Calcium phosphate growth beneath a polycationic monolayer at the air–water interface: effects of oscillating surface pressure on mineralization
Mathias
Junginger
ab,
Katrin
Bleek
a,
Katarzyna
Kita-Tokarczyk†
c,
Jürgen
Reiche
d,
Andriy
Shkilnyy‡
ab,
Felix
Schacher§
e,
Axel H. E.
Müller
e and
Andreas
Taubert
*ab
aInstitute of Chemistry, University of Potsdam, D-14476 Potsdam, Germany. E-mail: ataubert@uni-potsdam.de
bMax Planck Institute of Colloids and Interfaces, D-14476 Potsdam, Germany
cDepartment of Chemistry, University of Basel, CH-4056 Basel, Switzerland
dInstitute of Physics and Astronomy, University of Potsdam, D-14476 Potsdam, Germany
eMakromolekulare Chemie II, NW II, University of Bayreuth, D-95440 Bayreuth, Germany
First published on 13th September 2010
Abstract
The self-assembly of the amphiphilic block copolymer poly(butadiene)-block-poly[2-(dimethylamino)ethyl methacrylate] at the air–water interface and the mineralization of the monolayers with calcium phosphate was investigated at different pH values. As expected for polyelectrolytes, the subphase pH strongly affects the monolayer properties. The focus of the current study, however, is on the effect of an oscillating (instead of a static) polymer monolayer on calcium phosphate mineralization. Monitoring of the surface pressure vs. mineralization time shows that the monolayer is quite stable if the mineralization is performed at pH 8. In contrast, the monolayer at pH 5 shows a measurable decrease of the surface pressure already after ca. 2 h of mineralization. Transmission electron microscopy reveals that mineralization at low pH under constant oscillation leads to small particles, which are arranged in circular features and larger entities with holes of ca. 200 nm. The larger features with the holes disappear as the mineralization is continued in favor of the smaller particles. These grow with time and form necklace-like architectures of spherical particles with a uniform diameter. In contrast, mineralization at pH 8 leads to very uniform particle morphologies already after 2 h. The mineralization products consist of a circular feature with a dark dot in the center. The increasing contrast of the precipitates in the electron micrographs with mineralization time indicates an increasing degree of mineralization vs. reaction time. The study therefore shows that mechanical effects on mineralization at interfaces are quite complex.
Introduction
Biomimetic mineralization, that is, the synthesis of organic–inorganic or polymer–inorganic hybrid materials resembling materials of biological origin using “soft” synthesis protocols, has attracted tremendous interest. Calcium phosphate is one of the most important biominerals, as it is the main inorganic component of mammal bone and teeth.1,2 There have thus been numerous attempts at synthesizing calcium phosphate–polymer hybrid materials that mimic the structure and properties of, for example, natural bone.3–15The vast majority of these studies focused on mineralization in bulk solution with different additives, such as surfactants or polymers. As proteins associated with bone are mostly acidic,16–18 most of the work is concerned with acidic polymers such as poly(acrylic acid) (PAA). Several recent publications, however, have demonstrated that polycations are efficient growth modifiers for calcium phosphate as well.19,20
In many cases, mineral formation is controlled by interfaces and it is thus important to determine, quantify, and rationalize the parameters that make a certain surface or interface a good (or bad) interface for calcium phosphate growth. Surprisingly, only a few studies address the issue of surface effects on calcium phosphate mineralization. Ngankam et al.21 and Ball et al.22 showed that polymer multilayers affect calcium phosphate mineralization in several ways. Poly(sodium-4-styrenesulfonate) (PSS)/poly(allylamine hydrochloride) (PAH) multilayers on silica primarily affect the crystal phase selection.21 Multilayers of poly(L-glutamic acid) or poly(L-aspartic acid) and poly(L-lysine) affect the lag times before nucleation.22 Similarly, Spoerke et al. and Schweizer et al. demonstrated that polyelectrolyte coatings strongly modify titanium and nickel–titanium alloy surfaces.23,24
Using a polymer monolayer approach rather than polymers on a solid surface, Casse et al. demonstrated that polymer monolayers are useful model systems for the investigation of interface-controlled calcium phosphate mineralization.10 The study mainly reveals that small supersaturations, pH values above 8, and mechanical agitation of the subphase favor the formation of well-controlled, uniformly mineralized polymer monolayers.
In a recent study, Junginger et al. showed that, similar to bulk studies,19,20 cationic polymer monolayers strongly affect calcium phosphate mineralization.25 These authors demonstrated that also with a polycation, poly(n-butylmethacrylate)-block-poly[2-(dimethylamino)ethyl methacrylate] (PnBA-PDMAEMA), uniform calcium phosphate deposits can be obtained. In contrast to the earlier example,10 however, the most uniform mineral deposits were obtained at a relatively low pH of 5. Experiments at a higher pH of 8 yielded polydisperse, poorly defined, and large aggregates.
In agreement with the work by Casse et al.,10 gentle stirring of the subphase significantly improves the quality and uniformity of the mineralized film. This thus indicates that mechanical agitation is important for uniform and well-controlled mineralization, yet other modes of agitation have not been studied.
The current study focuses on the effect of oscillating cationic monolayers on the mineralization process. Instead of stirring the subphase, the monolayer was kept in motion by applying an oscillating barrier movement to the monolayer during mineralization to evaluate the role of a template in a non-equilibrium situation, “similar” to a biological surface, which often is in constant motion during mineralization.
Experimental
Chemicals
Tris(hydroxymethyl)aminomethane (Tris, Sigma-Aldrich), sodium acetate, chloroform (spectroscopic grade), hydrochloric acid, (Merck), acetic acid, and monosodium phosphate dihydrate (Roth) were used as received. Glassware was treated with a H2O2–H2SO4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) mixture and rinsed with bidistilled water. Water for Langmuir–Blodgett experiments was either double distilled from a dedicated quartz glass distillation setup or obtained from Millipore MilliQ® with a resistivity of 18.2 MΩ.
:2) mixture and rinsed with bidistilled water. Water for Langmuir–Blodgett experiments was either double distilled from a dedicated quartz glass distillation setup or obtained from Millipore MilliQ® with a resistivity of 18.2 MΩ.
For polymerization,26 tetrahydrofuran (Merck, p.a.) was purified by successive distillation over CaH2 and K and kept under dry nitrogen before use. Dimethylaminoethyl methacrylate (DMAEMA, Aldrich, p.a.) for anionic polymerization was degassed three times via freeze–pump–thaw cycles and stirred with trioctylaluminium prior to condensation. Afterwards, it was condensed into a glass reactor and stored over dibutylmagnesium. Prior to polymerization, the calculated amount of monomer was condensed into a pre-cooled burette. 1,1-Diphenylethylene (Aldrich, 97%) was purified by stirring with sec-BuLi under N2 followed by distillation. DMAEMA for RAFT polymerization was filtered over basic aluminium oxide to remove the inhibitor. Butadiene (Messer–Griesheim) was passed through columns filled with molecular sieves (4 Å) and basic aluminium oxide. The sec-BuLi (Acros, 1.3 M in cyclohexane–hexane: 92:8) was used as received. AIBN was recrystallized twice from ethanol. Dioxane was used as received after testing the water content, which was below 0.004%.
Synthesis of poly(butadiene)-block-poly[2-(dimethylamino)ethyl methacrylate] PB-PDMAEMA.26
Polymerization was carried out in a thermostated laboratory autoclave (Büchi) under dry nitrogen. The synthesis was accomplished in THF (500 mL) by sequential anionic polymerization of the corresponding monomers using sec-BuLi as initiator. sec-BuLi was added to THF at −70 °C followed by fast addition of butadiene. The conversion of butadiene was monitored via an NIR probe. After 10 h at −10 °C, 2 equiv. of 1,1-diphenylethylene were added via a syringe to end-cap the living chain ends. After 1 h, DMAEMA was added to the reaction mixture via a syringe and stirred for 2 h at −50 °C. The polymer was purified by precipitation in water. Under these conditions, mainly (∼85%) 1,2-polybutadiene is generated.Synthesis of PDMAEMA for titration experiments
A PDMAEMA homopolymer was synthesized via reversible addition fragmentation chain transfer (RAFT) polymerization. The RAFT agent S-1-dodecyl-S′-(α,α′ dimethyl-α′′-acetic acid)-trithio carbonate was synthesized according to Lai et al.27 Additionally, the initiator was recrystallized twice from n-hexane. PDMAEMA was synthesized by a modified protocol of Tan et al.28 DMAEMA and dodecyl trithiocarbonate were dissolved in 1,4-dioxane. The mixture was degassed and flushed with argon several times. AIBN was dissolved in 0.2 mL 1,4-dioxane and added to the mixture and the solution was placed in a preheated oil bath at 75 °C. After 17 h under nitrogen and steady stirring, the reaction was stopped by cooling the mixture to room temperature and adding 1,4-hydroquinone. The polymer was precipitated in diethyl ether, centrifuged (9000 rpm, 10 min), and redissolved in THF. The precipitation procedure was repeated twice. Mn = 23![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 300 g mol−1, PDI = 1.43.
300 g mol−1, PDI = 1.43.
Langmuir monolayer experiments
Surface pressure–area (π–A) isotherms of PB-PDMAEMA were recorded on a custom-made trough from KSV Finland with a total area of 440 cm2 and two moveable hydrophilic barriers at a temperature of T = 298 ± 1 K. Additional experiments were made on a Nima 611 (Nima Technology, UK) medium deposition trough with two symmetrically moveable hydrophobic barriers and a total area of 250 cm2. The stability measurements were performed on the Nima 611 LB trough. The troughs were placed on anti-vibration tables in a dust-free room. All experiments were done at room temperature.Monolayers were prepared by spreading aliquots of 54 μl (pH 5) and 49 μl (pH 8) of polymer solution (1.06 mg mL−1) in CHCl3 on aqueous monosodium phosphate solutions with pH 5 (acetate buffer) and 8 (Tris buffer). After spreading, a minimum of 10 min. was allowed for the solvent evaporation. The surface pressure π was recorded to ± 0.1 mN m−1 with a Wilhelmy plate (chromatography paper, ashless Whatman Chr1) connected to an electrobalance.
Mineralization
For mineralization experiments, PB-PDMAEMA was spread from chloroform (1.06 mg mL−1) on a 2 mM NaH2PO4 aqueous subphase buffered with acetic acid–sodium acetate for pH 5 and Tris/HCl for pH 8. After chloroform evaporation, a CaCl2 solution was injected into the subphase behind the barriers via a Hamilton syringe. The monolayers were kept in motion by applying a cycling compression–expansion program. The barrier speed was kept at 25 cm2 min−1 during the cycles. Cycle restrictions were: (1) a minimum surface area of 40 cm2, (2) a maximum surface area of 245 cm2, and (3) a maximum surface pressure of 30 mN m−1. Mineralization was stopped after 2, 4, 6, 8, and 17 h and the samples were transferred to TEM copper grids.Electron microscopy
Transmission electron microscopy was done on a CM100 electron microscope (Philips, Eindhoven, The Netherlands) operated at 80 kV. TEM copper grids were coated with Pioloform® before use. Particle statistics were done on 80–120 particles per sample.Titration
Titration experiments were done on a Mettler-Toledo T50 titrator with 0.04 M HCl and 0.5 M NaOH. NaOH solutions were freshly prepared from Riedel-de Häen Fixanal® standard solution using DI and distilled water. The electrode (MT DG 115-SC) was calibrated with three buffer standards (pH 4.0, 7.0, and 9.0). Titration was done in 0.1 mL steps with an equilibration time of 90 s between each addition. Evaluation of the titration data was done with MT LabX® software. Degrees of protonation were calculated from titration curves as described earlier.19Results
Fig. 1a shows the surface pressure–area (π–A) isotherms of PB-PDMAEMA at different pH values with and without H2PO4− ions in the subphase. The isotherms in the presence and absence of dihydrogen phosphate are virtually identical. For this reason, only one curve is shown. | ||
| Fig. 1 (a) π–A isotherms of PB-PDMAEMA on different subphases, (b) πvs. mineralization time at pH 5, and (c) πvs. mineralization time at pH 8. | ||
At pH 2 the isotherm shows only a gradual increase of the surface pressure up to a mean molecular area of 1000 Å2 followed by an expanded phase range with enhanced repulsive molecular interaction. At pH 5, additional repulsive forces lead to a clearly increased surface pressure in the intermediate isotherm range between 3000 Å2 and 500 Å2,while the collapse pressure is raised from 40 mN m−1 at pH 2 to 46 mN m−1 at pH 5.
A further increased pH of 7 or 8 results in a considerable gain of repulsive forces in the range between 3000 Å2 and 500 Å2, as indicated by the shift to higher surface pressure at comparable mean molecular areas. Close to the collapse of the expanded phase (for pH 7 and 8 at about 45 mN m−1), all isotherms converge, however, to almost the same mean molecular area, irrespective of the subphase pH.
Fig. 1b shows the surface pressure oscillation of PB-PDMAEMA at pH 5 during mineralization. The maximum value of π decreases and the minimum value of π increases with prolonged cycling and mineralization. This indicates an influence of the continuous expansion and compression of the films. In contrast, the isotherms of the oscillating films at pH 8 (Fig. 1c) show that the monolayer is more stable than at pH 5. The reproducible value of the maximum surface pressure (30 mN m−1) is due to the preset boundary conditions (see experimental section for details). Only after 14 h of mineralization does the maximum surface pressure decrease. The increase of the minimum surface pressure is slightly lower than at pH 5, which could be an artifact induced by water evaporation.
Fig. 2 shows representative TEM images of a sample mineralized with constant compression–expansion cycles and a film mineralized at a constant surface pressure of 30 mN m−1 at pH 8. The sample mineralized beneath the oscillating monolayer exhibits round features with a central dark dot and a large number of smaller particles in the background and on the circular features. The small particles are rather monodisperse and have a diameter of 4 ± 0.7 nm. The larger black dots have a diameter of 40 ± 6 nm and the circular features have a diameter between 110 and 140 nm.
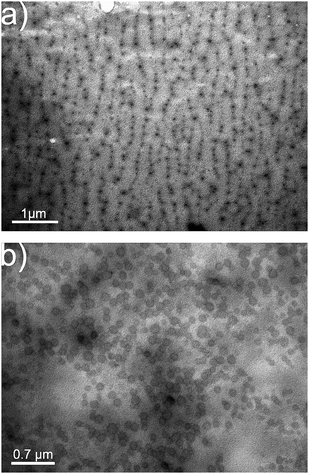 | ||
| Fig. 2 TEM images of precipitates obtained after 8 h at pH 8 beneath (a) an oscillating and (b) a static monolayer. | ||
In contrast, the calcium phosphate grown on the static monolayer shows features resembling hollow capsules or ring-like objects with a less electron-dense core. These features, although forming chain-like arrangements, do not appear oriented along a preferred direction. This observation thus suggests that the oscillating film does have an effect on the mineralization and at the same time rules out that the oriented features shown in Fig. 2a are transfer artifacts.
The origin of the different morphologies is unclear at the moment. One may speculate that the morphological differences between the precipitates (black dots vs. rings) are associated with polymer rearrangement during oscillation. The formation of defined polymer structures at the surface has at the moment not been proven and a final conclusion can therefore not be drawn. Further experiments to determine the behavior and structure of the polymer monolayer before, during, and after oscillation are on the way.
Fig. 3 shows the calcium phosphate morphology vs. mineralization time and pH. TEM shows that the morphological diversity is larger and more complex at pH 5 than at pH 8. After 2 h at pH 5, thin filaments form, similar to an earlier example.25 These filaments aggregate into ring- or circle-like entities with a diameter of ca. 800 nm. Besides these features, dark, larger particles with sizes between 500 and 700 nm form. These particles are not dense, but exhibit holes on the order of 200 nm.
 | ||
| Fig. 3 Morphology evolution of samples mineralized beneath oscillating monolayers at pH 5 (top row) and pH 8 (bottom row). The inset of 2 h at pH 5 shows the holey aggregates mentioned in the text (scale bar is 400 nm). Insets in the top row show a higher magnification of the holey objects mentioned in the text (2 h) and the crystal-looking features connected to the chains of spheres (17 h). The light line in the bottom row (17 h) is a crack in the film showing that the cracks very often pass through the centers of the features with the black dots. | ||
After 4 h at pH 5, the larger, holey particles grow further in the sense that they appear darker than after 2 h, but a quantification of the size changes is difficult because of their large diameter variation. The samples now also exhibit a uniform background of small particles with a diameter of 22 ± 5 nm, which often form ring-like features on the same order of the rings observed after 2 h. Similarly, after 6 h of mineralization, small particles and larger dark particles with holes are present in the sample, but the smaller particles are not arranged in circular features any more. However, they appear darker, implying that the electron density is somewhat higher than after 4 h.
A significant change in particle size and shape is observed after 8 h of mineralization. The particles agglomerate into larger objects, which form chain or necklace-like structures. Moreover, the individual particles are larger (55 ± 9 nm) and electron dense. The holey larger particles described above are less prominent. This development is even more pronounced after 17 h. Here, the spheres on the necklace-like aggregates have grown significantly from 55 ± 9 nm in diameter at 8 h to 116 ± 23 nm at 17 h. At some places of the necklace, crystals with well-defined edges are present (Fig. 3), but they disintegrate after a few seconds. The holey aggregates described above have disappeared.
At pH 8 significantly different morphologies are present. After 2 h, dark dots with a lighter, roughly circular surrounding are visible in TEM. The diameter of the central dots is 100 ± 23 nm. After 4 h, the matrix around the dark particles transforms into uniform circular features with diameters of 116 ± 19 nm. The shape is more uniform than after 2 h and the boundary of each feature is clearly visible as a dark line surrounding a light area. The boundary contains many small particles with diameters of only ca. 10 nm. After 6 h of mineralization, the features are identical to those observed after 4 h, but the contrast is higher. Moreover, the small particles that were observed after 4 h are now not only located on the ring-like features, but they are evenly distributed in the entire background of the samples. After 8 h, an orientation of the features can be observed. Unlike the features at shorter reaction times, they are now organized in short chains that are roughly parallel to one another. This orientation is however lost again at 17 h. At 17 h, the film is densely mineralized and cracks can often be observed in the films. The cracks appear to run through the centers of the dots. For better comparison between the different conditions, Fig. 4 gives a graphical representation of the morphologies vs. time and pH.
 | ||
| Fig. 4 Graphical representation of calcium phosphate formed beneath PB-PDMAEMA monolayers at pH 5 and 8. Gray levels of individual features give the relative contrast in the respective samples, as observed in the electron microsope (qualitative). The top row for both pH values represents lower magnification micrographs, the lower row represents higher magnification micrographs. | ||
It must be noted that the crystal phase and chemical composition of the precipitates are difficult to identify. As pointed out in our earlier publication25 the samples are very beam sensitive and the amount obtained from the experiments is not sufficient for spectroscopy or elemental analysis. Hence, we can not yet comment on the exact composition of the inorganic deposit on the film.
TEM shows that the subphase pH is a key parameter in the mineralization process. To quantify the pH response of the PDMAEMA block at different pH values, a PDMAEMA homopolymer with a molecular weight of Mn 23300 g mol−1 was titrated, Fig. 5. While the reference titration (no polymer, data not shown) shows textbook titration behavior for a strong acid with a strong base, the titration curve of the polymer solution in 0.04 M HCl shows two steps at 0.6 mL and 2.9 mL of base. According to the calculation of the degree of protonation from the titration curve, PDMAEMA is 100% protonated below a pH value of ca. 6. A rapid drop to a degree of protonation of ca. 40% is observed between pH 7 and 10. Above pH 11, a second drop in the degree of protonation is visible.
 | ||
| Fig. 5 Volumetric titration of a solution of 4.72 mg mL−1 of PDMAEMA in 0.04 M HCl vs. 0.5 M NaOH. | ||
Discussion
The study presented here investigates the role of monolayer oscillation (that is, direct mechanical action on the mineralization template) on calcium phosphate mineralization beneath a PB-PDMAEMA monolayer. The behavior of PB-PDMAEMA (Fig. 1) is comparable to poly(methyl methacrylate)-block-poly[-(2-dimethylamino)ethyl methacrylate] (PMMA-PDMAEMA) and poly(n-butyl acrylate)-block-poly[2-(dimethylamino)ethyl methacrylate] (PnBA-PDMAEMA).25,29 At pH 2, the monolayers only show a transition from gas to liquid expanded with a collapse pressure of 40 mN m−1. At this pH value roughly 100% of the DMAEMA side groups are protonated, implying that the PDMAEMA block extends well into the aqueous subphase.At pH 5, the isotherm shows a small plateau, which becomes more pronounced at pH 7 and higher. Rehfeldt et al.29 showed that the degree of protonation of PDMAEMA at pH 5 is about 85%, at pH 7 around 23%, and at pH 8 ca. 13%. This is slightly lower than our data (Fig. 5), but the overall behavior is identical. Both datasets thus show that the polymer becomes less hydrophilic and more compact at the interface. This is further supported by the qualitative observation that above pH ca. 11, the PDMAEMA solution used in the titration experiment is turbid, indicating polymer precipitation. Consequently, the surface pressure data vs. the mineralization time confirm that the monolayers are more stable at pH 8 than at pH 5.
At pH 5 the maximum surface pressure decreases rapidly during the first two hours from 30 mN m−1 to 25 mN m−1 (Fig. 1b). This can be attributed to material loss to the subphase. Alternatively, as the hydrophobic PB block is much larger than the PDMAEMA block, the oscillations could also lead to a rearrangement of the polymer chains at the surface and thereby optimize the mean molecular areas of the individual polymer chains. In contrast, oscillation experiments at pH 8 (Fig. 1c) show no decrease in surface pressure. This shows that the more compact and much less charged polymer present at pH 8 is not as likely to dissolve into the subphase. Alternatively, if the rearrangement argument from above holds true, one may argue that at pH 8, hydrogen bonds between charged groups, uncharged groups, and water molecules further stabilize the film. This reduces rearrangements of the polymer chains.
Overall, the surface study shows that the polymer behaves much like two earlier examples,25,29 yet we also show that mineralization can be performed under an oscillating monolayer, that is, under non-equilibrium conditions. Time-resolved TEM data (Fig. 2 and 3) show that the pH is a key parameter affecting the morphology, uniformity, and size of the calcium phosphate precipitates beneath the polymer monolayer. This is consistent with earlier studies.10,25,30 However, there are significant differences between the morphologies observed here and in the few other studies available.
Casse et al. showed that the mineralization beneath a monolayer of poly(n-butyl acrylate)-block-poly(acrylic acid) (PnBA-PAA) yields uniformly mineralized films between pH 5 and 11.10 However, the highest and most well-ordered films were obtained at high pH values, where most of the PAA block is deporotonated. Similarly, Junginger et al. showed that the most uniform precipitates beneath a PnBA-PDMAEMA monolayer form at low pH, where most of the PDMAEMA is protonated.25 This suggests that, for a mineralization reaction to occur in a well-controlled manner, the polymer monolayers need to be highly charged. This can be understood, because in the case of the anionic PnBA-PAA monolayer,10 positively charged calcium was present in the subphase, while in the case of the cationic PnBA-PDMAEMA monolayer,25 negatively charged phosphate was present in the subphase. In both cases, electrostatic interactions are thus the driving force for an initial pre-organization of the film, leading to a rather uniform mineralization after the injection of the second component, phosphate or calcium, respectively.
In contrast, the current study shows that the most well-organized and most uniform precipitates are observed at pH 8. This is interesting because at pH 8, the degree of protonation of the PDMAEMA block is below 30% (ref. 29 and Fig. 5) and most of the PDMAEMA chains are thus much less extended at the interface than at pH 5. One explanation can be found in the different block lengths. In the current study, the hydrophobic block is much larger than the hydrophilic block. In the preceding studies the polymers consisted of a large hydrophilic block10,25 or a hydrophilic block with approximately the same degree of polymerization as the hydrophobic block.29 As a result, the surface structures are quite different between the earlier studies and the current work. With a large hydrophobic block, such as the one used in the current study, the polymer may form (possibly kinetically frozen) surface aggregates with a more compact PDMAEMA core at pH 8. At pH 8 the PDMAEMA block is only slightly charged. Calcium phosphate nucleation and growth will therefore probably be less rapid, but more controlled, because the interaction between the calcium and phosphate ions with the polymer is less efficient. As the PDMAEMA block is confined at the center of the surface micelle, the dark dots observed in the center of the ringlike features can be assigned to mineralized surface aggregate cores. Further work to prove the presence of surface aggregates or polymer rearrangements are on the way.
In the earlier cases (where the hydrophilic blocks are equal to or much larger than the hydrophobic blocks), the most uniform films were obtained at pH 10 to 11 (with a polycarboxylate)10 and at pH 5 (with a polyamine).25 These findings suggest that polymers with a large hydrophilic block lead to the most uniform calcium phosphate layers if the blocks are highly charged and extend into the aqueous subphase. In these cases, the hydrophilic blocks have a large hydrodynamic radius and efficiently interact with the incoming calcium and phosphate ions. This leads to a uniform calcium phosphate nucleation and growth.
The current study further broadens this view and shows that polymers with a small hydrophilic block work best (i.e. form the most uniform films) if they are weakly charged and the hydrophilic block is less extended (presumably leading to surface aggregates, which are then initially only mineralized in the aggregate core).
Indeed, the mineralization at pH 5, where more than 85% of the PDMAEMA block is charged (ref. 29 and Fig. 5) is much more heterogeneous at first sight. However, there are in essence only two features present, small dots with diameters of 22 nm and larger aggregates 500–700 nm in size with ca. 200 nm sized holes. These aggregates disappear over time and the small particles prevail. This indicates that the larger aggregates are kinetic intermediates, similar to different, fibrillar features that have also been claimed to be kinetic intermediates.31 Unlike the earlier study, however, the current study shows that this hypothesis is at least partly true, because TEM shows their disappearance at longer reaction times.
Finally, the tiny black dots forming the circular features around the presumed mineralized surface aggregate cores at pH 8 must be discussed. These dots are, although very small, of a high electron contrast. This suggests that they are calcium phosphate nanoparticles which aggregate around a spherical feature at the air–water interface, but cannot penetrate into these features. This observation therefore further supports the notation of surface aggregates as mineralization templates at pH 8.
In summary, the current study further confirms that the pH is a key variable in calcium phosphate mineralization at the air–water interface. More interestingly, the study also shows that mechanical effects on mineralization are quite complex and will, in the future, need more attention. Finally, the data show that the correlation between the block-length ratio and the charge of the hydrophilic block could be another key parameter for mineralization control.
Conclusion
The current study focuses on the mineralization of PB-PDMAEMA monolayers with calcium phosphate at different pH in a non-equilibrium situation. The surface pressure–area isotherms show that the behavior of the block copolymer on a variety of subphases is essentially identical to two earlier studies.25,29 This is intriguing, because the polymer composition varies quite drastically between the different studies. One outcome of the current study is that the pH, similar to earlier work,10,25 is an important parameter. The new findings of this study, however, are the observations (1) that the block-length ratio and the degree of charging of the hydrophilic blocks are further important parameters (possibly more important than in bulk solution) and (2) that mineralization at non-equilibrium conditions may provide additional information on effects that occur at different length and time scales, but this will need to be evaluated in more detail in the future.Acknowledgements
We thank Dr H. Schlaad for useful discussion, Prof. O. Baumann for help with the TEM, the MPI of Colloids and Interfaces (Colloid Chemistry Department), the University of Potsdam, and the Swiss National Science Foundation, and the Deutsche Forschungsgemeinschaft for financial support. M. J. acknowledges a doctoral fellowship from the MPI of Colloids and Interfaces.References
- P. Behrens, E. Baeuerlein, in Handbook of Biomineralization, vol. 2, Wiley-VCH, Weinheim, 2007 Search PubMed.
- K. Gorna, R. Munoz-Espi, F. Gröhn and G. Wegner, Macromol. Biosci., 2007, 7, 163 CrossRef CAS.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef.
- S. Schweizer and A. Taubert, Macromol. Biosci., 2007, 7, 1085 CrossRef CAS.
- F. Variola, F. Vetrone, L. Richert, P. Jedrzejowski, J. H. Yi, S. Zalzal, S. Clair, A. Sarkissian, D. F. Perepichka, J. D. Wuest, F. Rosei and A. Nanci, Small, 2009, 5, 996 CrossRef CAS.
- N. A. J. M. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499 CrossRef CAS.
- R. Q. Song, H. Cölfen, A. W. Xu, J. Hartmann and M. Antonietti, ACS Nano, 2009, 3, 1966 CrossRef CAS.
- A. W. Xu, W. F. Dong, M. Antonietti and H. Cölfen, Adv. Funct. Mater., 2008, 18, 1307 CrossRef CAS.
- H. Cölfen, in Biomineralization II: Mineralization Using Synthetic Polymers and Templates, vol. 271, Springer-Verlag, Berlin, 2007, pp. 1 Search PubMed.
- O. Casse, O. Colombani, K. Kita-Tokarczyk, A. H. E. Müller, W. Meier and A. Taubert, Faraday Discuss., 2008, 139, 179 RSC.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS.
- Y. Politi, D. R. Batchelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Chem. Mater., 2010, 22, 161 CrossRef CAS.
- F. F. Amos, D. M. Sharbaugh, D. R. Talham, L. B. Gower, M. Fricke and D. Volkmer, Langmuir, 2007, 23, 1988 CrossRef CAS.
- E. DiMasi, V. M. Patel, M. Sivakumar, M. J. Olszta, Y. P. Yang and L. B. Gower, Langmuir, 2002, 18, 8902 CrossRef CAS.
- E. DiMasi, S. Y. Kwak, F. F. Amos, M. J. Olszta, D. Lush and L. B. Gower, Phys. Rev. Lett., 2006, 97, 045503 CrossRef.
- L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4110 CAS.
- M. J. Olszta, X. G. Cheng, S. S. Jee, R. Kumar, Y. Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77 CrossRef.
- M. Epple, E. Baeuerlein, in Handbook of Biomineralization, ed. M. Epple and E. Baeuerlein, Wiley-VCH, Weinheim, vol. 3 , 2007 Search PubMed.
- A. Shkilnyy, A. Friedrich, B. Tiersch, S. Schöne, M. Fechner, J. Kötz, C. W. Schlapfer and A. Taubert, Langmuir, 2008, 24, 2102 CrossRef CAS.
- A. Shkilnyy, J. Brandt, A. Mantion, O. Paris, H. Schlaad and A. Taubert, Chem. Mater., 2009, 21, 1572 CrossRef CAS.
- P. A. Ngankam, P. Lavalle, J. C. Voegel, L. Szyk, G. Decher, P. Schaaf and F. J. G. Cuisinier, J. Am. Chem. Soc., 2000, 122, 8998 CrossRef CAS.
- V. Ball, M. Michel, F. Boulmedais, J. Hemmerle, Y. Haikel, P. Schaaf and J. C. Voegel, Cryst. Growth Des., 2006, 6, 327 CrossRef CAS.
- E. D. Spoerke and S. I. Stupp, Biomaterials, 2005, 26, 5120 CrossRef CAS.
- S. Schweizer, T. Schuster, M. Junginger, G. Siekmeyer and A. Taubert, Macromol. Mater. Eng., 2010, 295, 535 CrossRef CAS.
- M. Junginger, K. Kita-Tokarczyk, T. Schuster, J. Reiche, F. Schacher, A. H. E. Müller, H. Cölfen and A. Taubert, Macromol. Biosci., 2010, 10, 1084 CAS.
- F. Schacher, M. Müllner, H. Schmalz and A. H. E. Müller, Macromol. Chem. Phys., 2009, 210, 256 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS.
- B. H. Tan, C. S. Gudipati, H. Hussain, C. B. He, Y. Liu and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 1002 CrossRef CAS.
- F. Rehfeldt, R. Steitz, S. P. Armes, R. von Klitzing, A. P. Gast and M. Tanaka, J. Phys. Chem. B, 2006, 110, 9171 CrossRef CAS.
- L.-J. Zhang, H.-G. Liu, X.-S. Feng, R.-J. Zhang, L. Zhang, Y.-D. Mu, J.-C. Hao, D.-J. Qian and Y.-F. Lou, Langmuir, 2004, 20, 2243 CrossRef CAS.
- A. Peytcheva, H. Cölfen, H. Schnablegger and M. Antonietti, Colloid Polym. Sci., 2002, 280, 218 CrossRef CAS.
Footnotes |
| † Present address: Centre for Molecular Nanoscience, University of Leeds, Leeds, LS2 9JT, UK |
| ‡ Present address: Department of Chemical and Biotechnological Engineering, Université de Sherbrooke, QC J1H 5N4, Canada |
| § Present address: Institute of Organic Chemistry and Macromolecular Chemistry, University of Jena, D-07743 Jena, Germany |
| This journal is © The Royal Society of Chemistry 2010 |