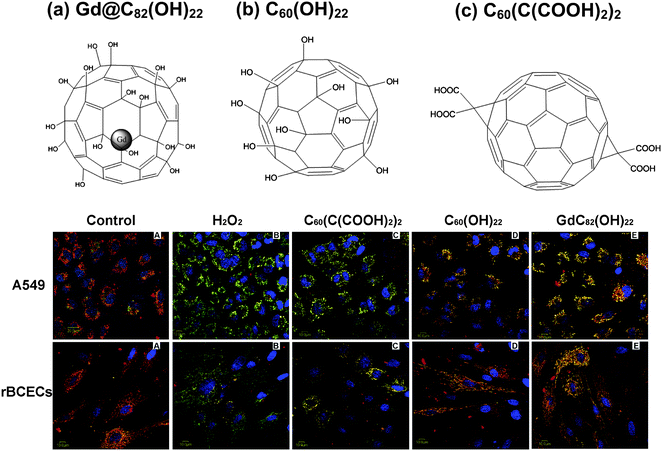 | ||
| Fig. 4 The differences in protective effects of mitochondria membrane in living cells by three different surface-modified fullerene nanomaterials C60(OH)22, and C60(C(COOH)2)2 and Gd@C82(OH)22. The images (lower panel) show potential and the integrity of mitochondria membrane of H2O2-induced A549 cells, and rBCECs cells (ref. 8). | ||
In analogy to fullerenols, carboxyfullerenes also have the ability to scavenge reactive oxygen species.162,165 For example, C60[C(COOH)2]2 could protect against NO-induced damage, inhibit NO-induced depolymerization of cytoskeleton, and had no effect on the growth of intact CMECs.137 It strongly scavenged reactive oxygen species and protected the cells from the H2O2-induced oxidative damage. However, compared to C60(OH)22, the potency of a different surface design C60[C(COOH)2]2 was relatively lower.8 Thus, the design of surface modification of C60 for its optimal function is essential. Recently, we found that the particulate form of water-soluble C60(C(COOH)2)2 (its nanoparticles) could selectively enter oxidized CMECs, and maintain the integrity of CMECs by attenuating H2O2-induced F-actin depolymerization. Moreover, the [C60(C(COOH)2)2]n nanoparticles protected the CMECs against H2O2-induced apoptosis, which related to the modulation of the JNK pathway.166 The [C60(C(COOH)2)2]n nanoparticles can readily enter living cells, mainly viaendocytosis, in a time-, temperature- and energy-dependent manner.167–169
What we have discussed above is how the surface-modifications change the biomedical functions of the fullerene derivatives themselves. Combining the surface-modified [60]fullerenes with drugs, one can create some excellent systems which can be designed to improve the pharmacological and therapeutic profiles of the drug molecule, and to solve current problems associated with the administration of the free drugs, such as the lack of solubility, poor-biocompatibility, lack of selectivity, and toxicity. The excellent stability and the spherical 3-D structure of fullerenes are most suitable for these purposes, they can be covalently attached with multiple drugs, which opens an avenue for creating novel drug delivery systems.
For example, [60]fullerene–paclitaxel conjugate was formulated.170 It is stable in aprotic organic solvents and aqueous media at a physiological pH, meanwhile the paclitaxel can be easily released from the conjugate when incubated with bovine plasma. Based on these properties, a slow-release system of paclitaxel was established, it greatly enhanced the therapeutic efficacy of paclitaxel against lung cancer in vivo. Besides drugs, biomacromolecules such as genes can be also delivered into cells by the formation of complex with [60]fullerene or its derivatives.171 For example, the complex of tetra(piperazino)fullerene epoxide which is stable and no acute organ toxicity could deliver more effectively the enhanced green fluorescent protein gene (EGFP) in vivo, as compared to lipofectin.172EGFP delivered by the fullerene complex on pregnant female ICR mice exhibited significant organ selectivity, and the complex can transfer the gene to liver and spleen, resulting in gene expression. Moreover, insulin gene delivered by tetra(piperazino) fullerene epoxide should reduce blood glucose levels. Thus, combining the surface-modified [60]fullerenes with drugs would create big chances to develop new therapeutic approaches, such as gene therapy, DNA vaccination, etc.
In general, there are two forms of medicines, one is traditional medicine, called molecular medicine, in which a drug exists as a molecular form, the other was proposed recently and is called particulate medicine,in which a drug exists in a particle form.173 It is worth to noting here that surface-modified fullerene derivatives can be both forms. We have discussed its particulate form (i.e., nanomedicine) in ref. 173. Actually, fullerene is also the base for creating multifunctional molecular medicines. As we described before, a set of molecular forms of [60]fullerene derivatives could interact efficiently with the active site of HIV-1 protease and inhibited the HIV-1 protease,174 acted as a strong inhibitor of HIV-1 and HIV-2 reverse transcriptase in the low micromolar concentration range,175 and were capable of the inhibition of HIV-RT.158,159 In scavenging reactive oxygen species in different model systems,176–178 the modified fullerenes likely increased the ROS-scavenging activity due to the attached groups improving the electronic affinity.179 Here, the aggregation and de-aggregation processes may result in the coexistence of both the particle and molecular forms in the system to scavenge free radicals.8,137
3. Low-toxic metallofullerene materials by surface-chemical design
Endohedral metallofullerenes possess unique properties that are different from empty fullerenes like [60]fullerene in physics, chemistry and biological activities.180,181 There are large numbers of endohedral metallofullerenes with formulations of M@C82, M@C80, M@C70, M@C60, etc. However, because of their relatively good solubility, M@C82 has become much more popular than others and has been much investigated, although the others may be formed with higher yields in the arc discharge process.182The surface chemistry of the fullerene cage provides a way to modulate the electronic configurations of the innermost metal atoms inside a nanospace.4 We investigated the chemical modification of the nano sheath to control the electronic properties of innermost metals using synchrotron X-ray photoelectron spectroscopy. Systematic variations in energy, intensity, and width of π* and σ* O 1s core level spectra, in absorption characteristics of C 1s → π* transition, in photoabsorption of pre-edge and resonance regions of the Gd 4d → 4f transition, were observed for Gd@C82, Gd@C82(OH)12, and Gd@C82(OH)22, and the reference materials Gd-DTPA and Gd2O3. A sandwich-type electronic interaction, as follows [outer modification group]–[nano sheaths]–[inner metallic atom], was proposed for fullerene cage modification.4 It demonstrated the possibility to control electron donation directions, either from the innermost metallic atom toward the outer nano sheaths or the reverse, opening a door to the realization of desired designs of the electronic and magnetic properties of metallofullerene nanomaterials.4,193–195
When designing chemical processes for surface modification of metallofullerene, the same question as we have faced with hollow fullerenes needs to be taken into account. There exist 30 unsaturated double bonds on the surface of one cage of a metallofullerene; a maximum number of 60 groups/molecules can be theoretically added onto a cage surface. However, as we have indicated, too many additional chemical groups/molecules will destroy the cage stability and integrity. We have discussed this issue in the previous section and the reader may refer to ref. 141. Two factors are of most importance for retaining the structural stability of the cage, one is the number of added chemical groups (less than 36), and the other is impure groups (the reduction of undesired impure groups that are coincidentally formed onto the cage surface during the chemical process of surface modification).
(1) Lowered toxicity responses. To explore the toxic responses of the unmodified metallofullerenes in vivo, radiochemical techniques again played a significant role.196 After 24 h from injection of the PVP solution of 140La-labled La encapsulated metallofullerenes into the rat, La@fullerene was mainly distributed in the liver and blood, with some retention in the brain. The surface modification of a metallofullerene can greatly change its biodistribution. For example, Hox@C82 was modified with hydroxyls to become water-soluble,183 the product 166Hox@C82(OH)y was then injected intravenously into BALB/c mice. After 48 h, they were accumulated in the liver and kidneys but with slow clearance, while a fraction of 166Hox@C82(OH)y cumulated in the bone, but could not be found in the brain and fat. The biodistribution and toxic response of a different surface-modified metallofullerene was also different from above two metallofullerenes. Gd@C60(C(COOH)2)10 in rodents rapidly reached the kidneys, with only minimal uptake in the liver.189 We found that when the Gd@C82(OH)22 aggregated as nanoparticles, [Gd@C82(OH)22]n nanoparticles could accumulate in the bone, kidneys, stomach, liver, spleen, pancreas and thymus.10 Importantly, the fullerene cage could not be destroyed during metabolic processes, and the Gd3+ did not diffuse out of the cage to directly interact with tissues in the living body. This greatly reduced the toxicity of Gd ions in vivo. Essentially, we found the nanoparticles had the capability to efficiently restore the damaged liver and kidney of the tumor-bearing mice. As there is little difference in the physicochemical properties of lanthanide elements, the surface modification plays a key roles in determining their biodistributions, which consequently alters their toxicities in vivo. Note that the biodistribution and toxic response of metallofullerenes and their derivatives are also dependent of the administration route and dose, etc.
(2) Enhanced biomedical functions. Among the biomedical functions of surface-modified metallofullerenes, anticancer activities and MRI imaging contrast agents are the most important, both are in development for practical use. The anticancer activity of surface-modified [Gd@C82(OH)22]n nanoparticles was first found in 2004. They exhibited a rather high antineoplastic efficiency in H22 hepatoma-implanted mice and exhibited almost no toxicity in vivo and in vitro.7 Compared to CTX and cisplatin which are widely clinically used as antineoplastic drugs, [Gd@C82(OH)22]n nanoparticles had a much higher inhibition efficacy. The most important point is that the high anticancer efficacy is not due to direct toxic effects on tumor cells; the nanoparticles did not kill either the tumor cells or the normal cells. This finding suggests that it possess an anticancer nature different from current anticancer drugs used clinically. The latter are mostly based on their capacity to effectively kill cells by toxicity.
Later, we found that [Gd@C82(OH)22]n nanoparticles possess strong antioxidative functions in tumor-bearing mice.10 In addition, they could also efficiently rehabilitate their hepatic and renal functions, and microstructures of the tumor-damaged liver and kidney tissues in vivo. [Gd@C82(OH)22]n nanoparticles in tumor-bearing mice were capable of scavenging the reactive oxygen species (ROS), and decreasing the activities of enzymes which were associated with the metabolism of ROS.197 The Gd@C82(OH)22 can scavenge, wiping out all types of ROS, depending on surface-modification. Among the water-soluble fullerenes, nanomaterials of C60(C(COOH)2)2, C60(OH)22, and Gd@C82(OH)22, Gd@C82(OH)22 possessed the best capacity of protecting cells against H2O2-induced damage, stabilizing the mitochondrial membrane potential and scavenging ROS.8 Further, the Gd@C82(OH)22 can also regulate the release of Th1/Th2 cytokines and TNF-α mediated cellular immunity on T cells and macrophages.9 These specifically immunomodulatory effects exhibited a good dose-dependent relationship.
More interestingly, we found that the surface-hydoxylated Gd@C82 could greatly enhance the performance of cisplatin, a chemotherapeutic drug commonly used in the clinic. Cisplatin has a strong acquired resistance which confines its application in chemotherapeutics. The [Gd@C82(OH)22]n nanoparticles can help overcome tumor resistance to cisplatin by increasing its intracellular accumulation through the mechanism of restoring defective endocytosis.12 When the CP-r PC-3-luc cells are exposed to cisplatin in the presence of nontoxic [Gd@C82(OH)22]n, we found it not only decreased the number of surviving CP-r cells but also inhibited growth of the CP-r tumors in athymic nude mice as measured by both MRI and optical imaging. These results demonstrated that pretreatment of the CPr PC-3-luc cells with [Gd@C82(OH)22]n enhanced intracellular accumulation of cisplatin and formation of cisplatin-DNA adducts by restoring the defective endocytosis of the CP-r cancer cells. This technology is expected to extend to other challenges related to multidrug resistance often found in cancer treatments.
In cancer treatment, antiangiogenesis is an effective strategy because uncontrollable tumor growth largely depends on tumor angiogenesis and sufficient blood supply. The traditional “molecular” angiogenesis inhibitors have a narrow inhibition spectrum, a “molecular” inhibitor usually targeted a few or even a single angiogenic factor among many angiogenic factors. This limits anticancer efficacy because these might initially be effective but ultimately lead to the failure of the treatment due to the induction of expression of other angiogenic factors. We found that the Gd@C82(OH)22 are capable of simultaneously downregulating more than 10 angiogenic factors in the mRNA level, which has been further confirmed at the protein level. A two-week treatment with the Gd@C82(OH)22 nanoparticles decreased ∼40% tumor microvessels density in nude mice and efficiently lowered the speed of blood supply to tumor tissues. Efficacy of the treatment using Gd@C82(OH)22 nanoparticles was comparable to the clinic anticancer drug paclitaxel, however, Gd@C82(OH)22 exhibited nontoxicity but paclitaxel has a very high toxicity. The metallofullerene with multiple hydroxyl groups can be a potent antiangiogenesis inhibitor which can simultaneously target multiple angiogenic factors.173
MRI is a clinic technique widely used for diseases diagnoses associated with contrast agents. The current contrast agents are mostly chelates of the Gd3+ ion. However, the Gd3+ ion can release from DTPA chelates and is highly toxic. As a result, the use of Gd3+ chelates as MRI agents may have potential risks. To this end, gadolinium-encapsulated metallofullerenes have been proposed and studied as a new generation of MRI contrast agents, because of the high stability of the fullerene cage, such as water-soluble Gd@C60[C(COOH)2]10,189 the ordered microstructure of Gd@C82(OH)22±2 nanoparticles,5 and Gd3N@C80[DiPEG5000(OH)x].190Gd@C82(OH)n have been used for in vivo MRI in the lung, liver, spleen and kidney of mice.198
More interestingly, the Gd in the fullerene cage can be replaced by radioactive metals.183,199–201 The obtained radioactive metallofullerenes can be used as radiotracers for X-ray diffraction contrast agents.202,203 The introduction of the metal inside the hollow fullerene cage and combination of the properties of the metal with the unique physicochemical characteristics of fullerene cages greatly increases the potential for applications of fullerene nanomaterials.
Compared to the metal chelate compounds, the much higher stability of metallofullerenes can enhance not only the safety (because the highly toxic metals cannot be released from fullerene cages and hence do not directly contact with the tissues of the body) but also the treatment effects. Metallofullerenes have been hence attracted great attention in nuclear medicine owing to many radiopharmaceuticals using heavy-metal isotopes.200 For this purpose, the metallofullerenes are usually modified with antibodies or proteins that can specifically bind to biomolecules which then transfer the radionuclide to the target organs.
The capacity to scavenge reactive radical species is also one of the most important functions of metallofullerenes with surface modifications.8,9,197 Free radicals are molecular species characterized by the presence of one or more unpaired electrons, and being highly reactive, easily react with major macromolecules like DNA, RNA, proteins, and lipids;, these thus lead to cellular toxicity. For example, reactive oxygen species (ROS) have been established as important molecules involved in the multistage process of cancer development. We showed that the surface-modified metallofullerenes could be employed as powerful antioxidants, protecting the cell against the H2O2-induced oxidative damage.8 We compared different surface modifications of fullerenes like C60[C(COOH)2]2, C60(OH)22, and Gd@C82(OH)22, the capacity of scavenging ROS is explicitly dependent on the surface-chemistry (Fig. 5).
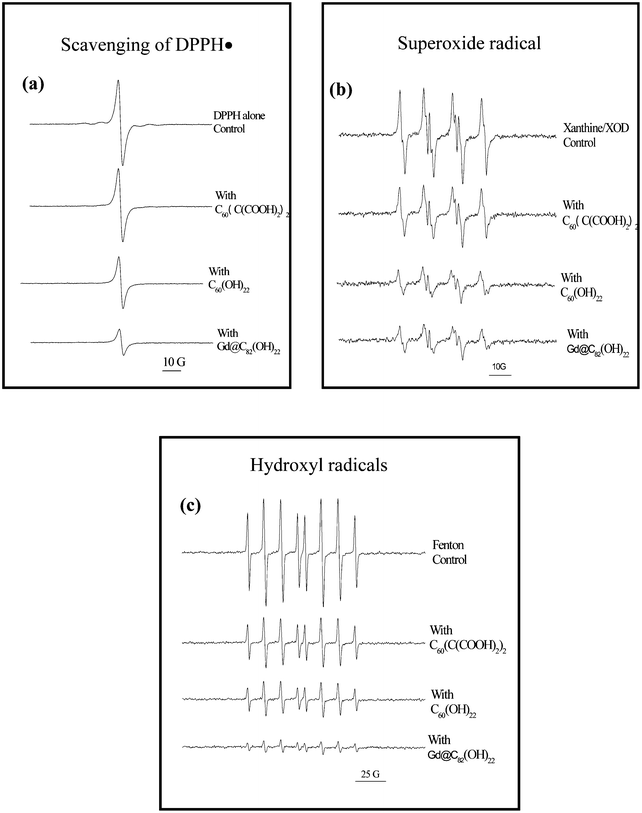 | ||
| Fig. 5 The differences in capacity of ROS scavenging by fullerene nanoparticles with different surface modifications. (a) Scavenging of DPPH˙ radicals by three different surface modifications of fullerenes, C60(C(COOH)2)2, C60(OH)22, and Gd@C82(OH)22. Samples contained 0.1 mM fullerenes, and 0.1 mM DPPH in 20% ethanol. ESR spectra were recorded 5 min after sample mixing at room temperature. Different surface chemistry results in different scavenging capacity of free radicals. This is further demonstrated by scavenging of superoxide radical anions (b) and hydroxyl radicals (c) (ref. 8,9). | ||
Moreover, we found that the efficacy of tumor growth inhibition dependent on the surface chemistry of the metallofullerenes.7,8,9,197 The metallofullerenes (especially Gd@C82) with diverse surface chemistry, because of their low-toxicity and high anticancer efficacy, may be developed as the next generation of anticancer drugs, which are much more advanced compared with conventional anticancer medicines, which are based on poisoning cells using highly toxic chemicals.
4. Low-toxicity graphene materials by surface-chemical design
Graphene, first isolated by Geim and co-workers at Manchester University,204 has attracted enormous interest in diverse fields owing to its extraordinary properties.205,206 Similar to other members in the family of carbon nanomaterials, its lack of solubility and difficult manipulation in solvent has greatly hindered its fundamental investigation and practical use. As-produced graphene is insoluble in organic solvents and agglomerates together in aqueous solutions. Thus, the modification of graphene becomes necessary for all applications.Graphene oxide can be obtained through two routes. One is to directly oxidize graphene with strong oxidants such as concentrated sulfuric acid, concentrated nitric acid, and potassium permanganate.207 The other is firstly oxidizing graphite through Hummer's method,208 Brodie's method,209 a modified Hummer's method,210 and so on. Then they can be exfoliated or thermally expanded to be the graphene oxide.210,211 In addition, lengthwise cutting and unraveling of multiwalled carbon nanotube (MWCNTs) side walls by an oxidative process is another way to gain graphene oxide.212 In contrast to graphene, graphene oxide is heavily oxygenated, bearing a widely variety of oxygen-containing functional groups on the basal plane and at the edges, such as carbonyls, carboxyls and hydroxyls. Therefore, graphene oxides are hydrophilic and hence become more easily modified by organic moieties or biomolecules.
The cycloaddition reaction is an important process for graphene modification. Similar to reactions with fullerenes213 and carbon nanotubes,214graphene reacted with nitrenes to form an aziridine adduct by the [2 + 1] cycloaddition reaction.215 The “click” reaction was also applied to graphene, by which alkyne-terminated molecules have been coupled to graphene sheets to form stable heterocyclic linkers under very mild reaction conditions.216 Taking into account the similar structure of the graphitic forms, some cycloadditions of fullerenes and CNTs, such as 1,3-dipolar cycloaddition,217 Bingel [2 + 1] cyclopropanation reaction,41 can be also employed for graphene modification. They can introduce many functional groups to the surface of graphene to modify its surface properties for diverse applications.
If one intends to adjust the solubility and achieve the possible manipulation of graphene in the variety of solvents, loading polymers onto the surface of graphene is a simple but very useful way. For example, PEG, DSPE-mPEG and PmPV have been loaded onto the graphene surface to obtain homogeneous suspensions.218,219 The products are readily soluble in water, PBS, cell, and serum. When the PEGylated nanographene oxide was mixed with SN38, a water-insoluble cancer drug, SN38 could be easily loaded onto the graphene surface in great quantities.220 More importantly, the PEG loaded nanographene oxide showed non-cytotoxicity.
(1) Lowered toxicity responses. Because of their amazing electronic properties, graphenes may have great potential in electronic devices, such as graphene circuits,221 thin-film transistors,222 solar cells,223etc. As a result, they have been scarcely studied so far to explore their biological effects. The cytotoxicity of graphene in neural phaeochromocytoma-derived PV12 cell was tested, and the cytotoxicity directly related to the shape and the agglomeration of the graphene nanomaterials.224 Both SWCNTs and graphene, which induced cytotoxic effects, show concentration- and shape- dependent effects. However, surprisingly, for graphenes, their toxicity was inversely proportional to the concentration. At a lower concentration, the graphene induced toxic response more intense than SWCNTs. Graphene had no effect on the release of lactate dehydrogenase at a lower concentration, and ROS was generated in a concentration- and time-dependent way after graphene exposure to PC12 cells. The understanding of the biological/toxic responses of graphenes is poor and needs further research.
(2) Enhanced biomedical functions. Because of its similar structure to carbon nanotubes, graphene may interact with biomolecules in a similar way, such as covalent or noncovalent conjugates. Biomolecules that have graphitic networks or high affinity can be loaded onto the graphene surface. The use of graphene is hence expected in the fabrication of next-generation of biotools, such as biobatteries, biomolecules recognition devices, biocarriers, and biomolecule sensors or biotransistors. The biomolecules tend to be adsorbed strongly on the basal plane. The richness of the functional groups on the surface make the modified graphene suitable as a substrate to detect and recognize particular biomolecules, such as DNA, amino acids and antibodies, etc.
The oxidized graphene showed the ability to detect sensitively and selectively DNA and proteinsvia measuring fluorescence from the dye.225 In this system the dye-labeled DNA was noncovalently loaded onto the graphene surface, the electron- or energy-transfer between graphene and dye results in fluorescence quenching. Using a similar process, a graphene oxide-based biosensor was fabricated to detect the ATP molecules in situ in living cells.226 A similar principle was used to fabricate a graphene-based molecular beacon to selectively detect specific DNA sequences. It possessed a relatively high signal-to-background ratio and hence greatly enhanced the detection sensitivity.227
Graphene-based biodevices and DNA transistors are other important applications of surface modified graphene. The Bacillus cereuscell could be adsorbed on the positively charged graphene amine immobilized on silica substrate via the electrostatic interaction between the positively charged graphitic surface due to the presence of an amine salt and the negatively charged surface of the Bacillus cereuscells due to the presence of the polyteichoic acid molecules.228DNA could be also covalently bound to graphene through an amine coupling reaction. The immobilized DNA could hybridize with complementary DNA, labeled with a dye. However, in this case, the fluorescence could not be completely quenched due to longer distance between the dye molecule and the graphene surface. It is worth noting that, in most processes, biological molecules randomly interact with the graphene surface, this renders both the interaction model and the detection and manipulation of the graphene–DNA complex difficult.
Because of their large specific area, the modified graphenes are expected to have promising applications as drug carriers.220,229 For instance, the graphene oxide modified with polyethylene glycol showed high aqueous solubility and stability in physiological solutions.220 The PEGylated graphene oxide was used as carrier to deliver the water-insoluble SN38, opening up a new window to deliver insoluble and aromatic drug molecules. In the same way, the anticancer drug doxorubicin was also loaded onto PEGylated nanographene oxide, which could selectively transport the doxorubicin into specific cancer cells to kill them.229 PEGylated nanographene oxide binding to rituxan was also used to image the B-cell and T-cell by detecting NIR photoluminescence, without a background due to the intrinsic photoluminescence of nanographene oxide sheets. Compared with CNTs, the nanographene oxide has some advantages, mainly including low cost, large spectral region, expedient functionalization and small size.
So far, there are only a few studies devoted to the biomedical properties of graphene-based materials. Undoubtedly, the development of graphene biology or graphene medical sciences can further motivate the development of graphene chemistry. For instance, electrochemical gate-controlled charge transport of graphene occurred at conditions similar to physiological surroundings.230 Also, similar to the process in which graphene oxide is modified by PEG and used as carrier to delivery aromatic, water-insoluble drugs, graphene oxide modified by diverse macromolecules of different functions is a broad area for future exploration, including the toxic responses of these modified graphene-based materials.
5. Conclusions, remarks and perspectives
We shown how to employ chemical modifications to design and change the biological and toxicological properties of carbon nanomaterials. These are implemented through changing the way in which the modified carbon nanomaterials interact with biological systems. Below, we summarize some significant aspects of the challenges for the field.a. Surface transformation. Modification by surface chemistry is an efficient way to transform the surface of carbon nanomaterials from hydrophobic to hydrophilic by attaching different water-soluble and functional moieties on the surfaces. It is also essential for controllable regulation of the biological behavior of carbon nanomaterials in microenvironments in vivo, e.g. the ADME of carbon nanomaterials. In most cases, this will reduce the toxic responses of the carbon nanomaterials.
b. Surface adsorbability. The surface adsorbability of nanoparticles is one of key factors in the toxic response of the carbon nanomaterials; in particular, protein adsorption was found to cause a severe obstruction of metabolism of carbon nanomaterials. Surface modification with designed chemical moieties can lower the protein adsorbability of carbon nanomaterials in vivo.
c. Nanosize. The size of nanoparticles directly relates to their penetration ability of the cell membrane, and also relates to their deposition in the organs. Surface modification can easily change the intensities of interparticulate interaction among carbon nanoparticles. Hence, this changes the size of the modified products in a given solvent. Usually, it is possible to determine an optimal size range that has the lowest toxic responses in vitro, using high throughput screening techniques for cellular tests of toxicity.
