Impact of the colloidal state on the oriented attachment growth mechanism
Cleocir José
Dalmaschio
a,
Caue
Ribeiro
b and
Edson Roberto
Leite
*a
aChemistry Department, Federal University of São Carlos, 13565-905São Carlos, SP, Brazil. E-mail: edson.leite@pq.cnpq.br; Tel: +55.16.3351.9567
bEMBRAPA Instrumentacao Agropecuária, 13569-970 São Carlos, SP, Brazil
First published on 8th September 2010
Abstract
In the last five years, several excellent reviews about oriented attachment (OA) have evidenced the advances achieved in this research area, detailing the growth mechanism and the kinetic models. The main focus of this review is to examine the dependence of the OA mechanism on the colloidal state and to demonstrate how the colloidal state modifies the OA mechanism. Basically, we can define two main possible approaches to achieve self-organization or mutual orientation of adjacent nanocrystals. One is the effective collision of particles with mutual orientation controlled by the number of collisions. This type of growth occurs in a well dispersed colloidal suspension and results in a statistical growth process. The second way is through coalescence induced by particle rotation. This mechanism must be dominant in a weakly flocculated colloidal state in which there is significant interaction among particles. This type of process leads to the formation of complex structures.
 Cleocir José Dalmaschio | Cleocir José Dalmaschio was born in 1983. He received his BSc degree in chemistry from the Federal Univeristy of Viçosa in 2006. Afterwards he joined the Chemistry Post-Graduation program at the Federal University of São Carlos (UFSCar), receiving his MSc degree in Physical Chemistry in 2008 under the supervision of Prof. Edson Roberto Leite. He is currently a doctoral candidate, in Physical Chemistry at UFSCar, researching the mechanisms and kinetics of oriented attachment. |
 Caue Ribeiro | Caue Ribeiro received his PhD in Physical Chemistry from the Federal University of São Carlos in 2005. After a period in the private sector, he joined the Brazilian Agricultural Research Corporation (Embrapa) as a full researcher, in the Agricultural Instrumentation Center in 2007. His primary research interests include the development of synthesis routes to produce heterogeneous catalysts (mainly inorganic nanoparticles) for water decontamination, investigation of the formation and crystallization mechanisms in nanoparticles and interactions of inorganic nanoparticles with plants and soil. |
 Edson Roberto Leite | Professor Edson Roberto Leite was born in 1965. He received his PhD in Material Science and Engineering in 1993 from the Materials Science and Engineering Department at the Federal University of São Carlos (with Prof. José Arana Varela). In 1998, he worked as visiting professor in the Materials Science and Engineering Department, Lehigh University. Currently, he is professor in the Chemistry Department, of the Federal University of São Carlos (Brazil). His research interest include the design and synthesis of nanostructured materials, fundamental research related to the nucleation and growth kinetics of nanocrystals and materials characterization by electron microscopy. |
1. Introduction
In the last two decades, the study and development of nanomaterials has made impressive progress, particularly in the chemical synthesis of inorganic nanocrystals via the colloidal process.1 This chemical approach allows nanocrystals with different sizes and morphologies to be obtained, as shown in Fig. 1a–b. Nanocrystals are not merely objects of nanoscale dimensions. Rather, nanocrystals are complex objects, as illustrated in Fig. 1c. We can observe that one of the constituents of an inorganic nanocrystal is a nanoparticle (with dimensions of 1 to 100 nm), with a single crystallographic domain. Moreover, due to their large proportion of surface atoms, nanocrystals present different types of organic and/or inorganic compounds or chemically or physically adsorbed ions. The presence of these compounds on the surface of nanocrystals is very important in controlling their size and shape, as well as in stabilizing and functionalizing the material.1,2 Another important point to be considered is the fact that during the genesis of nanocrystals by a colloidal approach, a phase-separation process takes place, which is generally associated with a nucleation and growth process.3–5 For didactic purposes, the nucleation and growth process can be divided into five steps, as illustrated in Fig. 2. The first step consists of the reaction of suitable precursors (monomers), which results in the desirable compound (i.e., metal atoms, a semiconductor or an oxide compound, for example). This compound will interact (step II), resulting in a cluster or monomer growth process. The growth mechanism that promotes the growth of the cluster is not yet established and steps I and II are reversible. When the cluster grows to a critical size (step III), the process becomes irreversible (thermodynamics condition) and crystal size can be controlled with the aid of stabilizers (step IV). To a first approximation, the shape of the crystal is controlled by energetic arguments (this is a thermodynamic condition). In fact, the cluster will grow in a geometric arrangement in order to minimize the surface