Destabilisation of complex hydrides through size effects†
Meganne
Christian
and
Kondo-Francois
Aguey-Zinsou
*
School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: f.aguey@unsw.edu.au; Tel: +61 (0)2 938 57970
First published on 1st October 2010
Abstract
Nanoparticles of NaAlH4, LiAlH4 and LiBH4 were prepared by encapsulating their respective hydrides within carbon nanotubes by a wet chemical approach. The resulting confinement had a profound effect on the overall hydrogen storage properties of these hydrides, with NaAlH4 and LiAlH4 releasing hydrogen from room temperature, for example.
Complex hydrides such as light metal aluminium hydrides and borohydrides are promising as hydrogen storage media due to their high hydrogen content. However, hydrogen desorption generally occurs in multi-step reactions at high temperatures with very slow kinetics, and reversibility requires pressures too high for practical applications.1–6 Efforts to improve the kinetics and thermodynamics of these hydrides have mainly revolved around two different approaches including doping/catalyst addition,7–11 and more recently destabilisation through Hδ+/Hδ− coupling,12,13 partial cation substitution14 or direct reaction with another hydride;2,15–17 in an attempt to substitute an energetically unfavorable dehydrogenation/hydrogenation reaction with other reactions involving the formation of less stable compounds with respect to hydrogen. Unfortunately, these approaches have resulted in little improvement in the overall performances of complex hydrides. Although thermodynamics have been shifted in some cases to more favorable conditions,2,6,15 the generation and absorption of hydrogen from complex hydrides remain seriously hindered by the need of high temperatures and pressures to overcome kinetic barriers. Complex hydrides undergo ionic transport across multiphase boundaries,12,18,19 and the activation of segregated reactive solid phases toward hydrogen when the system is dehydrogenated or hydrogenated. Overcoming such kinetic barriers is unlikely to occur unless reaction paths are shortened, active phases are stabilized in forms reactive enough to ease the dehydrogenation/hydrogenation processes and reactions are controlled at the molecular level. The solution to molecular control is most likely a nanodimension approach because many properties of nanomaterials are size dependent. Restricting the particle size of complex hydrides to the nanoscale, where size dependent effects appear, could facilitate the destabilisation of complex hydrides due to the excess of surface energy occurring in small particles.20,21 Such an approach would also permit simultaneously control over kinetics due to shorter diffusion paths and the manipulation of reactions with hydrogen at the molecular level.
Current attempts at a nanodimension approach are still in their infancy because of the difficulty of synthesizing and stabilizing nanoparticles of highly reactive materials such as complex hydrides. However, upon infiltration of NaAlH4,22–26 LiBH4,27–30 and Li3BN2H831 into or onto the mesoporosity of inorganic materials, initial results have indicated some improvements in thermodynamics and kinetics. The challenge still remains in a clear identification of a relationship between particle size and level of destabilization to be expected.
In order to broadly link size effects and destabilisation, a nano-confinement approach was chosen for three different complex hydrides, i.e. NaAlH4, LiAlH4 and LiBH4, of major interest for hydrogen storage purposes. The choice of a nano-confinement approach is motivated by the need for synthesising nanosized complex hydride particles with monodispersity and inhibiting reactions between nanoparticles. Carbon NanoTubes (CNTs) of 2 nm diameter (containing 10% of CNT with a diameter of 10 nm, see the ESI, Fig. S1†) were used for this purpose. Prior to hydride confinement the CNT were further purified and simultaneously cut by using conventional methods of refluxing in a concentrated acid solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 98% sulfuric acid and 70% nitric acid) at 70 °C for 3 h followed by ultrasonication, washing with high purity water and extraction into hexane to ease the collection of nanotubes (see the ESI†). Finally, the CNT were washed three times with ethanol and dried under vacuum at 140 °C. At the end of this treatment, the purity of the CNT was found to be >99.8% and the average length of the tubes 200 nm (Fig. 1). Further incorporation of the complex hydrides inside the CNT was achieved by wet impregnation with a 0.3 mol L−1 THF solution of the respective hydride. After 3 h, the solvent was removed and the CNT further dried under vacuum at −10 °C.
:1, 98% sulfuric acid and 70% nitric acid) at 70 °C for 3 h followed by ultrasonication, washing with high purity water and extraction into hexane to ease the collection of nanotubes (see the ESI†). Finally, the CNT were washed three times with ethanol and dried under vacuum at 140 °C. At the end of this treatment, the purity of the CNT was found to be >99.8% and the average length of the tubes 200 nm (Fig. 1). Further incorporation of the complex hydrides inside the CNT was achieved by wet impregnation with a 0.3 mol L−1 THF solution of the respective hydride. After 3 h, the solvent was removed and the CNT further dried under vacuum at −10 °C.
 | ||
| Fig. 1 Transmission electron microscopy (TEM) images of the CNT after incorporation of NaAlH4, LiAlH4 and LiBH4, respectively. | ||
According to elemental analysis the dark powder of CNT obtained at the end of the incorporation process, only contained a small amount (<6 mass%) of complex hydrides (Table 1). Further characterisation by TEM showed that mainly nanotubes with a diameter of 10 nm were successfully filled to some extent with large voids between the hydride particles (Fig. 1). Analysis by EDS confirmed that the encapsulated particles were composed of Na and Al for NaAlH4, and Al for LiAlH4 (ESI, Fig. S2†). The typical size of the encapsulated hydrides was found to be 10 nm in diameter and 15 nm in length (Fig. 2), with all the hydride particles contained within the CNT for LiAlH4 and LiBH4. However, in the case of NaAlH4 large particles with a size >25 nm were also found outside the CNT (Fig. 1a). The formation of such large particles may originate from the higher deposition of NaAlH4 that occurred during the impregnation (Table 1).
| Hydride confined in CNT | NaAlH4 | LiAlH4 | LiBH4 |
|---|---|---|---|
| Amount of hydride (mass%) | 5.68 ± 0.2 | 1.2 ± 0.2 | 1.5 ± 0.2 |
| Theoretical hydrogen content after complete decomposition (mass%) | 0.42 ± 0.03 | 0.11 ± 0.03 | 0.24 ± 0.03 |
| Hydrogen released (mass%) | 0.45 ± 0.05 | 0.19 ± 0.05 | 0.27 ± 0.05 |
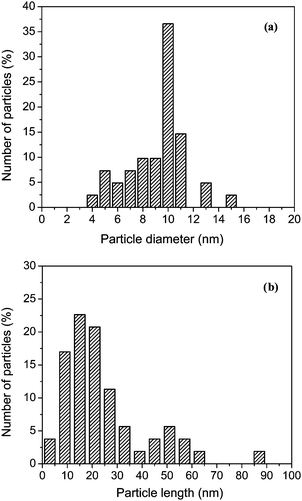 | ||
| Fig. 2 Average size distribution across a series of samples of lithium aluminium hydride nanoparticles encapsulated in CNT with respect to (a) particle diameter and (b) particle length along the nanotubes. Similar size distributions were found for the other nanoconfined hydrides. | ||
Following the encapsulation of the hydrides, hydrogen desorption behaviour was characterised by Temperature Programmed Desorption (TPD). Usually complex hydrides based on aluminium and boron release hydrogen following three main reaction steps, the last step corresponding to the decomposition of the remaining alkali hydride, i.e. NaH or LiH (Fig. 3a).2,6 Upon encapsulation in CNT, drastic modifications of the hydrogen desorption behaviour for NaAlH4, LiAlH4 and LiBH4 were observed (Fig. 3b). Hence, NaAlH4 encapsulated in CNT (NaAlH4–CNT) released hydrogen from room temperature and the hydrogen desorption profile of NaAlH4–CNT showed two main peaks, one at 60 °C and a broad peak at 220 °C. For LiAlH4 confined within CNT (LiAlH4–CNT), one hydrogen desorption peak was observed at 120 °C, i.e. far below the decomposition temperature of bulk LiAlH4. Furthermore, hydrogen release occurred from the sub-ambient condition and was fully completed by 250 °C. The most remarkable improvements were observed for LiBH4 encapsulated in CNT (LiBH4–CNT) with hydrogen release occurring between 75 and 250 °C, i.e. far below 500 °C generally needed to release hydrogen from bulk LiBH4. It is also noteworthy that for all hydrides encapsulated with CNT their decomposition starts well below the melting point of their bulk counterparts, i.e. 190, 170, and 120 °C for NaAlH4, LiAlH4 and LiBH4, respectively (ESI, Fig. S3–S5†). Considering the desorption behaviour of the three hydrides and the particle size distribution, the low temperature peak observed for NaAlH4 at 60 °C is believed to result from the encapsulated hydride particles, while the broad peak at the higher temperature would correspond to the full decomposition of the large particles of NaAlH4 found outside the CNT as NaH may also be destabilised by a carbon support32 (Fig. 1a).
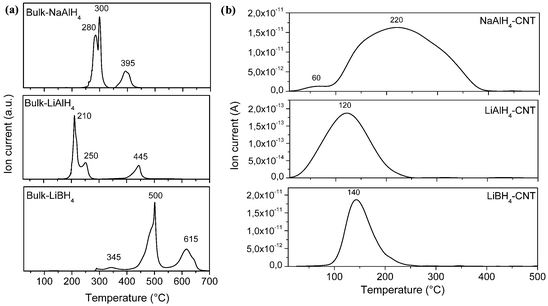 | ||
| Fig. 3 Temperature Programmed Desorption (TPD) of (a) the bulk hydrides NaAlH4, LiAlH4 and LiBH4 as received, and (b) after their respective incorporation in CNT. | ||
Further quantification of the total amount of hydrogen desorbed revealed that the full amount of hydrogen theoretically contained within the confined hydrides was released over the course of the TPD (Table 1). Therefore, there is a clear effect of size in facilitating the destabilisation of complex hydrides. Since the hydrogen desorption proceeded in a single step upon hydride confinement rather than the multiple steps observed for the bulk materials, significant modifications of the dehydrogenation mechanisms upon destabilisation through size effects may also occur. Lohstroh et al. also found a shift in the thermodynamic properties of NaAlH4 encapsulated within the mesoporosity of activated carbon, with the disappearance of the typical two step behaviour usually observed on the pressure composition isotherm of bulk NaAlH4.24 Once again, this may concur with an important alteration of thermodynamics. If the particle is sufficiently small, the increase in surface area would result in a low mean coordination number of surface atoms and thus an excess of surface energy (ΔWsurf) would lead to a modification of the thermodynamics and equilibrium pressure (Peq) according to the following equation:20
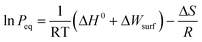
Hence, an excess of surface energy and/or the interaction of the hydride nanoparticle surface with the inner wall of the CNT could lead to a destabilisation of the complex hydride. An additional contribution from the entropy due to the increasing amount of disorder in smaller particles size could also result in a significant destabilisation of complex hydrides. Furthermore, such a change in thermodynamics may affect reaction paths at the nanoscale and lead to different reaction mechanisms that could explain the single desorption peak observed by TPD.
Further attempts to identify the effects of size on the thermodynamics of the reactions were unsuccessful due to the small amount of hydride encapsulated within the CNT. However, measuring the temperature at maximum desorption for each complex hydride at different heating rates allowed the activation energy (Ea) of the dehydrogenation process to be determined by the Kissinger method (ESI, Fig. S6†).33Ea for all three hydride materials were considerably lower than any observed before (Table 2). For the large and non-confined particles of NaAlH4 (>25 nm), Ea was found to be 122 ± 18 kJ mol−1, in good agreement with reported values for bulk NaAlH4 (120 kJ mol−1). Thus the large NaAlH4 particles behaved like the bulk hydride. However, a drastic decrease in Ea, i.e. 45 ± 2 kJ mol−1 instead of 120 kJ mol−1, was established for NaAlH4 particles confined within CNT. In fact, this value of Ea is even lower than the Ea of 58 kJ mol−1 reported for 2–10 nm NaAlH4 particles supported within the mesoporosity of carbon nanofibers.22 The difference is probably due to the confinement effect of the CNT or the stabilisation of different reactive planes at the surface of the NaAlH4 nanoparticles. Furthermore, Ea for hydrogen desorption from LiAlH4–CNT was calculated to be 64 ± 5 kJ mol−1, a decrease of 18 kJ mol−1 from 82 kJ mol−1 reported for bulk LiAlH4. The decrease in activation energy for LiBH4–CNT was more significant, i.e. 88 ± 5 kJ mol−1 compared to 146 kJ mol−1 for bulk lithium borohydride. Gross et al. found the activation energy for LiBH4 incorporated into the 13 nm porosity of a carbon aerogel to be 103 kJ mol−1.28 However, a temperature of at least 300 °C was still needed before hydrogen could be desorbed from this material.27,28,30 To the best of our knowledge, this is the first time that hydrogen has been released from LiAlH4 and LiBH4 at such low temperatures. This significant decrease in activation energy could have several origins including a possible catalytic effect of the surrounding carbon. However, this hypothesis should be ruled out because similar complex hydrides supported by carbon materials have not shown such a degree of destabilisation. Therefore, the enhanced desorption behaviour of NaAlH4, LiAlH4 and LiBH4 confined with CNT is believed to be due to the synergistic effects of size, resulting excess of surface energy, reduction in diffusion paths and possible confinement effects.
| E a/kJ mol−1 | |||
|---|---|---|---|
| Reaction 1: MAlH4 → 1/3M3AlH6 + 2/3Al + H2 | Reaction 2: M3AlH6 → 3MH + Al + 3/2H2 | ||
| a No data available, M = Na or Li. | |||
| NaAlH4 | Bulk | 11834,35 | 12034,35 |
| On carbon nanofibers (2–10 nm) | 5822 | ||
| In CNT | 45 ± 2 for the peak at 60 °C | 122 ± 18 for the peak at 220 °C | |
| LiAlH4 | Bulk | 82–1151,36 | 86–901,36 |
| In CNT | 64 ± 5 | ||
In conclusion, upon size restriction a drastic improvement of the desorption properties of NaAlH4, LiAlH4 and LiBH4 with respect to hydrogen was observed. Once in the 10 nm range, nanoparticles of these hydrides could release hydrogen from room temperature and at temperatures considerably below that of their bulk counterparts. Moreover, a drastic reduction of the activation energy for the dehydrogenation reaction was found to accompany size restriction of these hydrides. The physical-chemistry of complex hydrides at the nanoscale is uncertain and further experiments are pending to elucidate the improvements obtained. However, these findings validate the idea that the properties of complex hydrides are size dependent and that effective destabilisation of complex hydride can be solely achieved through size effects. Appropriate control of such size effects would ultimately lead to the next generation of materials for the practical storage of hydrogen.
Acknowledgements
This research was supported by UNSW Early Career Research Grant program (project PS17422) and under the Australian Research Council's Discovery Projects funding scheme (project DP1095209).References
- J. R. Ares, K. F. Aguey-Zinsou, M. Porcu, J. M. Sykes, A. Dornheim, T. Klassen and R. Bormann, Mater. Res. Bull., 2008, 43, 1263–1275 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schuth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS.
- J. Graetz, J. Wegrzyn and J. J. Reilly, J. Am. Chem. Soc., 2008, 130, 17790–17794 CrossRef CAS.
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- A. Zuttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, J. Power Sources, 2003, 118, 1–7 CrossRef CAS.
- B. Bogdanovic, M. Felderhoff and G. Streukens, J. Serb. Chem. Soc., 2009, 74, 183–196 CrossRef CAS.
- B. Bogdanovic and G. Sandrock, MRS Bull., 2002, 27, 712–716 CAS.
- V. P. Balema, J. W. Wiench, K. W. Dennis, M. Pruski and V. K. Pecharsky, J. Alloys Compd., 2001, 329, 108–114 CrossRef CAS.
- M. S. Wellons, P. A. Berseth and R. Zidan, Nanotechnology, 2009, 20, 204022 CrossRef.
- U. Bosenberg, J. W. Kim, D. Gosslar, N. Eigen, T. R. Jensen, J. M. B. von Colbe, Y. Zhou, M. Dahms, D. H. Kim, R. Gunther, Y. W. Cho, K. H. Oh, T. Klassen, R. Bormann and M. Dornheim, Acta Mater., 2010, 58, 3381–3389 CrossRef.
- J. Chen, N. Kuriyama, Q. Xu, H. T. Takeshita and T. Sakai, J. Phys. Chem. B, 2001, 105, 11214–11220 CrossRef CAS.
- K. F. Aguey-Zinsou, J. Yao and Z. X. Guo, J. Phys. Chem. B, 2007, 111, 12531–12536 CrossRef CAS.
- Z. T. Xiong, C. K. Yong, G. T. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138–141 CrossRef CAS.
- Y. Nakamori and S. Orimo, J. Alloys Compd., 2004, 370, 271–275 CrossRef CAS.
- F. E. Piinkerton, M. S. Meyer, G. P. Meisner, M. P. Balogh and J. J. Vajo, J. Phys. Chem. C, 2007, 111, 12881–12885 CrossRef CAS.
- X. B. Yu, Y. H. Guo, D. L. Sun, Z. X. Yang, A. Ranjbar, Z. P. Guo, H. K. Liu and S. X. Dou, J. Phys. Chem. C, 2010, 114, 4733–4737 CrossRef CAS.
- J. J. Vajo and G. L. Olson, Scr. Mater., 2007, 56, 829–834 CrossRef CAS.
- J. R. Ares, K. F. Aguey-Zinsou, F. Leardini, I. J. Ferrer, J. F. Fernandez, Z. X. Guo and C. Sanchez, J. Phys. Chem. C, 2009, 113, 6845–6851 CrossRef CAS.
- W. Lohstroh and M. Fichtner, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 184106 CrossRef.
- K. F. Aguey-Zinsou and J. R. Ares-Fernandez, Energy Environ. Sci., 2010, 3, 526–543 RSC.
- M. Fichtner, Nanotechnology, 2009, 20, 204009 CrossRef.
- C. P. Balde, B. P. C. Hereijgers, J. H. Bitter and K. P. de Jong, J. Am. Chem. Soc., 2008, 130, 6761–6765 CrossRef CAS.
- J. B. Gao, P. Adelhelm, M. H. W. Verkuijlen, C. Rongeat, M. Herrich, P. J. M. van Bentum, O. Gutfleisch, A. P. M. Kentgens, K. P. de Jong and P. E. de Jongh, J. Phys. Chem. C, 2010, 114, 4675–4682 CrossRef CAS.
- W. Lohstroh, A. Roth, H. Hahn and M. Fichtner, ChemPhysChem, 2010, 11, 789–792 CrossRef CAS.
- R. D. Stephens, A. F. Gross, S. L. Van Atta, J. J. Vajo and F. E. Pinkerton, Nanotechnology, 2009, 20, 204018 CrossRef.
- S. Y. Zheng, F. Fang, G. Y. Zhou, G. R. Chen, L. Z. Ouyang, M. Zhu and D. L. Sun, Chem. Mater., 2008, 20, 3954–3958 CrossRef CAS.
- Z. Z. Fang, P. Wang, T. E. Rufford, X. D. Kang, G. Q. Lu and H. M. Cheng, Acta Mater., 2008, 56, 6257–6263 CrossRef CAS.
- A. F. Gross, J. J. Vajo, S. L. Van Atta and G. L. Olson, J. Phys. Chem. C, 2008, 112, 5651–5657 CrossRef CAS.
- P. Ngene, P. Adelhelm, A. M. Beale, K. P. de Jong and P. E. de Jongh, J. Phys. Chem. C, 2010, 114, 6163–6168 CrossRef CAS.
- S. Cahen, J. B. Eymery, R. Janot and J. M. Tarascon, J. Power Sources, 2009, 189, 902–908 CrossRef CAS.
- H. Wu, W. Zhou, K. Wang, T. J. Udovic, J. J. Rush, T. Yildirim, L. A. Bendersky, A. F. Gross, S. L. Van Atta, J. J. Vajo, F. E. Pinkerton and M. S. Meyer, Nanotechnology, 2009, 20, 204002 CrossRef.
- P. Adelhelm, K. P. de Jong and P. E. de Jongh, Chem. Commun., 2009, 6261–6263 RSC.
- H. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- A. Andreasen, T. Vegge and A. S. Pedersen, J. Solid State Chem., 2005, 178, 3672–3678 CrossRef CAS.
- G. Sandrock, K. Gross and G. Thomas, J. Alloys Compd., 2002, 339, 299–308 CrossRef CAS.
- D. Blanchard, H. W. Brinks, B. C. Hauback, P. Norby and J. Muller, J. Alloys Compd., 2005, 404–406, 743–747 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; TEM of the CNT as received; EDS spectra of the CNT containing NaAlH4 and LiAlH4; thermal analysis and differential scanning calorimetry of the dehydrogenation reaction of the bulk hydrides; Kissinger plots for the hydrides incorporated in the CNT. See DOI: 10.1039/c0nr00418a |
| This journal is © The Royal Society of Chemistry 2010 |