d. Nanostructure . If the surface chemistry of carbon nanotubes is not anticipated, one can choose a safer nanostructure. Without an essential influence on the functions, using double-walled carbon nanotubes (DWCNTs) to replace single-walled carbon nanotubes (SWCNTs) in applications will achieve much safer products. Both MD theoretical and experimental results indicated that the DWCNTs showed much better biocompatibility and lower cytotoxicity than SWCNTs.
e. Aggregation or agglomeration. Easy aggregation or agglomeration is one of key factors that dominate the metabolic behavior of carbon nanomaterials in vivo. Aggregation in vivo mostly renders the nanoparticle difficult to clear from living organisms. Surface modification with a proper moiety can change the surface charges of the nanoparticles and hence change their aggregation/agglomeration state in vivo, and lower their toxicity.
 | ||
| Fig. 6 The knowledge gained so far allows the design of a safe carbon nanomaterial by surface modification. The key is to develop a controllable chemistry for surface modification, in order to reach the goal of reducing the toxicity, enhancing the bio-medical functions, realizing the delivery to targeted tissues, etc. On the other hand, to apply newly proposed chemical processes such as “click chemistry” or “green chemistry” to nanosurface modifications may become important topics in future for the creation of safe nanomaterials with designed surface chemistry. | ||
Acknowledgements
The authors are grateful for the support of the MOST 973 program (2011CB933403, 2010CB933904), and the CAS Knowledge Innovation Program.References
- Nanotoxicology, ed. Y. L. Zhao and H. S. Nalwa, American Scientific Publishers, Los Angeles, USA, 2006 Search PubMed.
- Y. L. Zhao, G. M. Xing and Z. F. Chai, Nat. Nanotechnol., 2008, 3, 191 CrossRef CAS.
- M. K. So, C. J. Xu, A. M. Loening, S. S. Gambhir and J. H. Rao, Nat. Biotechnol., 2006, 24, 339 CrossRef CAS.
- J. Tang, G. M. Xing, H. Yuan, W. B. Cao, L. Jing, X. F. Gao, L. Qu, Y. Cheng, C. Ye, Y. L. Zhao, Z. F. Chai, K. Ibrahim, H. J. Qian and R. Su, J. Phys. Chem. B, 2005, 109, 8779 CrossRef CAS.
- G. M. Xing, H. Yuan, R. He, X. Y. Gao, L. Jing, F. Zhao, Z. F. Chai and Y. L. Zhao, J. Phys. Chem. B, 2008, 112, 6288 CrossRef CAS.
- J. Tang, G. M. Xing, H. Yuan, X. F. Gao, L. Jing, S. Wang, Y. Cheng and Y. L. Zhao, J. Radioanal. Nucl. Chem., 2007, 272, 307 CrossRef CAS.
- C. Y. Chen, G. M. Xing, J. X. Wang, Y. L. Zhao, B. Li, J. Tang, G. Jia, T. C. Wang, J. Sun, L. Xing, H. Yuan, Y. X. Gao, H. Meng, Z. Chen, F. Zhao, Z. F. Chai and X. H. Fang, Nano Lett., 2005, 5, 2050 CrossRef CAS.
- J. J. Yin, F. Lao, P. P. Fu, W. G. Wamer, Y. L. Zhao, P. C. Wang, Y. Qiu, B. Y. Sun, G. M. Xing, J. Q. Dong, X. J. Liang and C. Y. Chen, Biomaterials, 2009, 30, 611 CrossRef CAS.
- Y. Liu, F. Jiao, Y. Qiu, W. Li, F. Lao, G. Q. Zhou, B. Y. Sun, G. M. Xing, J. Q. Dong, Y. L. Zhao, Z. F. Chai and C. Y. Chen, Biomaterials, 2009, 30, 3934 CrossRef CAS.
- J. X. Wang, C. Y. Chen, B. Li, H. W. Yu, Y. L. Zhao, J. Sun, Y. F. Li, G. M. Xing, H. Yuan, J. Tang, Z. Chen, H. Meng, Y. X. Gao, C. Ye, Z. F. Chai, C. F. Zhu, B. C. Ma, X. H. Fang and L. J. Wan, Biochem. Pharmacol., 2006, 71, 872 CrossRef CAS.
- D. Yang, Y. L. Zhao, H. Guo, Y. N. Li, P. Tewary, G. M. Xing, W. Hou, J. J. Oppenheim and N. Zhang, ACS Nano, 2010, 4, 1178 CrossRef CAS.
- X. J. Liang, H. Meng, Y. Z. Wang, H. Y. He, J. Meng, J. Lu, P. C. Wang, Y. L. Zhao, X. Y. Gao, B. Y. Sun, C. Y. Chen, G. M. Xing, D. W. Shen, M. M. Gottesman, Y. Wu, J. J. Yin and L. Jia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7449 CrossRef CAS.
- H. C. Wu, X. L. Chang, L. Liu, F. Zhao and Y. L. Zhao, J. Mater. Chem., 2010, 20, 1 RSC.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- Z. Liu, S. Tabakman, S. Sherlock, X. L. Li, Z. Chen, K. Jiang, S. S. Fan and H. J. Dai, Nano Res., 2010, 3, 222 CrossRef CAS.
- Z. Liu, X. L. Li, S. M. Tabakman, K. Jiang, S. S. Fan and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 13540 CrossRef CAS.
- Z. Liu, M. Winters, M. Holodniy and H. J. Dai, Angew. Chem., Int. Ed., 2007, 46, 2023 CrossRef CAS.
- D. X. Cui, F. R. Tian, C. S. Ozkan, M. Wang and H. J. Gao, Toxicol. Lett., 2005, 155, 73 CrossRef CAS.
- C. W. Lam, J. T. James, R. McCluskey and R. L. Hunter, Toxicol. Lett., 2004, 77, 126 CAS.
- M. Bottini, S. Bruckner, K. Nika, N. Bottini, S. Bellucci, A. Magrini, A. Bergamaschi and T. Mustelin, Toxicol. Lett., 2006, 160, 121 CrossRef CAS.
- D. B. Warheit, B. R. Laurence, K. L. Reed, D. H. Roach, G. A. M. Reynolds and T. R. Webb, Toxicol. Lett., 2004, 77, 117 CAS.
- C. A. Poland, R. Duffin, I. Kinloch, A. Maynard, W. A. H. Wallace, A. Seaton, V. Stone, S. Brown, W. MacNee and K. Donaldson, Nat. Nanotechnol., 2008, 3, 423 CrossRef CAS.
- L. H. Ding, J. Stilwell, T. T. Zhang, O. Elboudwarej, H. J. Jiang, J. P. Selegue, P. A. Cooke, J. W. Gray and F. Q. F. Chen, Nano Lett., 2005, 5, 2448 CrossRef CAS.
- S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Acc. Chem. Res., 2002, 35, 1105 CrossRef CAS.
- I. D. Rosca, F. Watari, M. Uo and T. Akaska, Carbon, 2005, 43, 3124 CrossRef CAS.
- S. C. Tsang, Y. K. Chen, P. J. F. Harris and M. L. H. Green, Nature, 1994, 372, 159 CrossRef CAS.
- J. Liu, A. G. Rinzler, H. J. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y. S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253 CrossRef CAS.
- S. Banerjee and S. S. Wong, J. Phys. Chem. B, 2002, 106, 12144 CrossRef CAS.
- L. L. Zeng, L. B. Alemany, C. L. Edwards and A. R. Barron, Nano Res., 2008, 1, 72 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. S. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95 CrossRef CAS.
- M. L. Schipper, N. Nakayama-Ratchford, C. R. Davis, N. W. S. Kam, P. Chu, Z. Liu, X. M. Sun, H. J. Dai and S. S. Gambhir, Nat. Nanotechnol., 2008, 3, 216 CrossRef CAS.
- T. Nakajima, S. Kasamatsu and Y. Matsuo, Eur. J. Solid State Inorg. Chem., 1996, 33, 831 CAS.
- A. Hamwi, H. Alvergnat, S. Bonnamy and F. Beguin, Carbon, 1997, 35, 723 CrossRef CAS.
- V. N. Khabashesku, W. E. Billups and J. L. Margrave, Acc. Chem. Res., 2002, 35, 1087 CrossRef CAS.
- R. K. Saini, I. W. Chiang, H. Peng, R. E. Smalley, W. E. Billups, R. H. Hauge and J. L. Margrave, J. Am. Chem. Soc., 2003, 125, 3617 CrossRef CAS.
- P. J. Boul, J. Liu, E. T. Mickelson, C. B. Huffman, L. M. Ericson, I. W. Chiang, K. A. Smith, D. T. Colbert, R. H. Hauge, J. L. Margrave and R. E. Smalley, Chem. Phys. Lett., 1999, 310, 367 CrossRef CAS.
- L. Zhang, V. U. Kiny, H. Q. Peng, J. Zhu, R. F. M. Lobo, J. L. Margrave and V. N. Khabashesku, Chem. Mater., 2004, 16.
- J. L. Stevens, A. Y. Huang, H. Q. Peng, I. W. Chiang, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2003, 3, 331 CrossRef CAS.
- M. J. Moghaddam, S. Taylor, M. Gao, S. M. Huang, L. M. Dai and M. J. McCall, Nano Lett., 2004, 4, 89 CrossRef CAS.
- K. S. Coleman, S. R. Bailey, S. Fogden and M. L. H. Green, J. Am. Chem. Soc., 2003, 125, 8722 CrossRef CAS.
- T. Umeyama, N. Tezuka, M. Fujita, Y. Matano, N. Takeda, K. Murakoshi, K. Yoshida, S. Isoda and H. Imahori, J. Phys. Chem. C, 2007, 111, 9734 CrossRef CAS.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760 CrossRef CAS.
- N. Tagmatarchis and M. Prato, J. Mater. Chem., 2004, 14, 437 RSC.
- A. Bianco and M. Prato, Adv. Mater., 2003, 15, 1765 CrossRef.
- D. Pantarotto, C. D. Partidos, R. Graff, J. Hoebeke, J. P. Briand, M. Prato and A. Bianco, J. Am. Chem. Soc., 2003, 125, 6160 CrossRef CAS.
- D. Pantarotto, C. D. Partidos, J. Hoebeke, F. Brown, E. Kramer, J. P. Briand, S. Muller, M. Prato and A. Bianco, Chem. Biol., 2003, 10, 961 CrossRef CAS.
- R. Singh, D. Pantarotto, D. McCarthy, O. Chaloin, J. Hoebeke, C. D. Partidos, J. P. Briand, M. Prato, A. Bianco and K. Kostarelos, J. Am. Chem. Soc., 2005, 127, 4388 CrossRef CAS.
- D. Pantarotto, J. P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16 RSC.
- G. Pastorin, W. Wu, S. Wieckowski, J. P. Briand, K. Kostarelos, M. Prato and A. Bianco, Chem. Commun., 2006, 1182 RSC.
- D. M. Guldi, H. Taieb, G. M. A. Rahman, N. Tagmatarchis and M. Prato, Adv. Mater., 2005, 17, 871 CrossRef CAS.
- N. Nakayama-Ratchford, S. Bangsaruntip, X. M. Sun, K. Welsher and H. J. Dai, J. Am. Chem. Soc., 2007, 129, 2448 CrossRef CAS.
- R. J. Chen, Y. G. Zhang, D. W. Wang and H. J. Dai, J. Am. Chem. Soc., 2001, 123, 3838 CrossRef CAS.
- J. Chen, H. Y. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034 CrossRef CAS.
- P. Wu, X. Chen, N. Hu, U. C. Tam, O. Blixt, A. Zettl and C. R. Bertozzi, Angew. Chem., Int. Ed., 2008, 47, 5022 CrossRef CAS.
- C. Richard, F. Balavoine, P. Schultz, T. W. Ebbesen and C. Mioskowski, Science, 2003, 300, 775 CrossRef CAS.
- R. J. Chen, S. Bangsaruntip, K. A. Drouvalakis, N. W. S. Kam, M. Shim, Y. M. Li, W. Kim, P. J. Utz and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4984 CrossRef CAS.
- P. Cherukuri, C. J. Gannon, T. K. Leeuw, H. K. Schmidt, R. E. Smalley, S. A. Curley and R. B. Weisman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18882 CrossRef CAS.
- Z. Liu, S. Tabakman, K. Welsher and H. J. Dai, Nano Res., 2009, 2, 85 CrossRef CAS.
- E. J. Wallace, R. S. G. D'Rozario, B. M. Sanchez and M. S. P. Sansom, Nanoscale, 2010, 2, 967 RSC.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 4783 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- L. Zhang, G. C. Zhao, X. W. Wei and Z. S. Yang, Electroanalysis, 2005, 17, 630 CrossRef CAS.
- R. P. Feazell, N. Nakayama-Ratchford, H. J. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438 CrossRef CAS.
- N. W. S. Kam, Z. Liu and H. J. Dai, J. Am. Chem. Soc., 2005, 127, 12492 CrossRef CAS.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Y. Chen, Q. W. Yang, D. W. Felsher and H. J. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668 CrossRef CAS.
- F. Balavoine, P. Schultz, C. Richard, V. Mallouh, T. W. Ebbesen and C. Mioskowski, Angew. Chem., Int. Ed., 1999, 38, 1912 CrossRef CAS.
- N. W. S. Kam, T. C. Jessop, P. A. Wender and H. J. Dai, J. Am. Chem. Soc., 2004, 126, 6850 CrossRef CAS.
- B. R. Azamian, J. J. Davis, K. S. Coleman, C. B. Bagshaw and M. L. H. Green, J. Am. Chem. Soc., 2002, 124, 12664 CrossRef CAS.
- H. K. Moon, C. I. Chang, D. K. Lee and H. C. Choi, Nano Res., 2008, 1, 351 CrossRef CAS.
- B. C. Thompson, J. Chen, S. E. Moulton and G. G. Wallace, Nanoscale, 2010, 2, 499 RSC.
- Z. Chen, H. Meng, G. M. Xing, C. Y. Chen and Y. L. Zhao, Int. J. Nanotechnol., 2007, 4, 179 Search PubMed.
- A.de L. Zerda, C. Zavaleta, S. Keren, S. Vaithilingam, S. Bodapati, Z. Liu, J. Levi, B. R. Smith, T. J. Ma, O. Oralkan, Z. Cheng, X. Y. Chen, H. J. Dai, B. T. Khuri-Yakub and S. S. Gambhir, Nat. Nanotechnol., 2008, 3, 556.
- G. Jia, H. F. Wang, L. Yan, X. G. Wang, R. J. Pei, T. Yan, Y. L. Zhao and X. B. Guo, Environ. Sci. Technol., 2005, 39, 1378 CrossRef CAS.
- A. Shvedova, V. Castranova, E. Kisin, D. Schwegler-Berry, A. R. Murray, V. Z. Gandelsman, A. Maynard and P. Baron, J. Toxicol. Environ. Health, Part A, 2003, 66, 1909 Search PubMed.
- A. D. Maynard, P. A. Baron, M. Foley, A. A. Shvedova, E. R. Kisin and V. Castranova, J. Toxicol. Environ. Health, Part A, 2004, 67, 87 Search PubMed.
- A. Huczko and H. Lange, Fullerene Sci. Technol., 2001, 9, 247 CrossRef CAS.
- A. R. Murray, E. Kisin, S. S. Leonard, S. H. Young, C. Kommineni, V. E. Kagan, V. Castranova and A. A. Shvedova, Toxicology, 2009, 257, 161 CrossRef CAS.
- S. Koyama, Y. A. Kim, T. Hayashi, K. Takeuchi, C. Fujii, N. Kuroiwa, H. Koyama, T. Tsukahara and M. Endo, Carbon, 2009, 47, 1365 CrossRef CAS.
- C. C. Ge, F. Lao, W. Li, Y. F. Li, C. Y. Chen, X. Y. Mao, B. Li, Z. F. Chai and Y. L. Zhao, Anal. Chem., 2008, 80, 9426 CrossRef CAS.
- J. Muller, F. Huaux, N. Moreau, P. Misson, J. F. Heilier, M. Delos, M. Arras, A. Fonseca, J. B. Nagy and D. Lison, Toxicol. Appl. Pharmacol., 2005, 207, 221 CAS.
- Z. Li, T. Hulderman, R. Salmen, R. Chapman, S. S. Leonard, S. H. Young, A. Shvedova, M. I. Luster and P. P. Simeonova, Environ. Health Perspect., 2006, 115, 377 CrossRef.
- A. A. Shvedova, E. R. Kisin, R. Mercer, A. R. Murray, V. J. Johnson, A. I. Potapovich, Y. Y. Tyurina, O. Gorelik, S. Arepalli, D. Schwegler-Berry, A. F. Hubbs, J. Antonini, D. E. Evans, B. K. Ku, D. Ramsey, A. Maynard, V. E. Kagan, V. Castranova and P. Baron, Am. J. Physiol.: Lung Cell. Mol. Phys., 2005, 289, 698 Search PubMed.
- A. Huczko, H. Lange, E. Całko, H. Grubek-Jaworska and P. Droszcz, Fullerene Sci. Technol., 2001, 9, 251 CrossRef CAS.
- A. Huczko, H. Lange, M. Bystrzejewski, P. Baranowski, H. Grubek-Jaworska, P. Nejman, T. Przybyłowski, K. Czumińska, J. Glapiński, D. R. M. Walton and H. W. Kroto, Fullerenes, Nanotubes, Carbon Nanostruct., 2005, 13, 141 Search PubMed.
- C. J. Driver, M. W. Ligotke, W. G. Landis, J. L. Downs, B. L. Tiller, E. B. Moore, Jr. and D. A. Cataldo, Environmental and Health Effects Review for Obscurant Graphite Flakes, U. S. Army Chemical and Biological Defense Agency, Pentagon Report ERDEC-CR-056, Washington DC, 1993.
- P. Cherukuri, S. M. Bachilo, S. H. Litovsky and R. B. Weisman, J. Am. Chem. Soc., 2004, 126, 15638 CrossRef CAS.
- A. E. Porter, M. Gass, K. Muller, J. N. Skepper, P. A. Midgley and M. Welland, Nat. Nanotechnol., 2007, 2, 713 CrossRef CAS.
- J. Muller, I. Decordier, P. H. Hoet, N. Lombaert, L. Thomassen, F. Huaux, D. Lison and M. Kirsch-Volders, Carcinogenesis, 2007, 29, 427 CrossRef.
- S. T. Yang, W. Guo, Y. Lin, X. Y. Deng, H. F. Wang, H. F. Sun, Y. F. Liu, X. Wang, W. Wang, M. Chen, Y. P. Huang and Y. P. Sun, J. Phys. Chem. C, 2007, 111, 17761 CrossRef CAS.
- H. F. Wang, J. Wang, X. Y. Deng, H. F. Sun, Z. J. Shi, Z. N. Gu, Y. F. Liu and Y. L. Zhao, J. Nanosci. Nanotechnol., 2004, 4, 1019 CrossRef CAS.
- J. Wang, X. Y. Deng, S. T. Yang, H. F. Wang, Y. L. Zhao and Y. F. Liu, Nanotoxicology, 2008, 2, 28 CrossRef CAS.
- Y. L. Zhao, Z. F. Chai, Nanotoxicology: the Bases for Safe Uses of Nanomaterials, Science Press, Beijing, China, 2010 Search PubMed.
- S. T. Yang, K. A. S. Fernando, J. H. Liu, J. Wang, H. F. Sun, Y. F. Liu, M. Chen, Y. P. Huang, X. Wang, H. F. Wang and Y. P. Sun, Small, 2008, 4, 940 CrossRef CAS.
- B. L. Allen, P. D. Kichambare, P. P. Gou, I. I. Vlasova, A. A. Kapralov, N. Konduru, V. E. Kagan and A. Star, Nano Lett., 2008, 8, 3899 CrossRef CAS.
- V. E. Kagan, N. V. Konduru, W. H. Feng, B. L. Allen, J. Conroy, Y. Volkov, I. I. Vlasova, N. A. Belikova, N. Yanamala, A. Kapralov, Y. Y. Tyurina, J. W. Shi, E. R. Kisin, A. R. Murray, J. Franks, D. Stolz, P. P. Gou, J. Klein-Seetharaman, B. Fadeel, A. Star and A. A. Shvedova, Nat. Nanotechnol., 2010, 5, 354 CrossRef CAS.
- G. Angelini, S. Boncompagni, P. D. Maria, M. D. Nardi, A. Fontana, C. Gasbarri and E. Menna, Carbon, 2007, 45, 2479 CrossRef CAS.
- X. Y. Deng, G. Jia, H. F. Wang, H. F. Sun, X. Wang, S. T. Yang, T. Wang and Y. F. Liu, Carbon, 2007, 45, 1419 CrossRef.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. P. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS.
- K. Welsher, Z. Liu, D. Daranciang and H. J. Dai, Nano Lett., 2008, 8, 586 CrossRef CAS.
- J. Crochet, M. Clemens and T. Hertel, J. Am. Chem. Soc., 2007, 129, 8058 CrossRef.
- S. Reich, M. Dworzak, A. Hoffmann, C. Thomsen and M. S. Strano, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 033402 CrossRef.
- L. Cognet, D. A. Tsyboulski, J. D. R. Rocha, C. D. Doyle, J. M. Tour and R. B. Weisman, Science, 2007, 316, 1465 CrossRef CAS.
- D. A. Heller, R. M. Mayrhofer, S. Baik, Y. V. Grinkova, M. L. Usrey and M. S. Strano, J. Am. Chem. Soc., 2004, 126, 14567 CrossRef CAS.
- Y. Lin, B. Zhou, R. B. Martin, K. B. Henbest, B. A. Harruff, J. E. Riggs, Z. X. Guo, L. F. Allard and Y. P. Sun, J. Phys. Chem. B, 2005, 109, 14779 CrossRef CAS.
- N. W. S. Kam, M. O'Connell, J. A. Wisdom and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600 CrossRef CAS.
- W. Wenseleers, I. I. Vlasov, E. Goovaerts, E. D. Obraztsova, A. S. Lobach and A. Bouwen, Adv. Funct. Mater., 2004, 14, 1105 CrossRef CAS.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. J. Dai, Nat. Nanotechnol., 2009, 4, 773 CrossRef CAS.
- D. A. Heller, S. Baik, T. E. Eurell and M. S. Strano, Adv. Mater., 2005, 17, 2793 CrossRef CAS.
- S. Keren, C. Zavaleta, Z. Cheng, A. D. L. Zerda, O. Gheysens and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5844 CrossRef CAS.
- M. R. McDevitt, D. Chattopadhyay, J. S. Jaggi, R. D. Finn, P. B. Zanzonico, C. Villa, D. Rey, J. Mendenhall, C. A. Batt, J. T. Njardarson and D. A. Scheinberg, PLoS One, 2007, 2, 907.
- Z. X. Xu, P. A. Hu, S. M. Wang and X. B. Wang, Appl. Surf. Sci., 2008, 254, 1915 CrossRef CAS.
- Z. Liu, K. Chen, C. Davis, S. Sherlock, Q. Z. Cao, X. Y. Chen and H. J. Dai, Cancer Res., 2008, 68, 6652 CrossRef CAS.
- S. Dhar, Z. Liu, J. Thomale, H. J. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467 CrossRef CAS.
- A. Galano, Nanoscale, 2010, 2, 373 RSC.
- J. Khanderi, R. C. Hoffmann and J. J. Schneider, Nanoscale, 2010, 2, 613 RSC.
- D. Pantarotto, J. P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16 RSC.
- N. W. S. Kam and H. J. Dai, J. Am. Chem. Soc., 2005, 127, 6021 CrossRef CAS.
- Y. Liu, D. C. Wu, W. D. Zhang, X. Jiang, C. B. He, T. S. Chung, S. H. Goh and K. W. Leong, Angew. Chem., Int. Ed., 2005, 44, 4782 CrossRef CAS.
- B. L. Allen, P. D. Kichambare and A. Star, Adv. Mater., 2007, 19, 1439 CrossRef CAS.
- S. N. Kim, J. F. Rusling and F. Papadimitrakopoulos, Adv. Mater., 2007, 19, 3214 CrossRef CAS.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. Cho and H. J. Dai, Science, 2000, 287, 622 CrossRef CAS.
- K. Bradley, M. Briman, A. Star and G. Grüner, Nano Lett., 2004, 4, 253 CrossRef CAS.
- R. J. Chen, H. C. Choi, S. Bangsaruntip, E. Yenilmez, X. W. Tang, Q. Wang, Y. L. Chang and H. J. Dai, J. Am. Chem. Soc., 2004, 126, 1563 CrossRef CAS.
- A. Star, E. Tu, J. Niemann, J. C. P. Gabriel, C. S. Joiner and C. Valcke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 921 CrossRef CAS.
- M. Sanles-Sobrido, L. Rodríguez-Lorenzo, S. Lorenzo-Abalde, Á. González-Fernández, M. A. Correa-Duarte, R. A. Alvarez-Puebla and L. M. Liz-Marzán, Nanoscale, 2009, 1, 153 RSC.
- P. Chakravarty, R. Marches, N. S. Zimmerman, A. D. E. Swafford, P. Bajaj, I. H. Musselman, P. Pantano, R. K. Draper and E. S. Vitetta, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8697 CrossRef CAS.
- R. Bullard-Dillard, K. E. Creek, W. A. Scrivens and J. M. Tour, Bioorg. Chem., 1996, 24, 376 CrossRef CAS.
- Z. Q. Ji, H. F. Sun, H. F. Wang, Q. Y. Xie, Y. F. Liu and Z. Wang, J. Nanopart. Res., 2006, 8, 53 CrossRef CAS.
- G. A. Burley, P. A. Keller and S. G. Pyne, Fullerenes, Nanotubes, Carbon Nanostruct., 1999, 7, 973 Search PubMed.
- C. Bingel, Chem. Ber., 1993, 126, 1957 CrossRef CAS.
- J. F. Nierengarten and J. F. François, Tetrahedron Lett., 1997, 38, 7737 CrossRef CAS.
- X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595 RSC.
- M. Brettreich and A. Hirsch, Tetrahedron Lett., 1998, 39, 2731 CrossRef CAS.
- M. Ferrari, Nat. Rev. Mol. Cell Biol., 2005, 5, 161 CAS.
- D. M. Guldi, Res. Chem. Intermed., 1997, 23, 653 CrossRef CAS.
- F. Lao, W. Li, D. Han, Y. Qu, Y. Liu, Y. L. Zhao and C. Y. Chen, Nanotechnology, 2009, 20, 225103 CrossRef.
- V. M. Rotello, J. B. Howard, T. Yadav, M. M. Conn, E. Viani, L. M. Giovane and A. L. Lafleur, Tetrahedron Lett., 1993, 34, 1561 CrossRef CAS.
- Y. Takaguchi, T. Tajima, K. Ohta, J. Motoyoshiya, H. Aoyama, T. Wakahara, T. Akasaka, M. Fujitsuka and O. Ito, Angew. Chem., Int. Ed., 2002, 41, 817 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798 CrossRef CAS.
- G. M. Xing, J. Zhang, Y. L. Zhao, J. Tang, B. Zhang, X. F. Gao, H. Yuan, L. Qu, W. B. Cao, Z. F. Chai, K. Ibrahim and R. Su, J. Phys. Chem. B, 2004, 108, 11473 CrossRef CAS.
- T. Moriguchi, K. Yano, S. Hokari and M. Sonoda, Fullerene Sci. Technol., 1999, 7, 179.
- A. Huczko, H. Lange and E. Calko, Fullerene Sci. Technol., 1999, 7, 935 CAS.
- T. Tsuchiya, I. Oguri, Y. N. Yamakoshi and N. Miyata, FEBS Lett., 1996, 393, 139 CrossRef.
- F. Moussa, F. Trivin, R. Céolin, M. Hadchoue, P. Y. Sizaret, V. Greugny, C. Fabre, A. Rassat and H. Szwrc, Fullerene Sci. Technol., 1996, 4, 21 CAS.
- J. Wong-Ekkabut, S. Baoukina, W. Triampo, I. M. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363 CrossRef CAS.
- R. Qiao, A. Roberts, A. S. Mount, S. J. Klaine and P. C. Ke, Nano Lett., 2007, 7, 614 CrossRef CAS.
- C. M. Sayes, J. D. Fortner, W. Guo, D. yon, A. M. Boyd, K. D. Ausman, Y. J. Tao, B. Sitharaman, L. J. Wilson, J. B. Hughes, J. L. West and V. L. Colvin, Nano Lett., 2004, 4, 1881 CrossRef CAS.
- T. Tsuchiya, Y. N. Yamakoshi and N. Miyata, Biochem. Biophys. Res. Commun., 1995, 206, 885 CrossRef CAS.
- J. P. Kamat, T. P. A. Devasagayam, K. I. Priyadarsini, H. Mohan and J. P. Mittal, Chem.-Biol. Interact., 1998, 114, 145 CrossRef CAS.
- T. Mashino, K. Okuda, T. Hirota, M. Hirobe, T. Nagano and M. Mochizuki, Fullerene Sci. Technol., 2001, 9, 191 CAS.
- D. J. Wolff, A. D. P. Papoiu, K. Mialkowski, C. F. Richardson, D. I. Schuster and S. R. Wilson, Arch. Biochem. Biophys., 2000, 378, 216 CrossRef CAS.
- D. J. Wolff, C. M. Barbieri, C. F. Richardson, D. I. Schuster and S. R. Wilson, Arch. Biochem. Biophys., 2002, 399, 130 CrossRef CAS.
- R. A. Kotelnikova, A. I. Kotelnikov, G. N. Bogdanov, V. S. Romanova, E. F. Kuleshova, Z. N. Parnes and M. E. Voľpin, FEBS Lett., 1996, 389, 111 CrossRef CAS.
- C. Toniolo, A. Bianco, M. Mabbini, G. Scorrano, M. Prato, M. Marastoni, R. Tomatis, S. Spisani, G. Paio and D. E. Blair, J. Med. Chem., 1994, 37, 4558 CrossRef CAS.
- S. Friedman, P. Ganapathi, Y. Rubin and G. Kenyon, J. Med. Chem., 1998, 41, 2424 CrossRef CAS.
- A. S. Boutorine, H. Tokuyama, M. Takasugi, H. Isobe, E. Nakamura and C. Hélène, Angew. Chem., Int. Ed. Engl., 1995, 33, 2462 CrossRef.
- S. Marchesan, T. D. Ros, G. Spalluto, J. Balzarini and M. Prato, Bioorg. Med. Chem. Lett., 2005, 15, 3615 CrossRef CAS.
- T. Mashino, K. Shimotohno, N. Ikegami, D. Nishikawa, K. Okuda, K. Takahashi, S. Nakamura and M. Mochizuki, Bioorg. Med. Chem. Lett., 2005, 15, 1107 CrossRef CAS.
- Y. W. Chen, K. C. Hwang, C. C. Yen and Y. L. Lai, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2004, 287, 21.
- A. M. Lin, T. Y. Luh, C. K. Chou and L. T. Ho, Ann. N. Y. Acad. Sci., 1999, 890, 340 CrossRef CAS.
- L. L. Dugan, J. K. Gabrielsen, S. P. Yu, T. S. Lin and D. W. Choi, Neurobiol. Dis., 1996, 3, 129 CrossRef CAS.
- Y. Liu, F. Jiao, Y. Qiu, W. Li, Y. Qu, C. X. Tian, Y. F. Li, R. Bai, F. Lao, Y. L. Zhao, Z. F. Chai and C. Y. Chen, Nanotechnology, 2009, 20, 415102 CrossRef.
- J. D. Zhu, Z. Q. Ji, J. Wang, R. H. Sun, X. Zhang, Y. Gao, H. F. Sun, Y. Y. Liu, Z. Wang, A. D. Li, J. Ma, T. C. Wang, G. Jia and Y. Q. Gu, Small, 2008, 4, 1168 CrossRef CAS.
- S. S. Ali, J. I. Hardt, K. L. Quick, J. S. Kim-Han, B. F. Erlanger, T. T. Huang, C. J. Epstein and L. L. Dugan, Free Radical Biol. Med., 2004, 37, 1191 CrossRef CAS.
- F. Lao, L. Chen, W. Li, C. C. Ge, Y. Qu, Q. M. Sun, Y. L. Zhao, D. Han and C. Y. Chen, ACS Nano, 2009, 3, 3358 CrossRef CAS.
- S. Foley, C. Crowley, M. Smaihi, C. Bonfils, B. F. Erlanger, P. Seta and C. Larroque, Biochem. Biophys. Res. Commun., 2002, 294.
- W. Li, C. Y. Chen, C. Ye, T. T. Wei, Y. L. Zhao, F. Lao, Z. Chen, H. Meng, Y. X. Gao, H. Yuan, G. M. Xing, F. Zhao, Z. F. Chai, X. J. Zhang, F. Y. Yang, D. Han, X. H. Tang and Y. G. Zhang, Nanotechnology, 2008, 19, 145102 CrossRef.
- C. Ye, C. Y. Chen, Z. Chen, H. Meng, L. Xing, Y. X. Jiang, H. Yuan, G. M. Xing, F. Zhao, Y. L. Zhao, Z. F. Chai, X. H. Fang, D. Han, L. Chen, C. Wang and T. T. Wei, Chin. Sci. Bull., 2006, 51, 1060 CrossRef CAS.
- T. Y. Zakharian, A. Seryshev, B. Sitharaman, B. E. Gilbert, V. Knight and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 12508 CrossRef CAS.
- C. Burger and B. Chu, Colloids Surf., B, 2007, 56, 134 CrossRef CAS.
- R. Maeda-Mamiyaa, E. Noirib, H. Isobec, W. Nakanishic, K. Okamotob, K. Doib, T. Sugayad, T. Izumie, T. Hommaa and E. Nakamura, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5339 CrossRef.
- H. Meng, G. M. Xing, B. Y. Sun, F. Zhao, H. Lei, W. Li, Y. Song, Z. Chen, H. Yuan, X. X. Wang, J. Long, C. Y. Chen, X. J. Liang, N. Zhang, Z. F. Chai and Y. L. Zhao, ACS Nano, 2010, 4, 2773 CrossRef CAS.
- S. H. Friedman, D. L. Decamp, R. P. Sijbesma, G. Srdanov, F. Wudl and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6506 CrossRef CAS.
- R. Sijbesma, G. Srdanov, F. Wudl, J. A. Castoro, C. Wilkins, S. H. Friedman, D. L. Decamp and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6510 CrossRef CAS.
- M. C. Tsai, Y. H. Chen and L. Y. Chiang, J. Pharm. Pharmacol., 1997, 49, 438 CAS.
- M. Bisaglia, B. Natalini, R. Pellicciari, E. Straface, W. Malorni, D. Monti, C. Franceschi and G. Schettini, J. Neurochem., 2000, 74, 1197 CrossRef CAS.
- H. S. Lai, W. J. Chen and L. Y. Chiang, World J. Surg., 2000, 24, 450 CrossRef CAS.
- T. Sun, Z. Jia and Z. Xu, Bioorg. Med. Chem. Lett., 2004, 14, 1779 CrossRef CAS.
- J. R. Heath, S. C. ÓBrien, Y. Liu, R. F. Curl, H. W. Kroto, F. K. Title and R. E. Smalley, J. Am. Chem. Soc., 1985, 107, 7779 CrossRef CAS.
- Y. Chai, T. Guo, C. Jin, R. E. Haufler, L. P. F. Chibante, J. Fure, L. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95, 7564 CrossRef CAS.
- M. D. Diener and J. M. Alford, Nature, 1998, 393, 668 CrossRef CAS.
- D. W. Cagle, S. J. Kennel, S. Mirzadeh, J. M. Alford and L. J. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5182 CrossRef CAS.
- R. D. ÉTóthBolskar, A. Borel, G. González, L. Helm, A. E. Merbach, B. Sitharaman and L. J. Wilson, J. Am. Chem. Soc., 2005, 127, 799 CrossRef CAS.
- S. Zhang, D. Sun, X. Li, F. Pei and S. Liu, Fullerene Sci. Technol., 1997, 5, 1635 CAS.
- Y. Iiduka, T. Wakahara, K. Nakajima, T. Nakahodo, T. Tsuchiya, Y. Maeda, T. Akasaka, K. Yoza, M. T. H. Liu, N. Mizorogi and S. Nagase, Angew. Chem., Int. Ed., 2007, 46, 5562 CrossRef CAS.
- H. Nikawa, T. Kikuchi, T. Wakahara, T. Nakahodo, T. Tsuchiya, G. M. A. Rahman, T. Akasaka, Y. Maeda, K. Yoza, E. Horn, K. Yamamoto, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 9684 CrossRef CAS.
- T. Akasaka, S. Nagase, K. Kobayashi, T. Suzuki, T. Kato, K. Kikuchi, Y. Achiba, K. Yamamoto, H. Funasaka and T. Takahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2139 CAS.
- R. D. Bolskar, A. F. Benedetto, L. O. Husebo, R. E. Price, E. F. Jackson, S. Wallace, L. J. Wilson and J. M. Alford, J. Am. Chem. Soc., 2003, 125, 5471 CrossRef CAS.
- P. P. Fatouros, F. D. Corwin, Z. J. Chen, W. C. Broaddus, J. L. Tatum, B. Kettenmann, Z. X. Ge, H. W. Gibson, J. L. Russ, A. P. Leonard, J. C. Duchamp and H. C. Dorn, Radiology, 2006, 240(3), 756 Search PubMed.
- C. Y. Shu, L. H. Gan, C. R. Wang, X. L. Pei and H. B. Han, Carbon, 2006, 44, 496 CrossRef CAS.
- D. M. Yue, X. J. Bai, S. X. Zhao, X. P. Miao, M. X. Li, J. Q. Dong, K. Ibrahim, J. O. Wang, Y. L. Zhao, H. Yuan, G. M. Xing and B. Y. Sun, J. Phys. Chem. C, 2010, 114, 7631 CrossRef CAS.
- J. Tang, G. M. Xing, Y. L. Zhao, L. Jing, X. F. Gao, Y. Cheng, H. Yuan, F. Zhao, Z. Chen, H. Meng, H. Zhang, H. J. Qian, R. Su and K. Ibrahim, Adv. Mater., 2006, 18, 1458 CrossRef CAS.
- J. Tang, G. M. Xing, Y. L. Zhao, L. Jing, H. Yuan, F. Zhao, X. Y. Gao, H. J. Qian, R. Su, K. Ibrahim, W. G. Chu, L. N. Zhang and K. Tanigaki, J. Phys. Chem. B, 2007, 111, 11929 CrossRef CAS.
- J. Tang, G. M. Xing, F. Zhao, H. Yuan and Y. L. Zhao, J. Nanosci. Nanotechnol., 2007, 7, 1085 CrossRef CAS.
- K. Kobayashi, M. Kuwano, K. Sueki, K. Kikuchi, Y. Achiba, H. Nakahara, N. Kananishi, M. Watanabe and K. Tomura, J. Radioanal. Nucl. Chem., 1995, 192, 81 CrossRef CAS.
- J. J. Yin, F. Lao, J. Meng, P. P. Fu, Y. L. Zhao, G. M. Xing, X. Y. Gao, B. Y. Sun, P. C. Wang, C. Y. Chen and X. J. Liang, Mol. Pharmacol., 2008, 74, 1132 CrossRef CAS.
- M. Mikawa, H. Kato, M. Okumura, M. Narazaki, Y. Kanazawa, N. Miwa and H. Shinohara, Bioconjugate Chem., 2001, 12, 510 CrossRef CAS.
- L. J. Wilson, D. W. Cagle, T. P. Thrash, S. J. Kennel, S. Mirzadeh, J. M. Alford and G. J. Ehrhardt, Coord. Chem. Rev., 1999, 190–192, 199 CrossRef CAS.
- T. P. Thrash, D. W. Cagle, J. M. Alford, K. Wright, G. J. Ehrhardt, S. Mirzadeh and L. J. Wilson, Chem. Phys. Lett., 1999, 308, 329 CrossRef CAS.
- K. Akiyama, Y. L. Zhao, K. Sueki, K. Tuskada, H. Haba, Y. Nagame, S. Suzuki, T. Otuski, M. Sakaguchi, K. Kikuchi, M. Katada and H. Nakahara, J. Am. Chem. Soc., 2001, 123, 181 CrossRef CAS.
- E. B. Lezzi, J. C. Duchamp, K. R. Fletcher, T. E. Glass and H. C. Dorn, Nano Lett., 2002, 2, 1187 CrossRef CAS.
- T. Wharton and L. J. WilsonBioorg, Bioorg. Med. Chem., 2002, 10, 3545 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- K. S. Novoselov, E. McCann, S. V. Morozov, V. I. Falko, M. I. Katsnelson, A. K. Geim, F. Schedin and D. Jiang, Nat. Phys., 2006, 2, 177 CrossRef.
- J. S. Bunch, A. M. van der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490 CrossRef CAS.
- K. S. Subrahmanyam, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2008, 18, 1517 RSC.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. C. Brodie, Ann. Chim. Phys., 1860, 59, 466 Search PubMed.
- S. Gilje, S. Han, M. Wang, K. L. wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201 CrossRef CAS.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872 CrossRef CAS.
- M. Yan, S. X. Cai and J. F. W. Keana, J. Org. Chem., 1994, 59, 5951 CrossRef CAS.
- A. Hirsch and O. Vostrowsky, Top. Curr. Chem., 2005, 245, 193 CAS.
- L. H. Liu and M. Yan, Nano Lett., 2009, 9, 3375 CrossRef CAS.
- A. Devadoss and C. E. D. Chidsey, J. Am. Chem. Soc., 2007, 129, 5370 CrossRef CAS.
- M. K. Bayazit and K. S. Coleman, J. Am. Chem. Soc., 2009, 131, 10670 CrossRef CAS.
- X. L. Li, G. Y. Zhang, X. D. Bai, X. M. Sun, X. R. Wang, E. Wang and H. J. Dai, Nat. Nanotechnol., 2008, 3, 538 CrossRef CAS.
- X. L. Li, X. R. Wang, L. Zhang, S. Lee and H. J. Dai, Science, 2008, 319, 1229 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS.
- D. A. Areshkin and C. T. White, Nano Lett., 2007, 7, 3253 CrossRef CAS.
- D. B. Farmer, R. Golizadeh-Mojarad, V. Perebeinos, Y. M. Lin, G. S. Tulevski, J. C. Tsang and P. Avouris, Nano Lett., 2009, 9, 388 CrossRef CAS.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2008, 8, 323 CrossRef CAS.
- Y. B. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. R. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181 CrossRef CAS.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785 CrossRef CAS.
- Y. Wang, Z. H. Li, D. H. Hu, C. T. Lin, J. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274 CrossRef CAS.
- F. Li, Y. Huang, Q. Yang, Z. T. Zhong, D. Li, L. H. Wang, S. P. Song and C. H. Fan, Nanoscale, 2010, 2, 1021 RSC.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469 CrossRef CAS.
- X. M. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. J. Dai, Nano Res., 2008, 1, 203 CrossRef CAS.
- F. Chen, Q. Qing, J. L. Xia, J. H. Li and N. J. Tao, J. Am. Chem. Soc., 2009, 131, 9908 CrossRef CAS.
Footnote |
| † Equal contribution |
| This journal is © The Royal Society of Chemistry 2011 |