energy. In general, the Wulff construction is an interesting way to predict the shape of a nanocrystal.6,7 In this step, one can interfere in the process by promoting, for instance, the preferential adsorption/desorption of a stabilizer, resulting in an anisotropic crystal.1,2,8 In fact, the size and shape of the nanocrystal can be controlled using a suitable combination of stabilizers and solvents. This process takes place in a kinetic condition. The last step (step V) consists of the interaction of the nanocrystals formed in step IV in order to produce larger structures. This nanocrystal-based self-assembly process is governed by particle–particle and particle–solvent interactions. In this step, the formation of an agglomerate (disordered assembly) of nanocrystals or even the formation of mesocrystals (mesoscopically structured crystals) may occur.9 Needless to say, the above described process is ideal. One of the major obstacles to achieving good control of the colloidal synthesis of nanocrystals is to separate the nucleation events from the growth process.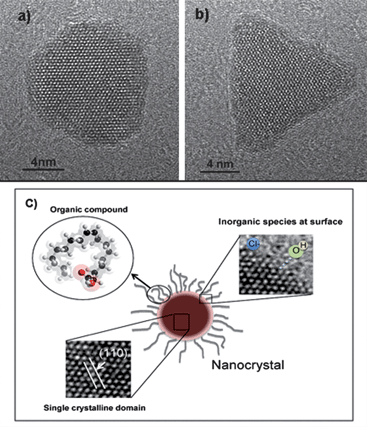 | ||
| Fig. 1 a) and b) High-magnification HRTEM images of magnetite nanocrystals with different morphologies. c) Schematic representation of a nanocrystal formed from an inorganic nanocrystal with an organic/inorganic compound bonded to its surface. | ||
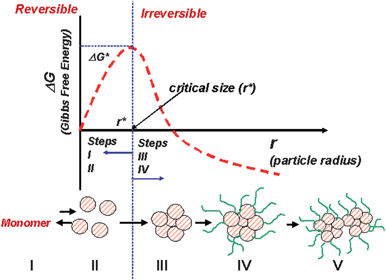 | ||
| Fig. 2 Schematic diagram of the nucleation and growth process showing the five stapes and the dependence of the Gibbs free energy (G) on the crystal size. Bellow a critical radius (r*), a reversible process occurs; for r > r* the process becomes irreversible. | ||
Based on this analysis, it is clear that the synthesis of a nanocrystal is not a simple task, because it requires control over the chemical composition and the purity, size, size distribution and shape of the crystallographic phase, as well as the chemical functionality of the nanocrystal surface.1,2,5 Today, the development of synthetic routes to produce nanocrystals with controlled size, size distribution and shape is directly correlated with the ability to control the nucleation and growth process.1,2,5,10
Detailed investigations of the growth process of nanocrystals in the colloidal state and an understanding of the parameters that affect crystal growth and shape development are fundamental to tailoring new types of nanostructured functional materials or even for the development of chemical protocols that allow for the processing of nanocrystals with high yield and reproducibility. The classical crystal growth mechanism, called the theory of Ostwald ripening (OR), is generally used to explain the crystal-growth process, in which the larger particles grow to the detriment of the smaller particles. The driving force of this process is the fact that the solubility of the particle (S) is related to its size, which can be described by the Ostwald–Freundlich equation:
S = S0![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) exp(4γSLVm/RTd) exp(4γSLVm/RTd) | (1) |
In the late 20th century, Penn and Banfield identified an important crystal growth mechanism in nanoparticles in colloidal dispersions. They called this new mechanism “oriented attachment (OA)” or “oriented aggregation”.13–15 The name adopted by the scientific community was OA. This mechanism is based on the spontaneous self-organization of adjacent nanocrystals, resulting in growth by the coalescence of solid particles that share a common crystallographic orientation. Nanocrystal growth via the OA mechanism generally leads to the formation of anisotropic nanocrystals and particles of irregular shapes,13–17 as well as nanocrystals with defects.15 Today, OA is a well established mechanism, which has been identified as the predominant growth mechanism during the synthesis or hydrothermal/solvothermal treatment of several materials.16,17
One of the characteristics of the OA mechanism not found in the OR mechanism is the presence of a solid–solid interface between the nanocrystals, indicating that the growth process begins only after contact is established between particles, as schematically illustrated in Fig. 3. A consequence of this characteristic is the strong dependence of the OA growth process on the colloidal state.
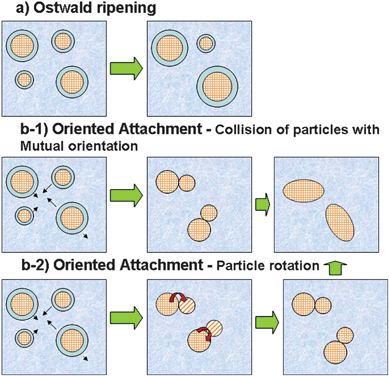 | ||
| Fig. 3 Schematic representation of nanocrystal growth controlled by a) OR and b) OA. | ||
Three different colloidal states18 can be defined, as illustrated in Fig. 4. In the dispersed state, the particles (or phase) in the suspension repel one another. In this state, the dominant potential energy among the particles is repulsive interaction. In weakly and strongly flocculated states, attractive interactions dominate the potential energy, and the difference between these states is the volume fraction of the dispersed phase. In the weakly flocculated state, particles aggregate in clusters or agglomerates in suspension at a volume fraction of particles (ϕ) below the gel point. In this case, it is possible to define an equilibrium separation distance between agglomerates or particles. In the strongly flocculated state, ϕ is higher than the gel point, resulting in a network of touching particles. In this state, strong sedimentation and phase separation is observed.
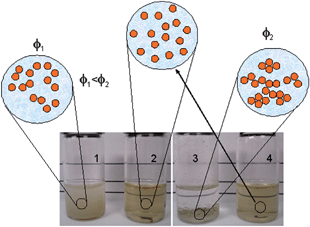 | ||
| Fig. 4 Schematic representation of the relationship between the visual aspect of the colloidal state and the resulting suspension structure. Vial 1 shows a weakly flocculated colloidal state, vials 2 and 4 show a dispersed colloidal state, and vial 3 shows a strongly flocculated state. | ||
Considering the above discussion, careful control of the interparticle forces can result in colloidal suspensions in one of the above described states. Thus, an analysis of these forces can help control the colloidal state.
Colloidal stability is controlled by the total interparticle potential energy (VT), which can be described as:
| VT = VvdW + Ve + Vsteric + Vstructural | (2) |
In the last five years, several excellent reviews about OA have described the advances achieved in this research area, detailing the growth mechanism and the kinetic models.16,19–21 The main focus of this review is to examine the dependence of the OA mechanism on the colloidal state and to demonstrate how the colloidal state modifies the OA mechanism. We also intend to establish energetic arguments that allow us to predict, to a first approximation through a qualitative and idealized description, the type of OA as well as the formation of mesocrystals.
A more in-depth analysis of growth mechanisms, such as OA, can shed light on the formation of anisotropic nanostructures as well as mesocrystals. The number of materials obtained by the OA process is growing rapidly16,19–21 and has become an attractive form of processing nanomaterials with anisotropic structures. Basically, we can define two main possible ways to bring about the self-organization or mutual orientation of adjacent nanocrystals. One is the effective collision of particles with mutual orientation, while the second is coalescence induced by particle rotation (see Fig. 3). The first situation must occur in the dispersed colloidal state, in which the number of collisions among particles is high. In the second situation, particle rotation must be dominant in a weakly flocculated colloidal state in which the interaction between particles is significant, although the nanoparticles still have rotational freedom.9,22 Here we observe that there is a close relationship between the OA mechanism and the colloidal state.
2. OA in the dispersed colloidal state
The dispersed colloidal state is kinetically stable and the repulsive forces are dominant. In this colloidal state, the OA growth rate is related to the effective collision rate among the nanocrystals in suspension and to the reduction of surface energy, aimed at minimizing the area of high-energy faces.23,24 An effective collision is one that occurs when particles collide, producing an irreversibly oriented attachment. This occurs only if their orientations at the time of collision achieve a congruent two-dimensional structure at the interface.25,26 Collision among particles with different crystallographic orientations will not be effective and will not result in irreversible attachment. The schematic in Fig. 5 shows the difference between effective and non-effective collision of nanocrystals in a dispersed colloid.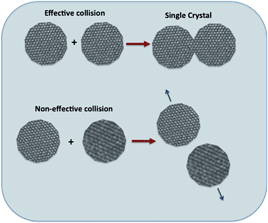 | ||
| Fig. 5 Schematic representation of effective and non-effective collisions between particles. | ||
It is well established that oriented attachment can promote the growth of zero-dimensional (0D), one-dimensional (1D), two-dimensional (2D) and three-dimensional self-assembled nanocrystals.27 In the case of OA controlled by collision, one will basically observe the growth of 0D (spheres, cubes or polyhedra) and 1D structures (rods and wires). In this situation, growth occurs through the interaction of 0D with 0D structures, or even through the interaction of 0D with 1D and 1D with 1D structures.9,28 The high-magnification HRTEM image in Fig. 6 illustrates the growth of a 1D structure from a 0D one, forming an anisotropic nanoparticle (a 1D rod formed by two primary particles) of Sb-doped SnO2 by OA. This process was observed in a low concentration of solids (organic solvent) in a well dispersed colloidal state.
 | ||
| Fig. 6 High-magnification HRTEM image showing the growth of a 1D structure from a 0D structure for Sb-doped SnO2 nanocrystals in the dispersed colloidal state (organic solvent). | ||
The OA process that occurs in the dispersed colloidal state can be considered to be a statistical process controlled by collision frequency. As a consequence of this statistical nature, during the growth process one can observe isotropic and anisotropic nanocrystals with different growth directions. For instance, in colloidal SnO2 nanocrystals, the formation of worm-like particles has been observed growing in the [001] and [110] directions, as well as the formation of equiaxed particles.22,23
The statistical nature of OA in the dispersed colloidal state is highly evident when the nanocrystals are faceted. For instance, the HRTEM image of Fig. 7 shows the formation of OA particles with different morphologies, which originate from the sharing of different crystal facets. Today, it can be stated that the OA mechanism controlled by effective collision is related basically to the number and area of the facets in a nanocrystal. In a recent work, Stroppa and co-authors29 showed a clear correlation between the number and area of facets in faceted nanocrystals of Sb-doped SnO2 and the OA process controlled by collisions in a dispersed system.
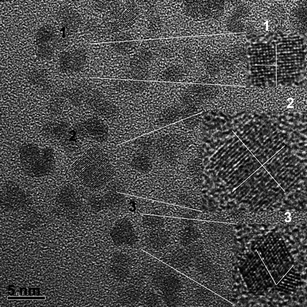 | ||
| Fig. 7 HRTEM image of Sb-doped SnO2 nanocrystals. The insets show in detail the growth process evolving different facets. | ||
2.1 Oriented attachment in the dispersed colloidal state: A quantitative description
To explain the growth behavior in SnO2 colloidal suspensions, Ribeiro et al.25 proposed that coalescence in dispersed suspensions may occur when particles with similar crystallographic orientations (or with slight differences) collide (effective collision). This mechanism is based on the assumption that nanoparticles dispersed in a liquid medium present a very high degree of freedom for rotational and translational motion. Hence, in suspensions in which agglomeration does not take place, growth by means of oriented collision should be more effective than by surface mechanisms (i.e., coalescence induced by relative rotations between particles in contact with each other). Well-dispersed nanoparticles should have a high velocity due to Brownian motion. Hence, it is expected that nanocrystals in suspension undergo a high frequency of collisions. Therefore, growth by coalescence may be interpreted statistically. This mechanism is similar to Smoluchowski's coagulation model,30,31 which is used extensively to explain polycrystalline colloidal growth and aggregation mechanisms in suspension.Assuming that all the above mentioned considerations are valid, the coalescence of two particles in suspension may be described by the following chemical eqn (3):
| A + A → B | (3) |
Based on the argument that nanoparticles act as molecules, it is important to evaluate the collision frequency of the particles. This can be evaluated by assuming that Brownian motion in dilute suspensions can be described by Maxwell–Boltzmann statistics. In this model, the collision frequency is evaluated analogously to the kinetics of gas molecules. It is important to analyze this frequency because it can give us the number of collisions. For instance, let us assume that the collision frequency (z) is given by:
| z = √(2πd2ūNV−1) | (4) |
| ū = √(3kBT/m) | (5) |
Based on the collision rate and defining a collision cross section as 3r, where r is the nanocrystal radius, we can write the following kinetics equation to describe the OA growth process in a well-dispersed colloidal suspension (a detailed description of this model can be found in ref. 25):
| r3eq − r3i = {[NkBT/η[A]0t]/[1 + NkBT/η[A]0t]} × r3i | (6) |
The model showed good applicability to SnO2 nanoparticles in hydrothermal conditions,25,40 since this system clearly grows only by this mechanism. However, in other systems such as CdSe or InAs,25 the equation did not fit the experimental data well. The deviations in the data may be explained, in part, by the presence of the OR mechanism associated to the OA mechanism in these nanomaterials. It should be pointed out that it is interesting to analyze the results of particle growth as a function of r3, since this enables an evaluation of the growth processes in terms of mean particle volumes. Consequently, it is reasonable to assume that the contributions to particle growth from Ostwald ripening and oriented attachment mechanisms should be correlated. Considering that coalescence predominates in initial stages and that Ostwald ripening can take place later, one can suppose that the initial radius for coarsening can be given by eqn (6), i.e., req = ri. Thus, particle growth may be described by:
| r3 − r3eq = [(8γV2mc∞)/(54aπηN)] × t | (7) |
In recent years, a multistep OA kinetics model has been developed.28 In this model, collision and coalescence may occur between any of two multilevel nanocrystals in the system. As the size of the nanocrystal increases, the collision cross-section of the particle enlarges and the particle velocity decreases. Putting these two facts together, a rapid decrease in the growth rate is observed, until finally the process ends. In the multistep kinetics model, the collision rate is an important parameter and consequently the well-dispersed colloidal state is a more appropriate environment to observe the occurrence of OA between multilevel particles. This model is also consistent with Smoluchowski's coagulation model and has been applied to describe the growth process of ZnS.
3. OA in the weakly flocculated colloidal state
In a weakly flocculated colloidal state, the OA process is not controlled by collisions between particles, but is dominated by the particle’s medium- and short-range interactions. In terms of energetic arguments, this is a condition in which the attractive forces between particles, such as van der Waals and structural forces, are higher than repulsive forces (electrostatic and steric mainly). This energetic condition is normally observed in weakly and strongly flocculated colloidal states. Since rotational freedom between particles is required to achieve crystallographic alignment, the weakly flocculated state is more appropriate.In these growth conditions, OA occurs between primary particles (0D with 0D interaction) resulting in 1D or 2D structures. In the second stage, the 1D and 2D structures can interact, forming 3D structures. In fact, the growth mechanism controlled by OA in this colloidal state follows a hierarchical process and can present mesocrystals as an intermediate state, i.e., a crystal formed by oriented assemblies of nanocrystals.49 This is a very interesting situation, because it enables one to grow mesocrystals with exposed facets not controlled by thermodynamic surface energy, or predicted by the Wulff construction, but controlled kinetically. Consequently, functional materials can be developed with different or even unique chemical, electrical and magnetic behavior. Fig. 9 describes a typical hierarchical growth process whereby TiO2 anatase nanocrystals assemble in larger agglomerates (Fig. 9a) via OA (see detail in the inset). With increased treatment time, one can observe the formation of well defined 3D mesocrystals (Fig. 9b) with low porosity. The inset of Fig. 9b shows the Fast-Fourer transform (FFT) of the image with reflections typical of a single crystal. It should be noted that this mesocrystal growth process occurs in a weakly flocculated colloidal state.
 | ||
| Fig. 9 HRTEM image showing the assembly of TiO2 nanocrystals resulting in the formation of anatase mesocrystals: a) short treatment time in solvothermal conditions. The inset shows features typical of the OA process; b) long treatment time in the same solvothermal conditions. The inset shows the FFT of the region indicated by the arrow. | ||
The statistical model used in a well-dispersed colloidal system cannot explain the mesocrystal growth process depicted in Fig. 9. Therefore, a different approach is needed in order to understand this ordered assembly of nanocrystals. By analogy with biological and molecular systems, the 1D, 2D and 3D morphologies attained by OA can be described by non-covalent interactions, i.e., by interactions with much weaker energy than that of covalent bonds (ca. 100–150kBT), in the range 0.1–10kBT. In the weakly flocculated state, the repulsive forces may be negligible; hence, the van der Waals (vdW) forces will be dominant. The vdW forces can be seen as resulting from three or more additive terms: the Keeson force, the Debye force, the charge–dipole interaction force and London dispersion (LD) forces. Table 1 lists common types of interaction forces between atoms, ions and molecules, and its potential energy dependence regarding the interparticle distance (L) and temperature (T).50
| Type of Interaction | Interparticle distance dependence of the potential energy | Temperature dependence of the potential energy |
|---|---|---|
| Charge–dipole | ∝L−4 | ∝T−1 |
| Dipole–dipole (Keeson) | ∝L−6 | ∝T−1 |
| Dipole-induced-dipole (Debye) | ∝L−6 | Not dependent |
| Induced dipole–induced dipole (LD) | ∝L−6 | ∝T |
In some molecules, as well as in ionic and covalent crystals, the atomic arrangement of the ions or atoms can induce a permanent dipole. In the case of crystals, this dipole can be associated with a specific crystallographic orientation. Permanent dipoles, charges or external electrical fields can also induce dipoles, for instance. These dipoles can interact not only with one another but also with charges, particularly in aqueous solution. It is not a simple task to analyze dipole–dipole and charge–dipole interactions; moreover, in solution, one must consider thermal fluctuations. However, the dipole–charge (〈V〉d–c) and dipole–dipole (〈V〉d–p) potentials (Keeson) can be described in a simplified way as:
| 〈V〉d–c = −c2q2p2/3kBTL4 | (8) |
| 〈V〉d–p = −⅔(c2p21p22/3kBTL6) | (9) |
A direct correlation between dipole interactions and nanocrystal and mesocrystal growth has been demonstrated in the literature.53,54 In their classic work,53 Giersig and co-authors described the spontaneous organization of single CdTe nanocrystals into 1D nanostructures (nanowires), attributing this spontaneous interaction to dipole–dipole attraction. In their work, they estimated the potential attractive energy to be ∼10 kJ mol−1. One of the key steps in the preparation of the 1D nanostructure was the removal of excessive stabilization through the precipitation of nanocrystals by the addition of methanol. This experiment was performed at room temperature. In a recent study, Kotov and co-authors54 showed that the dipole moment, a small positive charge, and hydrophobic attraction are the driving forces for the self-organization of CdTe into 2D structures resembling the assembly of surface layer (S-layer) proteins.54
Leite and coworkers55 demonstrated that the use of microwave (MW) heating during the growth process reduces the treatment time required to obtain anisotropic nanostructures. They reported that OA is the dominant mechanism responsible for the growth process, implying that it could be possible to introduce MW irradiation as a parameter to control the anisotropic growth of crystals.55 The authors attributed the shorter treatment time to the increase of the effective collision rate, i.e., the presence of attractive forces. A plausible explanation for the increase of the effective collision rate under microwave irradiation is the increase in the collision cross-section (σ) of the particle. In a previous article,25 Leite's group made a detailed kinetic analysis of the OA mechanism, in which it was assumed that a non-interactive collision occurred with a collision cross-section (σ), σ = 3r, where r is the particle's radius. Under microwave irradiation, it was assumed that an interactive collision occurred which introduced a steric factor (P), expressing the interactive cross-section (σ*) as a multiple of σ:
| σ* = Pσ | (10) |
From the above discussion, it is clear that the colloidal state strongly influences the OA growth process. For a better description of this dependence, one can use an energetic argument to define when OA will be controlled by the collision rate or by interacting nanoparticles. For a simplified description of the interparticle interaction and thermal fluctuation, we will consider only the kinetic energy (KE) of an ideal gas and the attractive potential. At first glance, these considerations may be too drastic for a real system; however, our goal is to come up with a description that allows for a qualitative and phenomenological analysis of the OA process.
Considering eqn (5), we can estimate the KE of one particle, using the following equation:
| KE = 1.5kBT | (11) |
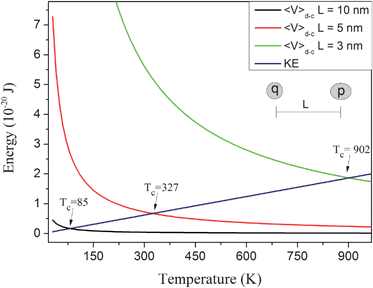 | ||
| Fig. 10 KE and the modulus of 〈V〉d–c as function of temperature, considering different L values. | ||
Analyzing Fig. 10, one can also see that different interparticle distances (L) result in different Tc. In fact, Tc is a term that must be dependent on other parameters besides L. Tc can be defined as:
| Tc = (cpq)/√1.5kBL2 | (12) |
| Tc = C′σ*[A]n′/r | (13) |
This qualitative analysis is consistent with experimental results obtained recently in our laboratory for the synthesis of gadolinium-doped cerium oxide (GCO) nanorods. As the scanning electron micrographs in Fig. 11 indicate, decreasing the growth temperature increases the yield of nanorods. At 40 °C one can observe a large number of nanorods (Fig. 11a). Upon changing the temperature, one can see differences in rod size and concentration. At 200 °C, one can no longer see the formation of rods, but only equiaxed nanoparticles. A HRTEM characterization of GCO nanorod, growth at 40 °C, shows features typical of OA process (Fig. 12). In this figure, we can observe the presence of a mesocrystal-like structure, with a clear presence of the boundary between oriented primary particles, as shown in the reconstructed lattice image of the Fig. 12c. We can observe also an irregular rod surface. Fig. 13 shows HRTEM images of the materials treated at higher temperature. Increasing the growth temperature to 130 °C, the presence of a grain boundary is not evident, indicating the formation of single-crystalline nanorods with a smooth surface (a self-recrystallization process) (Fig. 13a). Actually, this observation indicates that the temperature is an important parameter to promote grain boundary elimination and surface reconstruction, suggesting that a thermally activated process, such as diffusion, controls both phenomena. High-magnification HRTEM images of the GCO growth at 200 °C (Fig. 13b) show features typical of the OA process induced by effective collision between nanocrystals.
 | ||
| Fig. 11 Secondary electron image of the GCO nanorods growth under MW irradiation at several temperatures (for experimental details about the synthesis, see ref. 55). a) at 40 °C, b) at 130 °C, c) at 200 °C. | ||
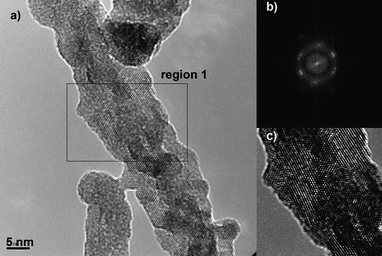 | ||
| Fig. 12 HRTEM images of GCO growth at 40 °C. a) High-magnification HRTEM image and b) FFT of region 1, indicated in a). c) reconstructed lattice image of region 1. | ||
 | ||
| Fig. 13 High-magnification HRTEM images of the GCO nanorods. a) growth at 130 °C (the inset shows a FFT of region A), b) growth at 200 °C (the arrows indicate typical features of the OA process). | ||
Since the initial concentration of nanocrystal, treatment time, ionic strength and pH were kept constant during the growth experiment; the morphological modifications are related mostly to temperature variations. Based on these experimental results, we can postulate that increasing the temperature causes the thermal fluctuation to increase (mainly the KE), resulting in a statistical growth process controlled by the collision rate. Low temperatures favor the predominance of attractive interactions, leading to the formation of nanorods (mesocrystals). This analysis is entirely consistent with the idea of a transition temperature between a growth regime controlled by interaction and a regime controlled by statistical collision.
The next challenger in the study of OA is the proposal of a quantitative kinetic model to describe this growth mechanism in a weakly flocculated state. Of course, this it is not a simple endeavor since such a model must include the attractive potential between nanocrystals. In fact, the first step in this direction has already been taken. In his paper about a kinetic model, Penn26 proposed to correlate the rate constant with an attractive potential between nanoparticles. This proposal may be a good starting point, but the model requires further improvement in order to obtain a representative kinetic equation.
4. Summary
This paper presented a new vision of the OA mechanism, including the classification of OA into two different types of process. Basically, we can define two main possible approaches to achieve the self-organization or mutual orientation of adjacent nanocrystals. One is an effective collision of particles with mutual orientation controlled by the number of collisions. This type of growth occurs in a well dispersed colloidal suspension (a kinetically stable suspension) and results in a statistical growth process with a low level of control. The temperature controls basically the collision frequency and consequently the coalescence between nanoparticles. In the second approach, coalescence is induced by particle rotation. The particle rotation must be dominant in a weakly flocculated colloidal state in which particle interaction is significant, although the nanoparticles still have rotational freedom. In this type of process, we will observe the formation of complex structures and a good level of control over their shape and size. In the weakly flocculated state, the temperature is important to promote the grain boundary migration and surface reconstruction, after the attachment of particles with the same crystallographic orientation.Acknowledgements
Financial support for this study was provided by FAPESP, FINEP and CNPq, all Brazilian agencies. The authors also thank many of their colleagues for stimulating discussions and collaboration, especially Prof. Elson Longo and Prof. Jose Arana Varela.References
- Y. Yin and A. P. Alivisatos, Colloidal nanocrystal synthesis and the organic–inorganic interface, Nature, 2005, 437, 664–670 CrossRef CAS.
- I.-W. Jun, J.-S. Choi and J. Cheon, Shape control of semiconductor and metal oxide nanocrystals through nonhydrolytic colloidal routes, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS.
- V. K. Lamer and R. H. Dinegar, Theory, Production and Mechanism of Formation of Monodispersed Hydrosols, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- J. Turkevich and G. Kim, Palladium - Preparation and Catalytic Properties of Particles of Uniform Size, Science, 1970, 169, 873–879 CrossRef CAS.
- E. R. Leite, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, Stevenson Ranch, 2004, vol. 6, pp 537–554 Search PubMed.
- S. M. Allen, E. L. Thomas, The Structure of Materials, John Wiley & Sons, New York, 1999, p 447 Search PubMed.
- C. Herring, Some Theorems on the Free Energies of Crystal Surfaces, Phys. Rev., 1951, 82, 87–93 CrossRef CAS.
- J. Polleux, N. Pinna, M. Antonietti and M. Niederberger, Ligand-directed assembly of preformed titania nanocrystals into highly anisotropic nanostructures, Adv. Mater., 2004, 16, 436–439 CrossRef CAS.
- H. Colfen, M. Antonietti, Mesocrystals and Nonclassical Crystallization, John Wiley & Sons, Hoboken, 2008, p 288 Search PubMed.
- G. Garnweitner and M. Niederberger, Organic chemistry in inorganic nanomaterials synthesis, J. Mater. Chem., 2008, 18, 1171–1182 RSC.
- C. Wagner, Theorie Der Alterung Von Niederschlagen Durch Umlosen (Ostwald–Reifung), Z. Elektrochem., 1961, 65, 581–591 CAS.
- I. M. Lifshitz and V. V. Slyozov, The Kinetics of Precipitation from Supersaturated Solid Solutions, J. Phys. Chem. Solids, 1961, 19, 35–50 CrossRef.
- R. L. Penn and J. F. Banfield, Oriented attachment and growth, twinning, polytypism, and formation of metastable phases: Insights from nanocrystalline TiO2, Am. Mineral., 1998, 83, 1077–1082 CAS.
- R. L. Penn and J. F. Banfield, Morphology development and crystal growth in nanocrystalline aggregates under hydrothermal conditions: Insights from titania, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Imperfect oriented attachment: Dislocation generation in defect-free nanocrystals, Science, 1998, 281, 969–971 CrossRef CAS.
- H. Cölfen and M. Antonietti, Mesocrystals: Inorganic superstructures made by highly parallel crystallization and controlled alignment, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef.
- M. Niederberger and H. Colfen, Oriented attachment and mesocrystals: Non-classical crystallization mechanisms based on nanoparticle assembly, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- J. A. Lewis, Colloidal processing of ceramics, J. Am. Ceram. Soc., 2004, 83, 2341–2359 CrossRef.
- Q. Zhang, S. J. Liu and S. H. Yu, Recent advances in oriented attachment growth and synthesis of functional materials: concept, evidence, mechanism, and future, J. Mater. Chem., 2009, 19, 191–207 RSC.
- J. Zhang, F. Huang and Z. Lin, Progress of nanocrystalline growth kinetics based on oriented attachment, Nanoscale, 2010, 2, 18–34 RSC.
- N. D. Burrows, V. M. Yuwono and R. L. Penn, Quantifying the Kinetics of Crystal Growth by Oriented Aggregation, MRS Bulletin, 2010, 35, 133–137 CAS.
- E. J. H. Lee, C. Ribeiro, E. Longo and E. R. Leite, Oriented attachment: An effective mechanism in the formation of anisotropic nanocrystals, J. Phys. Chem. B, 2005, 109, 20842–20846 CrossRef CAS.
- E. R. Leite, T. R. Giraldi, F. M. Pontes, E. Longo, A. Beltran and J. Andres, Crystal growth in colloidal tin oxide nanocrystals induced by coalescence at room temperature, Appl. Phys. Lett., 2003, 83, 1566–1568 CrossRef CAS.
- J. Polleux, N. Pinna, M. Antonietti, C. Hess, U. Wild, R. Schlogl and M. Niederberger, Ligand functionality as a versatile tool to control the assembly behavior of preformed titania nanocrystals, Chem.–Eur. J., 2005, 11, 3541–3551 CrossRef CAS.
- C. Ribeiro, E. J. H. Lee, E. Longo and E. R. Leite, A kinetic model to describe nanocrystal growth by the oriented attachment mechanism, ChemPhysChem, 2005, 6, 690–696 CrossRef CAS.
- R. L. Penn, Kinetics of oriented aggregation, J. Phys. Chem. B, 2004, 108, 12707–12712 CrossRef CAS.
- H. G. Yang and H. C. Zeng, Self-construction of hollow SnO2 octahedra based on two-dimensional aggregation of nanocrystallites, Angew. Chem., Int. Ed., 2004, 43, 5930–5933 CrossRef CAS.
- J. Zhang, Z. Lin, Y. Z. Lan, G. Q. Ren, D. G. Chen, F. Huang and M. C. Hong, A multistep oriented attachment kinetics: Coarsening of ZnS nanoparticle in concentrated NaOH, J. Am. Chem. Soc., 2006, 128, 12981–12987 CrossRef CAS.
- D. G. Stroppa, L. A. Montoro, A. Beltran, T. G. Conti, R. O. da Silva, J. Andres, E. Longo, E. R. Leite and A. J. Ramirez, , Unveiling the Chemical and Morphological Features of Sb-SnO2 Nanocrystals by the Combined Use of High-Resolution Transmission Electron Microscopy and ab Initio Surface Energy Calculations, J. Am. Chem. Soc., 2009, 131, 14544–14548 CrossRef CAS.
- M. von Smoluchowski, Versuch einer Mathematischen Theorie der Koagulations-Kinetiks. Kolloider Lösungen, Z. Physik. Chem., 1917, 92, 129–168 Search PubMed.
- D. Mozyrsky and V. Privman, Diffusional growth of colloids, J. Chem. Phys., 1999, 110, 9254–9258 CrossRef CAS.
- F. Huang, H. Z. Zhang and J. F. Banfield, The role of oriented attachment crystal growth in hydrothermal coarsening of nanocrystalline ZnS, J. Phys. Chem. B, 2003, 107, 10470–10475 CrossRef CAS.
- F. Huang, H. Z. Zhang and J. F. Banfield, Two-stage crystal-growth kinetics observed during hydrothermal coarsening of nanocrystalline ZnS, Nano Lett., 2003, 3, 373–378 CrossRef CAS.
- F. Huang, B. Gilbert, H. Zhang, M. P. Finnegan, J. R. Rustad, C. S. Kim, G. A. Waychunas and J. F. Banfield, Interface interactions in nanoparticle aggregates, Geochim. Cosmochim. Acta, 2004, 68, 127–254 CrossRef.
- T. O. Drews, M. A. Katsoulakis and M. Tsapatsis, A mathematical model for crystal growth by aggregation of precursor metastable nanoparticles, J. Phys. Chem. B, 2005, 109, 23879–23887 CrossRef CAS.
- T. O. Drews and M. Tsapatsis, Model of the evolution of nanoparticles to crystals via an aggregative growth mechanism, Microporous Mesoporous Mater., 2007, 101, 97–107 CrossRef CAS.
- C. Ribeiro, E. J. H. Lee, E. Longo and E. R. Leite, Oriented attachment mechanism in anisotropic nanocrystals: A “polymerization” approach, ChemPhysChem, 2006, 7, 664–670 CrossRef CAS.
- H. Z. Zhang and J. F. Banfield, Kinetics of crystallization and crystal growth of nanocrystalline anatase in nanometer-sized amorphous titania, Chem. Mater., 2002, 14, 4145–4154 CrossRef CAS.
- E. R. Leite, C. Vila, J. Bettini and E. Longo, Synthesis of niobia nanocrystals with controlled morphology, J. Phys. Chem. B, 2006, 110, 18088–18090 CrossRef CAS.
- E. J. H. Lee, C. Ribeiro, E. Longo and E. R. Leite, Growth kinetics of tin oxide nanocrystals in colloidal suspensions under hydrothermal conditions, Chem. Phys., 2006, 328, 229–235 CrossRef CAS.
- S. A. Kukushkin and A. V. Osipov, Kinetics of first-order phase transitions in the asymptotic stage, J. Exp. Theor. Phys., 1998, 86, 1201–1208 CrossRef.
- N. S. Pesika, Z. S. Hu, K. J. Stebe and P. C. Searson, Quenching of growth of ZnO nanoparticles by adsorption of octanethiol, J. Phys. Chem. B, 2002, 106, 6985–6990 CrossRef CAS.
- G. Oskam, Z. S. Hu, R. L. Penn, N. Pesika and P. C. Searson, Coarsening of metal oxide nanoparticles, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 66, 011403–4 CrossRef.
- Z. S. Hu, G. Oskam, R. L. Penn, N. Pesika and P. C. Searson, The influence of anion on the coarsening kinetics of ZnO nanoparticles, J. Phys. Chem. B, 2003, 107, 3124–3130 CrossRef CAS.
- G. Oskam, A. Nellore, R. L. Penn and P. C. Searson, The growth kinetics of TiO2 nanoparticles from titanium(IV) alkoxide at high water/titanium ratio, J. Phys. Chem. B, 2003, 107, 1734–1738 CrossRef CAS.
- Z. S. Hu, G. Oskam and P. C. Searson, Influence of solvent on the growth of ZnO nanoparticles, J. Colloid Interface Sci., 2003, 263, 454–460 CrossRef CAS.
- Z. S. Hu, D. J. E. Ramirez, B. E. H. Cervera, G. Oskam and P. C. Searson, Synthesis of ZnO nanoparticles in 2-propanol by reaction with water, J. Phys. Chem. B, 2005, 109, 11209–11214 CrossRef CAS.
- H. M. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Observation of Single Colloidal Platinum Nanocrystal Growth Trajectories, Science, 2009, 324, 1309–1312 CrossRef CAS.
- V. M. Yuwono, N. D. Burrows, J. A. Soltis and R. L. Penn, Oriented Aggregation: Formation and Transformation of Mesocrystal Intermediates Revealed, J. Am. Chem. Soc., 2010, 132, 2163–2165 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 1992 Search PubMed.
- V. A. Parsegian and B. W. Ninham, Temperature-Dependent Vanderwaals Forces, Biophys. J., 1970, 10, 664–673 CrossRef CAS.
- L. Bergstrom, Hamaker constants of inorganic materials, Adv. Colloid Interface Sci., 1997, 70, 125–169 CrossRef CAS.
- Z. Y. Tang, N. A. Kotov and M. Giersig, Spontaneous organization of single CdTe nanoparticles into luminescent nanowires, Science, 2002, 297, 237–240 CrossRef CAS.
- Z. Y. Tang, Z. L. Zhang, Y. Wang, S. C. Glotzer and N. A. Kotov, Self-assembly of CdTe nanocrystals into free-floating sheets, Science, 2006, 314, 274–278 CrossRef CAS.
- M. Godinho, C. Ribeiro, E. Longo and E. R. Leite, Influence of microwave heating on the growth of gadolinium-doped cerium oxide nanorods, Cryst. Growth Des., 2008, 8, 384–386 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |