Controlled formation of calcium-phosphate-based hybrid mesocrystals by organic–inorganic co-assembly
Halei
Zhai
a,
Xiaobin
Chu
a,
Li
Li
a,
Xurong
Xu
ab and
Ruikang
Tang
*ab
aCentre for Biomaterials and Biopathways, Zhejiang University, Hangzhou, Zhejiang 310027, China. E-mail: rtang@zju.edu.cn; Fax: +86-571-87953736; Tel: +86-571-87953736
bState Key Laboratory of Silicon Materials, Zhejiang University, Hangzhou, Zhejiang 310027, China
First published on 13th October 2010
Abstract
An understanding of controlled formation of biomimetic mesocrystals is of great importance in materials chemistry and engineering. Here we report that organic–inorganic hybrid plates and even mesocrystals can be conveniently synthesized using a one-pot reaction in a mixed system of protein (bovine serum albumin (BSA)), surfactant (sodium bis(2-ethylhexyl) sulfosuccinate (AOT)) and supersaturated calcium phosphate solution. The morphologies of calcium-phosphate-based products are analogous to the general inorganic crystals but they have abnormal and interesting substructures. The hybrids are constructed by the alternate stacking of organic layer (thickness of 1.31 nm) and well-crystallized inorganic mineral layer (thickness of 2.13 nm) at the nanoscale. Their morphologies (spindle, rhomboid and round) and sizes (200 nm–2 μm) can be tuned gradually by changing BSA, AOT and calcium phosphate concentrations. This modulation effect can be explained by a competition between the anisotropic and isotropic assembly of the ultrathin plate-like units. The anisotropic assembly confers mesocrystal characteristics on the hybrids while the round ones are the results of isotropic assembly. However, the basic lamellar organic–inorganic substructure remains unchanged during the hybrid formation, which is a key factor to ensure the self-assembly from molecule to micrometre scale. A morphological ternary diagram of BSA–AOT–calcium phosphate is used to describe this controlled formation process, providing a feasible strategy to prepare the required materials. This study highlights the cooperative effect of macromolecule (frame structure), small biomolecule (binding sites) and mineral phase (main component) on the generation and regulation of biomimetic hybrid mesocrystals.
Introduction
Scientists are eager to mimic nature's ability to design functional materials whose properties are often superior to the synthetic ones. In nature, biominerals are widely produced by bacteria, protists, plants, invertebrates and vertebrates, including humankind.1 These biological materials are featured by a smart combination of multi-components especially in the form of integrated organic–inorganic hybrid materials, in which the organic parts are often proteins and low-molecular-mass molecules.2 They are constructed by using organic components to control the nucleation, growth, organization and transformation of inorganic phases. Interactions between organic and inorganic phases at the molecular level, although complex, are common occurrences to determine the size, shape, and properties of the resulting products.1,3 Different from the synthesized ones, the functions of biominerals depend to a large extent on the ordered association of biomolecules with mineral phases. The organized hybrid materials, unlike the single components, can be tailored into different compositions and morphologies, e.g. bone,4 tooth5 and mollusc shells6etc., to ensure the optimal mechanical and physicochemical characteristics.The controls that determine the sizes, shapes, and properties of crystals are a key to addressing numerous challenges in material designs and applications. It has been revealed that organic molecules can influence the shape and properties of inorganic crystals.7 However, it is difficult for the two distinct organic and inorganic phases to spontaneously assemble into highly ordered structures. In living organisms, biological mineralization is able to combine particular building blocks or entities into functional hybrid composites. An understanding of these biochemical controls is essential and important, not only to study biomineralization mechanisms further, but also to design novel hybrid materials and processing technologies. Despite the complicated hierarchical structures of biominerals, their basic building blocks are frequently the nano-sized organic–inorganic composites.8 Therefore, an ordered and periodic assembly of organic and inorganic nanophases at the nanoscale is crucial to biomimetically synthesize hybrid materials. But, how can we design ordered hybrid composites and how can we conveniently control their structures, sizes and morphologies under mild conditions?
Although organic–inorganic hybrid materials have been approached by various methods such as layer-by-layer (LbL)9 and template-directed crystallization,10 the bottom-up fabrication from ions or molecules is still a great challenge in the laboratory since the control of periodic deposition is difficult to achieve at hierarchical scales. In conventional biomimetic crystallization studies, organic molecules, which act as structure-directing agents, modulate the crystal morphology by their selective absorption onto crystal faces, altering crystal facet stability and growth kinetics.7,11 Recently, a non-classical crystal growth pathway based upon nano assembly has received considerable attention.12 The nanoparticles, which are directed by specific organic additives, can act as the basic building units to assemble into superstructures or mesocrystals. During such a process, the organic molecules (especially macromolecules) selectively absorb and interact with primary nanocrystals. The assembly process follows programmed arrangement into high order hybrid structures.13 The morphology can be tuned by varying the interactions between different organic and inorganic phases. However, the one step bottom-up process, which starts from the molecular level rather than from preformed nanoparticle precursors, may be readily able to control the orientation and order of assembly processes to form integrated hybrid nanocomposite. But this strategy requires a precise and spontaneous co-assembly of both organic and inorganic phases alternately at both the molecular level and the nanoscale.14
In this paper, we reported an easy but effective method for direct synthesis of organic–inorganic hybrid mesocrystals by a emergent co-assembly process of protein (bovine serum albumin (BSA)) and surfactant (sodium bis(2-ethylhexyl) sulfosuccinate (AOT)) in a supersaturated calcium phosphate solution. The calcium-phosphate-based hybrid crystals with lamellar structure have different properties from conventional ones. Here we emphasize that the size and morphology of the resulting hybrids could be regulated readily by varying BSA, AOT and calcium phosphate concentrations according to a suggested morphological ternary diagram. This study provided a novel pathway to one-pot preparation of functional hybrid crystal materials with tuneable size and morphologies by organic–inorganic co-assembly.
Results and discussion
It is believed that functional organic molecules can interact with calcium species at the organic–inorganic interfaces to modulate the growth and assemble of inorganic crystals. BSA is one of the most studied proteins but this biological macromolecule is not an effective modifier in calcium phosphate crystallization.15 It has been previously confirmed that the interaction between BSA and calcium or phosphate ions in aqueous solutions is poor.16 BSA itself is inert in mineral deposition. In contrast, many surfactant molecules are widely used as effective promoters and templates in biomimetic calcium mineralization since their hydrophilic groups (especially the sulfonate and carboxylate groups) provide active binding sites to calcium ions. AOT is one among typical agents that can modulate calcium phosphate precipitation significantly. AOT molecules have a strong binding effect with calcium ions due to their highly charged -SO32− groups.16,17 However, hierarchical or complicated biomineral-like structures cannot be achieved by using this small molecule due to the lack of higher-order structures. In our control experiments, only poor crystalline HAP was obtained if BSA was added into the supersaturated calcium phosphate solutions; AOT alone produced the conventional rod-like HAP crystals without any organized hybrid structure. These results matched the previous studies and understandings well. However, the cooperative effect of BSA and AOT in the calcium phosphate solution could lead to the formation of unique hybrids in a one-pot reaction.Under an experimental condition of 2 mM AOT, 1 mg ml−1 BSA and 1.25 mM calcium ions (the molar ratio of calcium to phosphate was fixed at 1.67 in all experiments), the uniform rhombic plates precipitated spontaneously as shown by scanning electron microscopy (SEM, Fig. 1(A)). Their size distribution was homogeneous. The typical rhombic plates were 1.23 ± 0.21 and 0.91 ± 0.18 μm along their long and short axes, respectively (statistical results from ∼100 plates); the aspect ratio was about 1.4. The thickness of the plates was 130 ± 20 nm. These rhombic plates had exactly same morphology (Fig. 1(B)) and this characteristic was similar to the general inorganic crystals. However, the chemical compositions of the obtained plates were relatively complicated. Besides the elements of calcium and phosphorus, the element of sulfur was detected in the solids by using energy-dispersive X-ray spectroscopy (EDS). This result indicated the presence of AOT (-SO32−) in the hybrid plates. It was also revealed that inorganic part in the plates was a kind of calcium phosphate minerals with Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P molar ratio of 1.5–1.6. The coexistence of organic–inorganic components was also confirmed by Fourier transform infrared spectroscopy (FT-IR, Fig. 1(C)). The peaks at 1737, 1459 and 1419 cm−1 were the characteristic signals of AOT, while the bands at 1656 (amide I) and 1555 cm−1 (amide II) showed the involvement of BSA in the solids.18 The broad peaks at 1022 and 564 cm−1 were assigned to the inorganic phosphate groups.19 Thermogravimetric analysis (TGA) showed that the mineral phase was the main composition in the solids. The weight loss of 38% between 100 and 500 °C was corresponded predominantly to removal of the organic phase, while the weight contents of the inorganic phases were 62%. In addition, the plates became ‘crimped-paper’-like after calcinations at 500 °C in air for 2 h. Without the organic frame, the solids became brittle and the structures were collapsed readily into small pieces under an ultrasonic condition. Many previous studies suggested that the organic compounds play a regulation role in inorganic mineralization rather than being involved in structural recombination. However, the current results implied that BSA and AOT were the key components in the hybrid construction. Thus, these solids were different from the other precipitated inorganic crystals in the presence of organic additives.
:P molar ratio of 1.5–1.6. The coexistence of organic–inorganic components was also confirmed by Fourier transform infrared spectroscopy (FT-IR, Fig. 1(C)). The peaks at 1737, 1459 and 1419 cm−1 were the characteristic signals of AOT, while the bands at 1656 (amide I) and 1555 cm−1 (amide II) showed the involvement of BSA in the solids.18 The broad peaks at 1022 and 564 cm−1 were assigned to the inorganic phosphate groups.19 Thermogravimetric analysis (TGA) showed that the mineral phase was the main composition in the solids. The weight loss of 38% between 100 and 500 °C was corresponded predominantly to removal of the organic phase, while the weight contents of the inorganic phases were 62%. In addition, the plates became ‘crimped-paper’-like after calcinations at 500 °C in air for 2 h. Without the organic frame, the solids became brittle and the structures were collapsed readily into small pieces under an ultrasonic condition. Many previous studies suggested that the organic compounds play a regulation role in inorganic mineralization rather than being involved in structural recombination. However, the current results implied that BSA and AOT were the key components in the hybrid construction. Thus, these solids were different from the other precipitated inorganic crystals in the presence of organic additives.
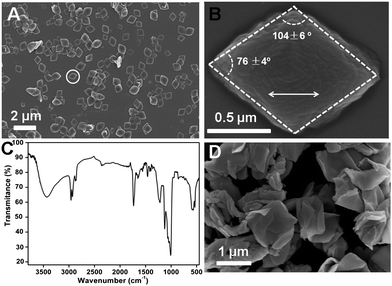 | ||
| Fig. 1 (A) SEM image of the rhombic plates. (B) Enlarged image of the rhombic plate in the white circle; the double-headed arrow shows the extended orientation. (C) FT-IR pattern of the products. (D) The rhombic plates after calcination at 500 °C in air. | ||
The resulting rhombic plates shared the same size and anisotropic morphology similar as general inorganic crystals. However, in-depth examination revealed that they were distinct from the conventional calcium phosphate crystals.20 The rhombic plates were examined by wide angle X-ray diffraction (WAXD, Fig. 2(A)) and as expectated, the crystalline HAP-like calcium phosphate phase was detected. The WAXD pattern was very similar to that of pure HAP but small peak shifts were also observed. We suggested that the binding effect between the organic component and calcium ions would cause the lattice distortion. The lattice structure of the inorganic phase could be revealed at the atomic scale by using high resolution transmission electron microscopy from a top view of the plates (HRTEM, Fig. 2(B)). This image represents a typical ultrathin inorganic crystal layer embedded in the rhombic plates. However, another independent set of diffraction peaks was found in the X-ray diffraction (XRD) pattern, which revealed that a superstructure was present in the hybrids. The characteristic peaks of lamellar structure (interspacing distance, d = 3.43 nm) could be found from both small angle X-ray diffraction (SAXD) and WAXD (Fig. 2(A)), indicating an ordered arrangement of subunits along a crystallographic direction rather than a simple mixture of the organic and inorganic phases. A side view of the ultra-thin sectioned samples under transmission electron microscopy (TEM, Fig. 2(C)) confirmed the internal structure: the organic layers (light, 1.31 nm) and the inorganic layers (dark, 2.13 nm) alternately stacked at the nanoscale to form the compact hybrid structure. Thus, the organic molecules (BSA and AOT) were well organized to form the layered organic phase. Each organic–inorganic ultra-thin unit had a thickness of 3.44 nm, which agreed with the XRD data, 3.43 nm, and the individual inorganic layer was a calcium phosphate crystal plate with a thickness of only 2.13 nm. These nanoplates acted as the building blocks that could self-assemble together with the organic layers to generate the lamellar complex. Additionally, a wave-like superficial texture of the hybrids could be observed (Fig. 1(B)) and the profiles were similar to the hybrid crystal morphology. This phenomenon indicated that the assembly might be an anisotropic process.
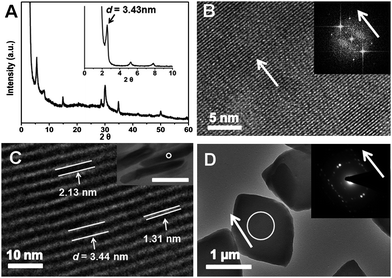 | ||
| Fig. 2 (A) WAXD and SAXD (insert) patterns of the rhombi; (B) HRTEM of a rhombus (top view). Insert: FFT simulation result; (C) TEM image of ultra-thin sectioned rhomb from side view. The values of 2.13, 1.31 and 3.44 nm corresponded to the thicknesses of inorganic (dark), organic (light) and organic–inorganic complex layers, respectively. Insert: TEM image of the side view of the ultra-thin sections of the plates, bar is 0.5μm. (C) is the enlargement of the region within the white circle; (D) TEM image of the hybrids. Insert was the SEAD pattern (white circle area). The HRTEM image in (B) was also obtained on the same area by the in situ technique. Arrows showed that each individual inorganic plate in the hybrid shared the same crystallographic orientation, which was the long axis of the rhombus. | ||
In order to understand the orientation of each inorganic layer, selected area electron diffraction study (SAED, Fig. 2(D)) was applied. It was noted that the anisotropic diffraction dots rather than the isotropic diffraction rings were obtained during the examination of a whole rhombic crystal, which represented a similar characteristic of single crystal. It was interesting that the orientation reflected by these dots (arrow in the insert image) was exactly same as the long axis of the examined rhombic crystal. Such a coincidence implied that all the ultrathin inorganic crystal layers within the hybrid plates should share the same crystallographic orientation. Additionally, the experimental diffractions dots of the whole crystal were almost same as the fast Fourier transform (FFT) result (Fig. 2(B)) of an individual crystal layer. Therefore, the formed hybrid crystal exhibited similar features to a single crystal; however, it had additional superlattice structure. Since the rhombic plates had a specific morphology while they were not constructed as the conventional single crystals, these hybrids could be considered as a kind of artificial mesocrystal.12,21 However, the imperfect dots on Fig. 2 (D) might indicate that the misaligned orientation still occurred during nano assembly. Since the material was constructed by ultrathin calcium phosphate units, it was interesting that flexible and elastic features were conferred onto the mesocrystal along the lamellar packing direction in spite of that; its main composition was a brittle ceramic phase. These mechanical properties of the hybrids had been characterized by our previous study,16 demonstrating the advantages of organized assembly for formation of mesocrystals in material functionalization.
The convenient control of the size and morphology of the organic–inorganic hybrids and mesocrystals is a challenge, although those for single hybrid crystals are nowadays sophisticated. In our experiments, the calcium phosphate–BSA–AOT hybrid mesocrystals with different size and morphology could be feasibly regulated within a simple reaction system by changing the reactant concentrations. We fixed BSA and calcium concentration at 0.5 mg ml−1 and 1.25 mM, respectively. When the AOT concentration was 1.00 mM, the obtained hybrid plates were not rhombic plates any more. Their shapes became spindle-like. The hybrid plates changed into a round shape when the AOT concentration was increased to 4.00 mM. However, the further decreasing or increasing of AOT concentration result into the disappearance of the co-assembly or hybrid in the system. In this experiment, their morphologies were gradually adjustable from spindle, to rhombus to round by increasing the AOT concentration from 1.00 to 4.00 mM (Fig. 3). During the evolution process, the length along the short axis of the formed hybrid plates did not change significantly, it was maintained at 300–400 nm. However, the long axis kept on decreasing from 1.50 μm to 300–400 nm with increasing AOT concentration. Accordingly, the hybrid morphology became isotropic. This phenomenon implied that AOT component was an important factor to control the a degree of anisotropic co-assembly of the hybrids.
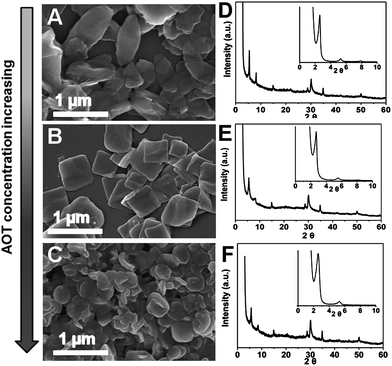 | ||
| Fig. 3 SEM images of the hybrids synthesised at AOT concentrations of 1.00 (A), 2.00 (B) and 4.00 mM (C). (D)–(F) are the corresponding XRD patterns of (A)–(C), respectively. | ||
Although the morphologies and sizes of the resulted hybrid plates were influenced remarkably by the changing of AOT concentrations in the reaction solutions, the internal organic–inorganic subunit remained. The WAXD and SAXD patterns of the spindles, rhombi and rounds were exactly same without any change. But the misalignments of each individual inorganic layer in the hybrid increased with the increasing of AOT concentration. The crystallographic mismatch of the inorganic layers could be examined by using SEAD. During the evolution from the regular rhombi to round shapes, the diffraction dots disappeared gradually while the diffraction rings existed (Fig. 4). This tendency indicated that the preferred orientation of the thin calcium phosphate planes in the hybrid was weakened. Although AOT itself could result in aggregates in solution to induce calcium mineralization, the aggregation was simple and isotropic due to the lack of complicated configuration. Therefore, it was reasonable that the excessive AOT could destroy the anisotropic assembly of the ultrathin mineral plates in the hybrid rhombi. Although the inorganic and organic layers were still packed layer-by-layer strictly along the thickness direction, the crystallographic directions of the inorganic crystal planes in the hybrids became disordered. The anisotropic assembly transformed the orientation of the long axes into the isotropic mode with increasing AOT concentration; thus, the round plates were finally yielded at 4.00 mM AOT and the hybrid was not mesocrystalline any more. Besides, it should be mentioned that the percentages of organic and inorganic contents in the hybrid solids was not changed significantly during the morphology modulation; in which the inorganic content was kept within a range of 69–72% from the spindles to the rounds.
 | ||
| Fig. 4 During the morphology change from rhombus (A) to round (B), anisotropic diffraction dots became isotropic rings in the corresponding SEAD pattern. | ||
Besides the AOT concentrations, the formation of hybrid crystals could be also adjustable by BSA concentration. In this examination, the concentrations of AOT and calcium were maintained at 2 mM and 1.25 mM, respectively, and the BSA concentrations were increased from 0.25 to 2.00 mg ml−1. It was noted that the morphologies of hybrid plates underwent another gradual evolution from the irregular quadrilaterals to rhombi and then to plump spindles (Fig. 5). The sizes and aspect ratios of the hybrids increased from 200 nm to 2 μm and 1.1 to 2.0, respectively, during the modulation. Although the hybrid width increased along the short axis, the more extended length along the long axis indicated that the anisotropy assembly process was affected significantly by the protein concentration. It was noticed that in biomineralization, the complicated hierarchical building structures of biominerals are frequently contributed by the ordered aggregates of proteins. Again, the basic organic–inorganic units and their ordered packing behaviours were not changed during the morphology and size regulations. It was mentioned that, when the BSA concentration increased, the role of AOT in the synthesis decreased. Therefore, the ratio or the cooperative effect of BSA and AOT was another key factor in mesocrystal formation and regulation.
 | ||
| Fig. 5 SEM images of the hybrids at BSA concentration of 0.25 (A), 1.13 (B) and 2.00 mg ml−1 (C). | ||
It was known that the co-assembly could not occur in the absence of the inorganic phase. Thereby, it was reasonable that the concentrations of calcium and phosphate could control the mesocrystal formation too (Fig. 6). Under BSA and AOT concentrations of 1.00 mg ml−1 and 2.00 mM, respectively, the resulting rhombi shared the same intermediate state with increasing calcium and phosphate concentration in the reaction solution. If calcium concentration was decreased to 0.63 mM, the poly-dispersed quadrilaterals-like plates (size of 400–800 nm) formed with the small aspect ratio of 1.1. If the concentration was increased to 2.50 mM, the slender spindle-like plates were obtained and their size distribution was 1.8–2.3 μm with an aspect ratio was 2.5. From the evolution from quadrilaterals, rhombi to slender spindles, it could be seen that the anisotropic co-assembly process was enhanced.
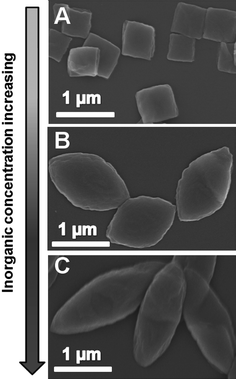 | ||
| Fig. 6 SEM images of the hybrids at calcium concentration of 0.63 (A), 1.56 (B) and 2.50 mM. In all experiments, the ratio of calcium to phosphate in the reaction solution was maintained at 1.67. | ||
The previous studies of biomimetic fabrication of hybrid materials with artificial molecules such as peptide-amphiphile,22 block copolymer,23 and amphiphilic dendro-calixarene,24 suggested that the specific sites and sterically constrained effect may control the assembly of the organic template and then the size and morphology of the final hybrid materials. Different from the above-mentioned understanding, under our experimental conditions, the change of BSA and AOT concentrations were directly related to the different modification state of BSA. The BSA protein, which was constituted by a single chain of 583 amino acid residues, acted as a stable and relatively rigid fragment connected with the special motif (AOT aggregates).25 The hydrophilic groups of aggregates exposed to aqueous solutions and their configuration can be adjusted. The highly charged group (–SO32−) in AOT could greatly interact with calcium ions and then modulated calcium phosphate precipitation significantly, which had been demonstrated experimentally in many works and in our previous paper.16,26 However, the binding ability of BSA with calcium ions is weak and the controlling effect on the mineral formation is relatively poor. As a result, BSA acted as structural frame while the AOT aggregates provided the nucleation sites of mineral during the co-assembly process. In the current study, BSA macromolcules combined with smaller AOT molecules to form a BSA–AOT complex and such a modified protein could effective control the crysallization and assembly of the calcium phosphate mineral. To some extent, this method provides an efficient way to turn a non-mineralization protein into a mineralization protein by using surfactants. The conformation of the macromolecules restricts the assembly only along certain specific directions. However, the larger concentration of AOT is accompanied by an increase in the amount and size of AOT aggregates, offering more sites for the assembly process.27 As a result, the controlling effect from the protein was counteracted and the assembly process could happen at more directions to form the isotropic rounds. Furthermore, increasing the amount or the relative amount of BSA concentrations partly restricted the assembly process in specific preferential orientations by spatial configuration to form the anisotropic hybrids or mesocrystals.28 Thus, the co-assembly process preferred to occur in certain directions, especially along the long axis of the hybrid plates rather than the short axis. Although the short axis partly extended under some experimental cases, the greatly increase along the long axis resulted into the spindles-like mesocrystal formation. The competitive controlling effect of BSA and AOT led to the transformation of an isotropic and anisotropic assembly process during hybrid crystal construction. Thus, the formation of different hybrids and mesocrystals with tuneable size and morphologies could be achieved.
An anisotropic co-assembly process could also be promoted by increasing the mineral ion concentrations. In the formation process of mesocrystals, the inorganic precursor controlled the size and morphology of the final product by tuning the amounts, size and shapes of the nano-sized building blocks.29 Under our experimental conditions, the controlling role of mesocrystal growth became dominant in greater saturation to decide the product size and structure. As the preferred orientation of the calcium phosphate crystal plates is parallel to the long axis of the rhombic plates, the fast growth of the calcium phosphate plate crystals along this preferred orientation promoted the formation of the slender spindle-like plates with larger aspect ratios during the co-assembly process. However, the interaction between BSA-AOT complex and calcium phosphate crystal was also responsible for the co-assembly of the organic and inorganic phase to form highly ordered hybrid materials and maintain their internal structure.
Actually, the generation of hybrid material via the cooperative effect of macromolecules (mainly proteins), small biomolecules and the mineral phase is a common strategy in natural biomineralization.30 In the biological construction, high-molecular-weight macromolecules, such as collagen, act as support matrix to provide a structural frame for the mineralization, the biomineralization proteins themselves have nucleation sites but most matrices receive mineralization function by binding and stabilizing functional motifs that are carboxylate- or sulfonate-rich. Thus, the combination of organic–inorganic mineralization interfaces and the organized organic matrices can concentrate the mineral ions to induce the deposition as well as to regulate the size, morphology and orientation of the inorganic building blocks to form integrated organic–inorganic hybrid composites with complicated structure. We suggest that in this system, BSA is the structural frame to control the anisotropic assembly; the adsorption of AOT onto BSA enhances the mineralization ability of the protein; and the mineral acts as an inorganic conjunction phase to solidify the organic–inorganic hybrid structure. In the experiments, the increase of BSA promoted the formation of larger hybrid plates with increased aspect ratio, while AOT exhibited the opposite controlling effect. The increasing of inorganic concentrations preferred the formation of slender hybrid plates with a larger size. In order to show the controlling effect of the reactant concentrations, the simplified morphological maps in the form of solution ternary diagrams was proposed (Fig. 7). The biomimetic formation hybrid and mesocrystals could be yielded in the grey region. In the specific regions, the formed hybrid plates had a similar size and morphology. From points A to B, the increase of aspect ratio was preferred as the hybrid rounds transformed into the spindle ones. Since the anisotropic assembly behaviour was enhanced, this evolution implied that the resulting mesocrystals became more organized and the mismatch degrees of the inorganic layers in the hybrids could be reduced. From points A to C, both the size and aspect ratio of the resulted hybrids were increased and their morphologies were changed from rounds to spindles. From points B to C, the hybrids turned from wide spindles to slender spindles with increased size and aspect ratio too. By using this morphological ternary diagram, we could design readily hybrids and mesocrystals with the required size and morphology.
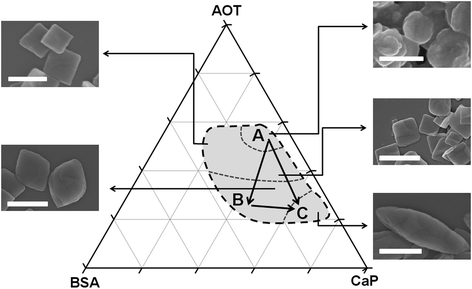 | ||
| Fig. 7 Controlled synthesis of hybrids by a morphological ternary diagram. The co-assembly occurred within the grey area and the formation of mesocrystals was preferred in its left and bottom sections. The typical morphology of the final products were also demonstrated. Bar = 1μm. | ||
Conclusions
We demonstrate that the ordered and uniform hybrids or mesocrystals can be biomimetically synthesized by the co-assembly of proteins, small functional molecules and minerals using a simple one-pot reaction. Their size distributions and morphologies can be adjusted by varying the component concentration in reaction solutions. The anisotropic co-assembly of the BSA–AOT complex and ultrathin calcium phosphate crystal plates is a key to the control of mesocrystal formation. A morphological ternary diagram can be used to design different hybrid materials as requireed. This work may give another inspiration to the assembly of multi components into one integrated hybrid material with a highly ordered structure. Furthermore, the bottom-up pathway of controlled fabrication may be developed as a simple and effective strategy to prepare feasibly functional hybrid and mesocrystal materials.Experimental
Materials
Triply distilled water was used in all the experiments. Ca(NO3)2 and (NH4)2HPO4 were of analytical and their solution were filtered twice using 0.22μm Millipore films prior to use. BSA (Albumin Bovine fraction V, BR, purity > 98%, LABMAX) and AOT (Aldrich) were used without any further purification.Hybrid plate preparation
Using a typical experiment as an example, 100 ml aqueous solution containing 4 mM AOT and 0.20 g BSA was mixed with 50 ml Ca(NO3)2 solution (5mM). The solution pH was adjusted to 10.0 ± 0.5 at room temperature by 3 M ammonia solution. Then 50 ml (NH4)2HPO4 solution (3 mM, pH = 10.0 ± 0.5) was added dropwise at a rate of 1.5 ml min−1. The reaction solution contained 2.00 mM AOT, 1.00 mg ml−1 BSA, 1.25 mM Ca(NO3)2 and 0.75 mM (NH4)2HPO4. The mixture was gently stirred at 30 ± 1 °C for 24 h. The precipitated solids were collected by centrifugation at 6000 rpm. The solid were washed by water for three times and were vacuum-dried at 35 ± 1 °C. In order to examine the controlling effect of reactant concentrations on hybrid formation, different concentrations of AOT, BSA and calcium phosphate ions were used and all the experimental processes were the same.Characterizations
SEM was performed by using a HITACHI S-4800 at a typical acceleration voltage of 5 kV. FT-IR spectra (Nicolet Nexus 670) were applied to analysis the hybrid compositions. WAXD and SAXD were characterized by a Rigaku D/max-2550pc with monochromatized Cu-Kα radiation; the scanning step was 0.02°. TGA was performed by a TA Instrument SDT Q600. The experiment was measured in a temperature range from room temperature to 1000 °C under nitrogen atmosphere. TEM observations were performed by a CM200UT TEM (Philips) at an acceleration voltage of 160 kV. During the ultra-thin sectioned TEM examination, rhombi were embedded in epoxy. The mixture was solidified at 80 °C for 12 h and then carefully microtomed by a Reichert-Jung Ultracut E using a diamond knife.Acknowledgements
We thank Jieru Wang, Xinting Cong, Xiaomin Tang, Yin Xu and Linshen Chen for their help with characterization, Haihua Pan and Yuan Su for discussions. This work was supported by the Fundamental Research Funds for the Central Universities, National Natural Science Foundation of China (20871102), Zhejiang Provincial Natural Science Foundation (R407087) and Daming Biomineralization Foundation.Notes and references
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, 2001 Search PubMed.
- L. Bédouet, F. Rusconi, M. Rousseau, D. Duplat, A. Marie, L. Dubost, K. Le Ny, S. Berland, J. Péduzzi and E. Lopez, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2006, 144, 532–543 CrossRef; J. L. Arias and M.a. S. Fernaćndez, Chem. Rev., 2008, 108, 4475–4482 CrossRef CAS.
- C. E. Killian and F. H. Wilt, Chem. Rev., 2008, 108, 4463–4474 CrossRef CAS; J. S. Evans, Chem. Rev., 2008, 108, 4455–4462 CrossRef CAS.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1998, 28, 271–298 CrossRef.
- S. Busch, U. Schwarz and R. Kniep, Chem. Mater., 2001, 13, 3260–3271 CrossRef CAS.
- N. Watabe, J. Ultrastruct. Res., 1965, 12, 351–370 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS.
- R. Z. Wang, Z. Suo, A. G. Evans, N. Yao and I. A. Aksay, J. Mater. Res., 2001, 16, 2485–2493 CAS; H. J. Gao, B. H. Ji, I. L. Jager, E. Arzt and P. Fratzl, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5597–5600 CrossRef CAS.
- Z. Tang, N. A. Kotov, S. Magonov and B. Ozturk, Nat. Mater., 2003, 2, 413–418 CrossRef CAS; P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80–83 CrossRef CAS.
- N. Gehrke, N. Nassif, N. Pinna, M. Antonietti, H. S. Gupta and H. Cölfen, Chem. Mater., 2005, 17, 6514–6516 CrossRef CAS; P. H. Kithva, L. Grondahl, R. Kumar, D. Martin and M. Trau, Nanoscale, 2009, 1, 229–232 RSC.
- N. A. J. M. Sommerdijk and G. d. With, Chem. Rev., 2008, 108, 4499–4550 CrossRef CAS.
- R. Q. Song and H. Cölfen, Adv. Mater., 2010, 22, 1301–1330 CrossRef CAS.
- M. Li, H. Cölfen and S. Mann, J. Mater. Chem., 2004, 14, 2269–2276 RSC.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS.
- R. I. Martin and P. W. Brown, J. Mater. Sci.: Mater. Med., 1994, 5, 96–102 CrossRef CAS; K. L. Yadav and P. W. Brown, J. Biomed. Mater. Res., 2003, 65a, 158–163 Search PubMed.
- H. Zhai, W. Jiang, J. Tao, S. Lin, X. Chu, X. Xu and R. Tang, Adv. Mater., 2010, 22, 3729–3734 CrossRef CAS.
- C. E. Fowler, M. Li, S. Mann and H. C. Margolis, J. Mater. Chem., 2005, 15, 3317–3325 RSC.
- G. Falini, S. Weiner and L. Addadi, Calcif. Tissue Int., 2003, 72, 548–554 CrossRef CAS.
- S. J. Gadaleta, E. P. Paschalis, F. Betts, R. Mendelsohn and A. L. Boskey, Calcif. Tissue Int., 1996, 58, 9–16 CrossRef CAS.
- J. Song, V. Malathong and C. R. Bertozzi, J. Am. Chem. Soc., 2005, 127, 3366–3372 CrossRef CAS; A. Ethirajan, U. Ziener, A. Chuvilin, U. Kaiser, H. Cölfen and K. Landfester, Adv. Funct. Mater., 2008, 18, 2221–2227 CrossRef CAS; Y. Zhang and J. Lu, Cryst. Growth Des., 2008, 8, 2101–2107 CrossRef CAS.
- A.-W. Xu, M. Antonietti, S.-H. Yu and H. Cölfen, Adv. Mater., 2008, 20, 1333–1338 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS; V. M. Yuwono and J. D. Hartgerink, Langmuir, 2007, 23, 5033–5038 CrossRef CAS.
- Z. H. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS; H. Wang, A. J. Patil, K. Liu, S. Petrov, S. Mann, M. A. Winnik and I. Manners, Adv. Mater., 2009, 21, 1805–1808 CrossRef CAS.
- M. Kellermann, W. Bauer, A. Hirsch, B. Schade, K. Ludwig and C. Böttcher, Angew. Chem., Int. Ed., 2004, 43, 2959–2962 CrossRef CAS.
- N. J. Turro, X. G. Lei, K. P. Ananthapadmanabhan and M. Aronson, Langmuir, 1995, 11, 2525–2533 CrossRef CAS; S. De, A. Girigoswami and S. Das, J. Colloid Interface Sci., 2005, 285, 562–573 CrossRef CAS.
- S. Sarda, M. Heughebaert and A. Lebugle, Chem. Mater., 1999, 11, 2722–2727 CrossRef CAS.
- C. K. Ober and G. Wegner, Adv. Mater., 1997, 9, 17–31 CrossRef CAS.
- H.-A. Klok, J. F. Langenwalter and S. Lecommandoux, Macromolecules, 2000, 33, 7819–7826 CrossRef CAS; X. Kong and S. A. Jenekhe, Macromolecules, 2004, 37, 8180–8183 CrossRef CAS; L. Rubatat, X. Kong, S. A. Jenekhe, J. Ruokolainen, M. Hojeij and R. Mezzenga, Macromolecules, 2008, 41, 1846–1852 CrossRef CAS; A. Saćnchez-Ferrer and R. Mezzenga, Macromolecules, 2010, 43, 1093–1100 CrossRef.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef.
- N. Kroger, R. Deutzmann, C. Bergsdorf and M. Sumper, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14133–14138 CrossRef CAS; L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |